
Uterine Microbiota Composition in Dairy Cows with Different Vaginal Discharge Scores: Suggesting Caviibacter as a Potential Pathogen in Mild Purulent Metritis
 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Feeding Management
2.2. Test Grouping
2.3. Sample Collection
2.4. DNA Extraction and Sequencing
2.5. Bioinformatics Analysis
3. Results
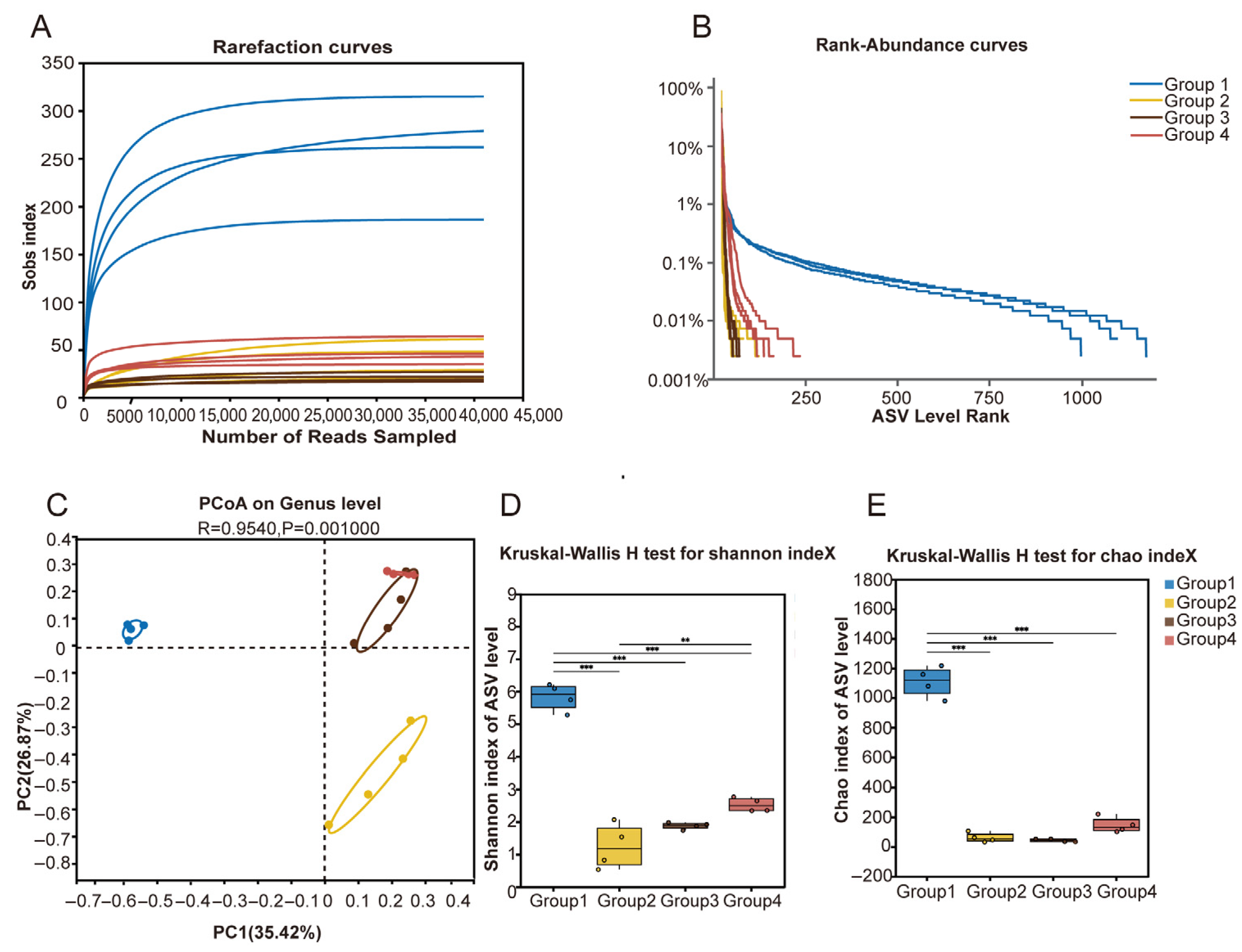
3.1. Differences in Uterine Microbial Diversity and Richness Across Different Vaginal Discharge Scores
3.2. Differences in Uterine Microbial Composition Across Different Vaginal Discharge Scores
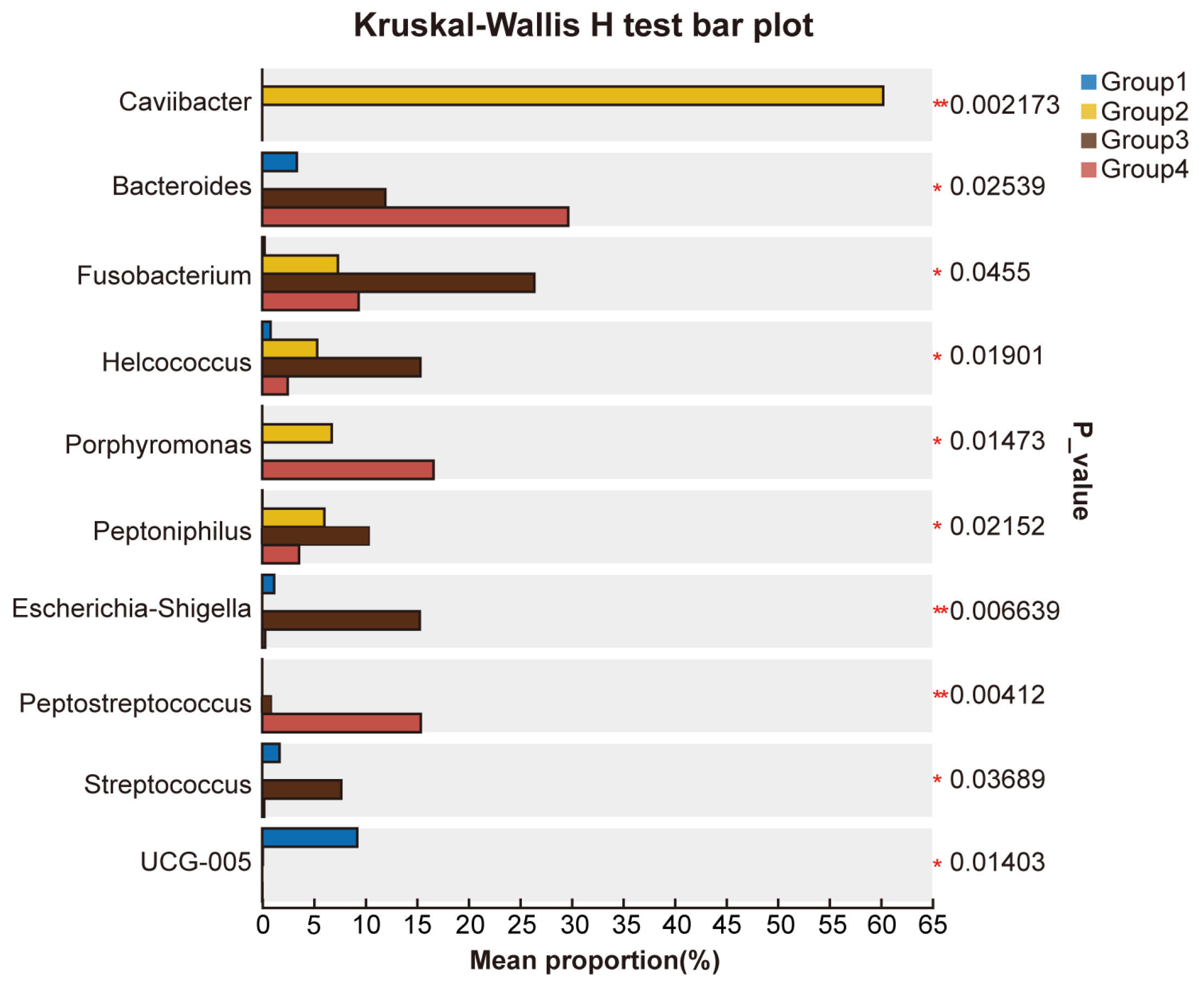
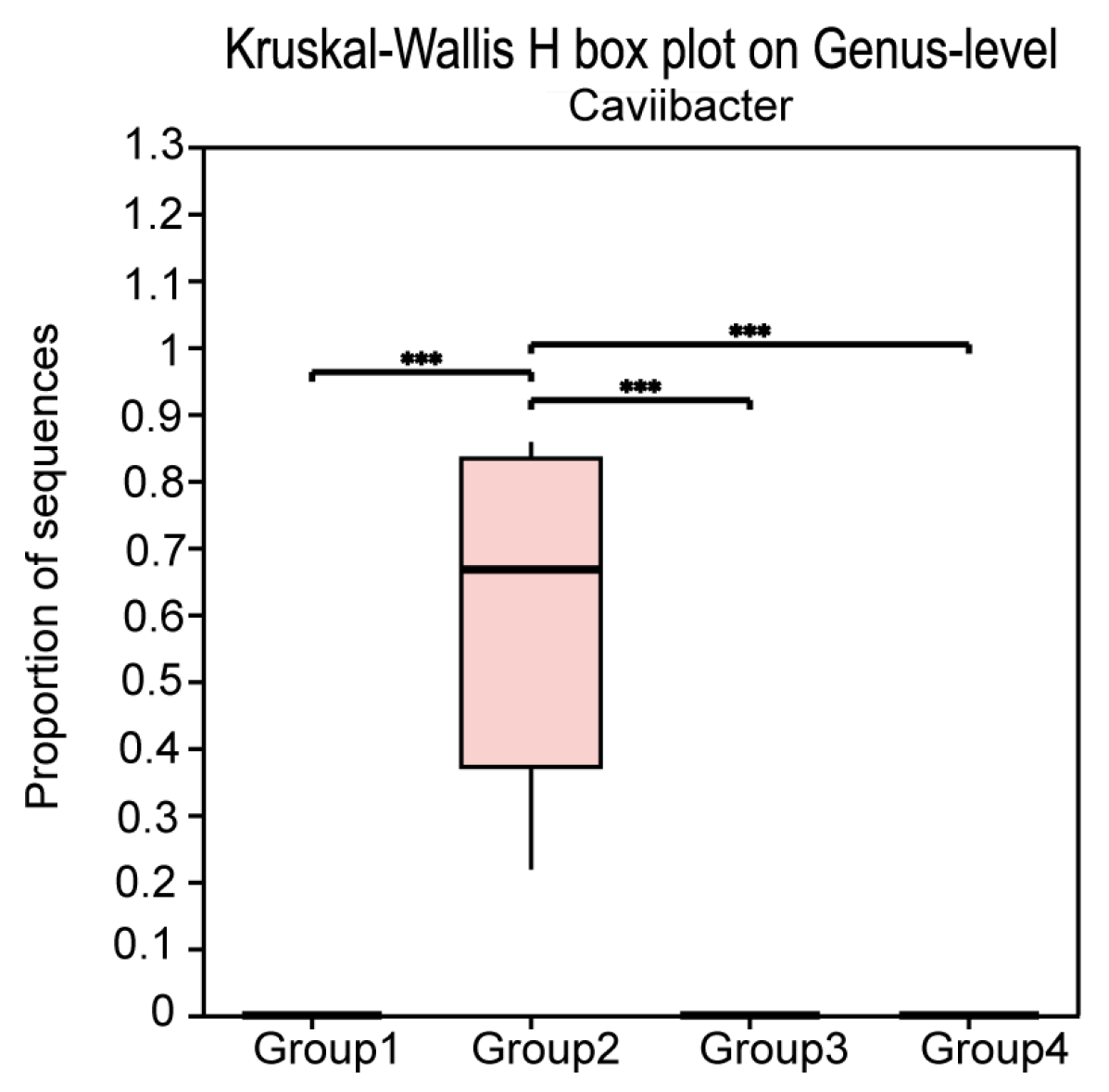
3.3. Differential Microbial Communities in the Uterus of Cows with Different Vaginal Discharge Scores
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pascottini, O.B.; LeBlanc, S.J.; Gnemi, G.; Leroy, J.L.; Opsomer, G. Genesis of clinical and subclinical endometritis in dairy cows. Reproduction 2023, 166, R15–R24. [Google Scholar] [CrossRef] [PubMed]
- Miranda-CasoLuengo, R.; Lu, J.; Williams, E.J.; Miranda-CasoLuengo, A.A.; Carrington, S.D.; Evans, A.C.; Meijer, W.G. Delayed differentiation of vaginal and uterine microbiomes in dairy cows developing postpartum endometritis. PLoS ONE 2019, 14, e0200974. [Google Scholar] [CrossRef] [PubMed]
- Druker, S.A.; Sicsic, R.; van Straten, M.; Goshen, T.; Kedmi, M.; Raz, T. Cytological endometritis diagnosis in primiparous versus multiparous dairy cows. J. Dairy Sci. 2022, 105, 665–683. [Google Scholar] [CrossRef] [PubMed]
- Baker, J.M.; Chase, D.M.; Herbst-Kralovetz, M.M. Uterine microbiota: Residents, tourists, or invaders? Front. Immunol. 2018, 9, 208. [Google Scholar] [CrossRef] [PubMed]
- Figueiredo, C.C.; Monteiro, H.F.; Cunha, F.; Bisinotto, D.Z.; Ruiz, A.R.; Duarte, G.A.; Ge, Y.; Lima, F.S.; Mohamadzadeh, M.; Galvão, K.N.; et al. Shifts in uterine microbiome associated with pregnancy outcomes at first insemination and clinical cure in dairy cows with metritis. Sci. Rep. 2024, 14, 11864. [Google Scholar] [CrossRef] [PubMed]
- Moore, S.; Ericsson, A.; Poock, S.; Melendez, P.; Lucy, M. Hot topic: 16S rRNA gene sequencing reveals the microbiome of the virgin and pregnant bovine uterus. J. Dairy Sci. 2017, 100, 4953–4960. [Google Scholar] [CrossRef] [PubMed]
- Tasara, T.; Meier, A.B.; Wambui, J.; Whiston, R.; Stevens, M.; Chapwanya, A.; Bleul, U. Interrogating the diversity of vaginal, endometrial, and fecal microbiomes in healthy and metritis dairy cattle. Animals 2023, 13, 1221. [Google Scholar] [CrossRef] [PubMed]
- Galvão, K.N.; Bicalho, R.C.; Jeon, S.J. Symposium review: The uterine microbiome associated with the development of uterine disease in dairy cows. J. Dairy Sci. 2019, 102, 11786–11797. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Wang, Y.; Hang, S.; Zhu, W. Microbial diversity in uterus of healthy and metritic postpartum Holstein dairy cows. Folia Microbiol. 2013, 58, 593–600. [Google Scholar] [CrossRef] [PubMed]
- Bicalho, M.; Machado, V.; Higgins, C.; Lima, F.; Bicalho, R. Genetic and functional analysis of the bovine uterine microbiota. Part I: Metritis versus healthy cows. J. Dairy Sci. 2017, 100, 3850–3862. [Google Scholar] [CrossRef] [PubMed]
- Silva, J.C.; Caldeira, M.O.; Moraes, J.G.; Sellmer Ramos, I.; Gull, T.; Ericsson, A.C.; Poock, S.E.; Spencer, T.E.; Lucy, M.C. Metritis and the uterine disease microbiome are associated with long-term changes in the endometrium of dairy cows. Biol. Reprod. 2024, 111, 332–350. [Google Scholar] [CrossRef] [PubMed]
- Sicsic, R.; Goshen, T.; Dutta, R.; Kedem-Vaanunu, N.; Kaplan-Shabtai, V.; Pasternak, Z.; Gottlieb, Y.; Shpigel, N.Y.; Raz, T. Microbial communities and inflammatory response in the endometrium differ between normal and metritic dairy cows at 5–10 days post-partum. Vet. Res. 2018, 49, 77. [Google Scholar] [CrossRef] [PubMed]
- Bicalho, M.; Santin, T.; Rodrigues, M.; Marques, C.; Lima, S.; Bicalho, R. Dynamics of the microbiota found in the vaginas of dairy cows during the transition period: Associations with uterine diseases and reproductive outcome. J. Dairy Sci. 2017, 100, 3043–3058. [Google Scholar] [CrossRef] [PubMed]
- Santos, T.M.; Bicalho, R.C. Diversity and succession of bacterial communities in the uterine fluid of postpartum metritic, endometritic and healthy dairy cows. PLoS ONE 2012, 7, e53048. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Fu, K.; Pang, B.; Wang, J.; Li, H.; Jiang, Z.; Feng, Y.; Tian, W.; Cao, R. Determination of uterine bacterial community in postpartum dairy cows with metritis based on 16S rDNA sequencing. Vet. Anim. Sci. 2020, 10, 100102. [Google Scholar] [CrossRef] [PubMed]
- Levy, M.; Kolodziejczyk, A.A.; Thaiss, C.A.; Elinav, E. Dysbiosis and the immune system. Nat. Rev. Immunol. 2017, 17, 219–232. [Google Scholar] [CrossRef] [PubMed]
- Abusleme, L.; Hoare, A.; Hong, B.Y.; Diaz, P.I. Microbial signatures of health, gingivitis, and periodontitis. Periodontology 2000 2021, 86, 57–78. [Google Scholar] [CrossRef] [PubMed]
- David, L.A.; Weil, A.; Ryan, E.T.; Calderwood, S.B.; Harris, J.B.; Chowdhury, F.; Begum, Y.; Qadri, F.; LaRocque, R.C.; Turnbaugh, P.J. Gut microbial succession follows acute secretory diarrhea in humans. mBio 2015, 6, e00381-15. [Google Scholar] [CrossRef] [PubMed]
- Eisenberg, T.; Glaeser, S.P.; Ewers, C.; Semmler, T.; Drescher, B.; Kämpfer, P. Caviibacter abscessus gen. nov., sp. nov., a member of the family Leptotrichiaceae isolated from guinea pigs (Cavia porcellus). Int. J. Syst. Evol. Microbiol. 2016, 66, 1652–1659. [Google Scholar] [CrossRef] [PubMed]
- Bemis, D.A.; Johnson, B.H.; Bryant, M.J.; Jones, R.D.; McCleery, B.V.; Greenacre, C.B.; Perreten, V.; Kania, S.A. Isolation and identification of Caviibacter abscessus from cervical abscesses in a series of pet guinea pigs (Cavia porcellus). J. Vet. Diagn. Investig. 2016, 28, 763–769. [Google Scholar] [CrossRef] [PubMed]
- Moore, S.; Feehily, C.; Doyle, R.; Buckley, F.; Lonergan, P.; Cotter, P.; Butler, S. Associations between the postpartum uterine and vaginal microbiota and the subsequent development of purulent vaginal discharge vary with dairy cow breed and parity. J. Dairy Sci. 2023, 106, 8133–8151. [Google Scholar] [CrossRef] [PubMed]
- Vaccari, J.; Borsanelli, A.C.; Athayde, F.R.; Saraiva, J.R.; Ramos, T.N.; Dutra, I.S. Gingivitis-and periodontitis-associated microbiota in bovine deciduous incisor teeth–A preliminary study. Microbes Infect. 2024, 27, 105468. [Google Scholar] [CrossRef] [PubMed]
- Ni, J.; Wang, J.; Zhao, K.; Chen, Y.; Xia, S.; Lai, S. Vaginal microbiome dynamics of cows in different parities. Animals 2023, 13, 2880. [Google Scholar] [CrossRef] [PubMed]
- Hofer, U. Fusobacterium orchestrates oral biofilms. Nat. Rev. Microbiol. 2022, 20, 576. [Google Scholar] [CrossRef] [PubMed]
- Zafar, H.; Saier, M.H., Jr. Gut Bacteroides species in health and disease. Gut Microbes 2021, 13, 1848158. [Google Scholar] [CrossRef] [PubMed]
- Acuna-Amador, L.; Barloy-Hubler, F. Porphyromonas spp. have an extensive host range in ill and healthy individuals and an unexpected environmental distribution: A systematic review and meta-analysis. Anaerobe 2020, 66, 102280. [Google Scholar] [CrossRef] [PubMed]
- Moreno, C.G.; Luque, A.T.; Galvão, K.N.; Otero, M.C. Bacterial communities from vagina of dairy healthy heifers and cows with impaired reproductive performance. Res. Vet. Sci. 2022, 142, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Basbas, C.; Garzon, A.; Schlesener, C.; van Heule, M.; Profeta, R.; Weimer, B.C.; Silva-del-Rio, N.; Byrne, B.A.; Karle, B.; Aly, S.S.; et al. Unveiling the microbiome during post-partum uterine infection: A deep shotgun sequencing approach to characterize the dairy cow uterine microbiome. Anim. Microbiome 2023, 5, 59. [Google Scholar] [CrossRef] [PubMed]
- LeBlanc, S. Review: Postpartum reproductive disease and fertility in dairy cows. Animal 2023, 17 (Suppl. 1), 100781. [Google Scholar] [CrossRef] [PubMed]
- Casaro, S.; Prim, J.; Gonzalez, T.; Cunha, F.; Silva, A.; Yu, H.; Bisinotto, R.; Chebel, R.; Santos, J.; Nelson, C.; et al. Multi-omics integration and immune profiling identify possible causal networks leading to uterine microbiome dysbiosis in dairy cows that develop metritis. Anim. Microbiome 2025, 7, 4. [Google Scholar] [CrossRef] [PubMed]
- Jeon, S.J.; Galvão, K.N. An advanced understanding of uterine microbial ecology associated with metritis in dairy cows. Genom. Inform. 2018, 16, e21. [Google Scholar] [CrossRef] [PubMed]
- Cunha, F.; Jeon, S.J.; Daetz, R.; Vieira-Neto, A.; Laporta, J.; Jeong, K.C.; Barbet, A.F.; Risco, C.A.; Galvão, K.N. Quantifying known and emerging uterine pathogens, and evaluating their association with metritis and fever in dairy cows. Theriogenology 2018, 114, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Deng, Z.; Zhang, L.; Gu, X.; Liu, G.; Liu, Y.; Chen, P.; Gao, J.; Han, B.; Qu, W. Biological characteristics and pathogenicity of Helcococcus ovis isolated from clinical bovine mastitis in a Chinese dairy herd. Front. Vet. Sci. 2022, 8, 756438. [Google Scholar] [CrossRef] [PubMed]
- Jianfei, E.; Wang, D.; Wei, W.; Chen, Z. Umbilical abscess caused by Helcococcus kunzii: A case report and literature review. J. Infect. Dev. Ctries. 2024, 18, 1632–1635. [Google Scholar] [CrossRef] [PubMed]
- Mouro, M.; Frade, J.; Chaves, C.; Velez, J. Helcococcus kunzii, a skin-colonising microorganism with pathogenic capacity: A case of polymicrobial intracranial empyema. BMJ Case Rep. CP 2021, 14, e244600. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Liu, Y.; Chen, W.; Chen, S.; Su, M.; Zheng, Y.; Liu, W.; Li, L.; Zeng, L.; Shi, Q.; et al. Uterine Commensal Peptostreptococcus Species Contribute to IDO1 Induction in Endometrial Cancer via Indoleacrylic Acid. Biomedicines 2024, 12, 573. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
He, X.; Wang, J.; Jiang, L.; Wang, X.; Wang, Y.; Liu, Y.; Cheng, Y.; Xu, F.; Li, X. Uterine Microbiota Composition in Dairy Cows with Different Vaginal Discharge Scores: Suggesting Caviibacter as a Potential Pathogen in Mild Purulent Metritis. Microorganisms 2025, 13, 1728. https://doi.org/10.3390/microorganisms13081728
He X, Wang J, Jiang L, Wang X, Wang Y, Liu Y, Cheng Y, Xu F, Li X. Uterine Microbiota Composition in Dairy Cows with Different Vaginal Discharge Scores: Suggesting Caviibacter as a Potential Pathogen in Mild Purulent Metritis. Microorganisms. 2025; 13(8):1728. https://doi.org/10.3390/microorganisms13081728
Chicago/Turabian StyleHe, Xiaolei, Jiajia Wang, Lin Jiang, Xinyu Wang, Yuxing Wang, Yang Liu, Yanping Cheng, Fei Xu, and Xiubo Li. 2025. "Uterine Microbiota Composition in Dairy Cows with Different Vaginal Discharge Scores: Suggesting Caviibacter as a Potential Pathogen in Mild Purulent Metritis" Microorganisms 13, no. 8: 1728. https://doi.org/10.3390/microorganisms13081728
APA StyleHe, X., Wang, J., Jiang, L., Wang, X., Wang, Y., Liu, Y., Cheng, Y., Xu, F., & Li, X. (2025). Uterine Microbiota Composition in Dairy Cows with Different Vaginal Discharge Scores: Suggesting Caviibacter as a Potential Pathogen in Mild Purulent Metritis. Microorganisms, 13(8), 1728. https://doi.org/10.3390/microorganisms13081728