
Effects of Salinity Fluctuation on Antimicrobial Resistance and Virulence Factor Genes of Low and High Nucleic Acid-Content Bacteria in a Marine Environment

Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Sample Collection and Physicochemical Parameters
2.2. Flow Cytometry, Fluorescent Staining and Filtration Protocol
2.3. Continuous Experimental Design
2.4. DNA Extraction, 16S rRNA Amplicon Sequencing, and Sequence Processing
2.5. Data Analysis
3. Results
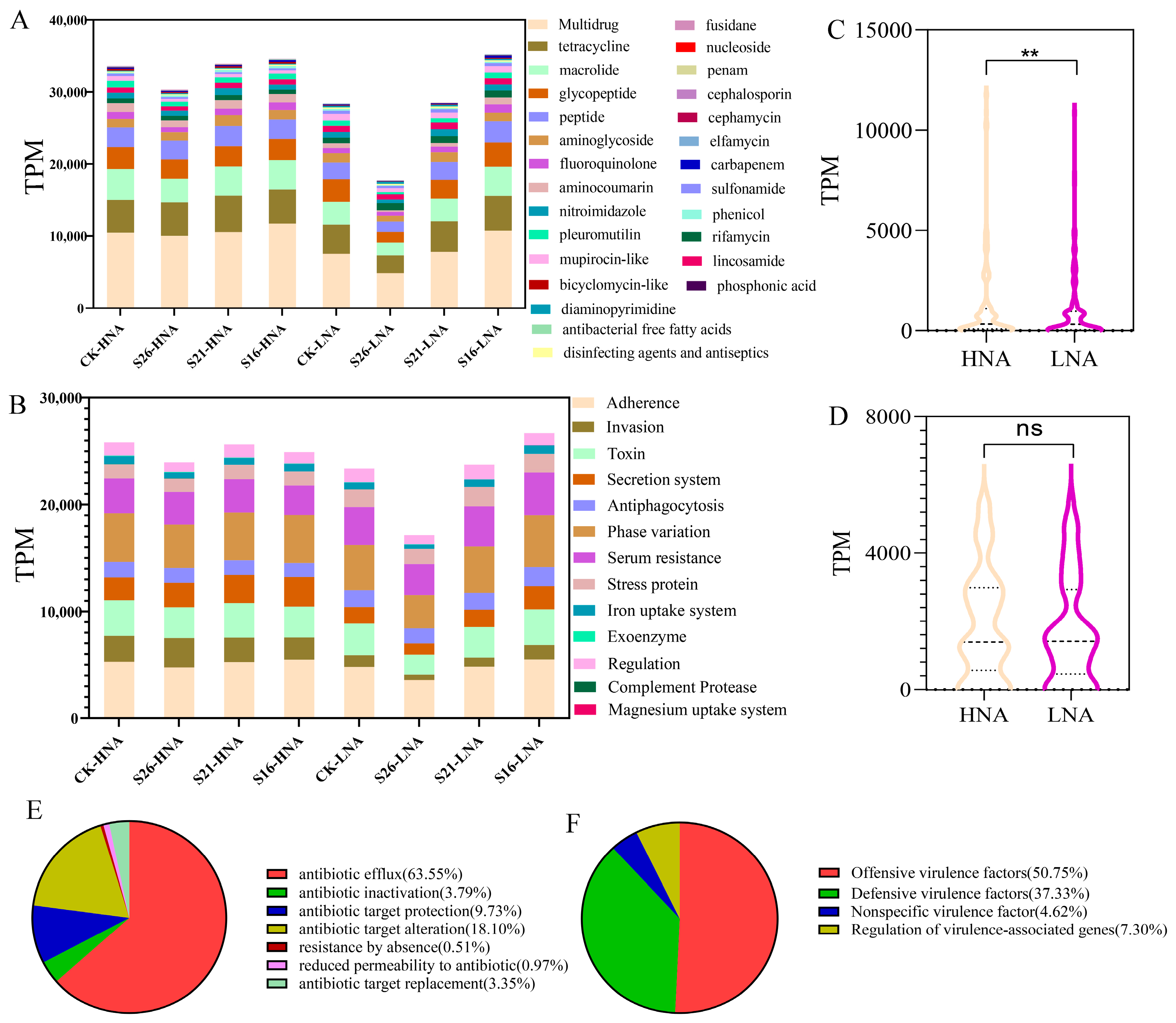
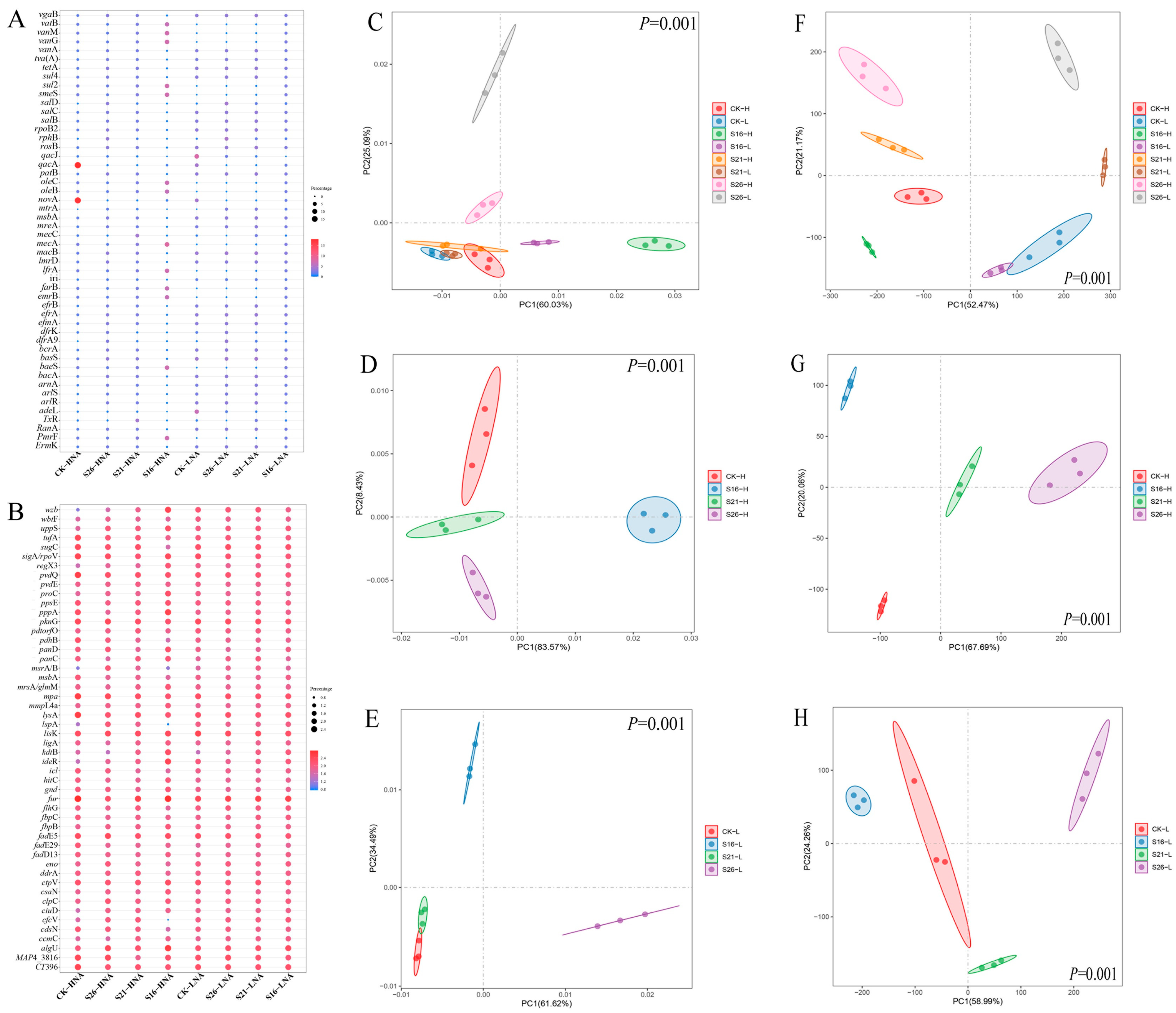
3.1. Prevalence of ARGs and VFGs in HNA and LNA Bacteria Revealed Using Metagenomics
3.2. Effects of Decreased Salinity on the Abundance of ARGs and VFGs
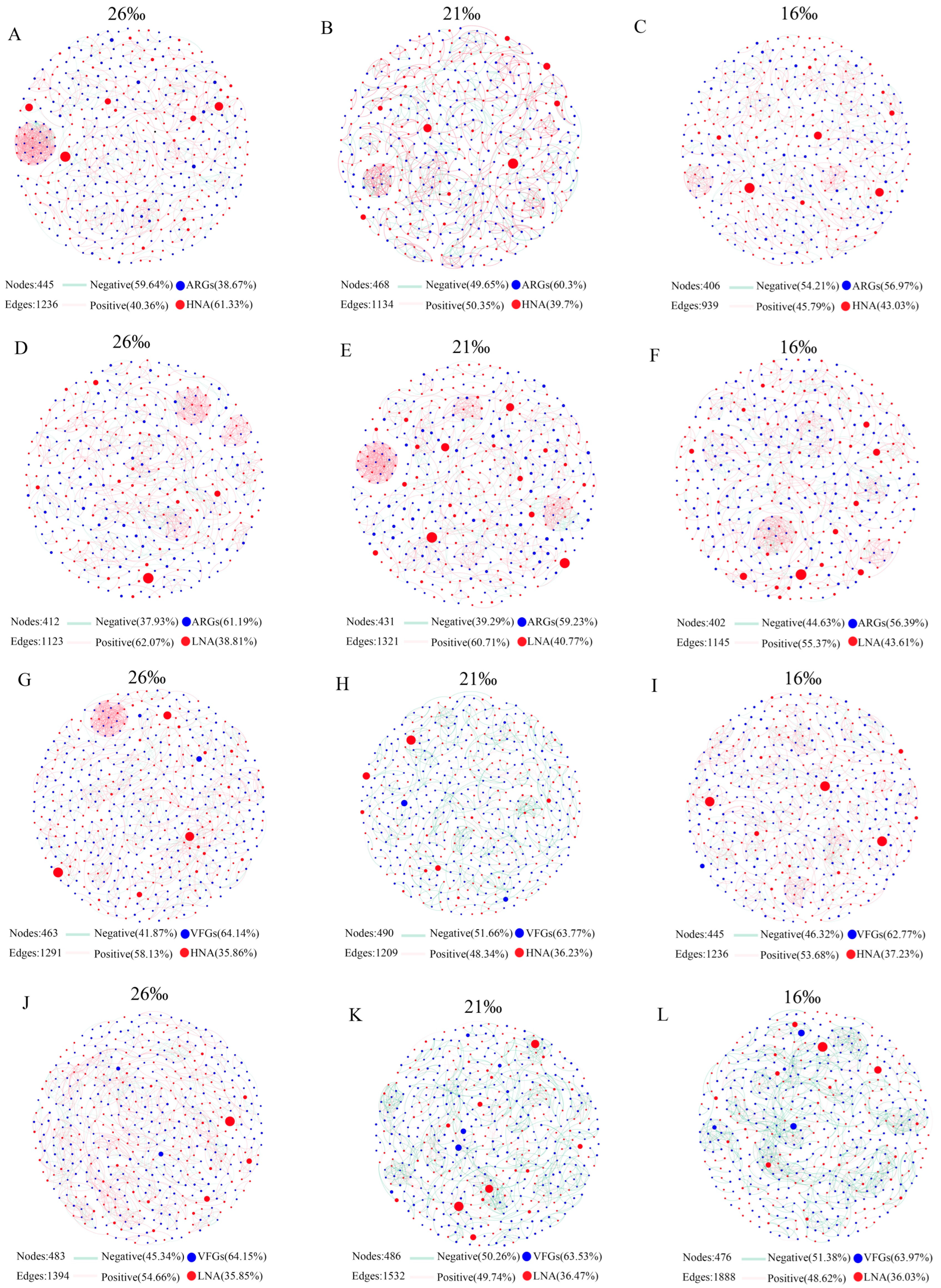
3.3. Co-Occurrence Network Analysis of ARGs and VFGs in the Bacteria
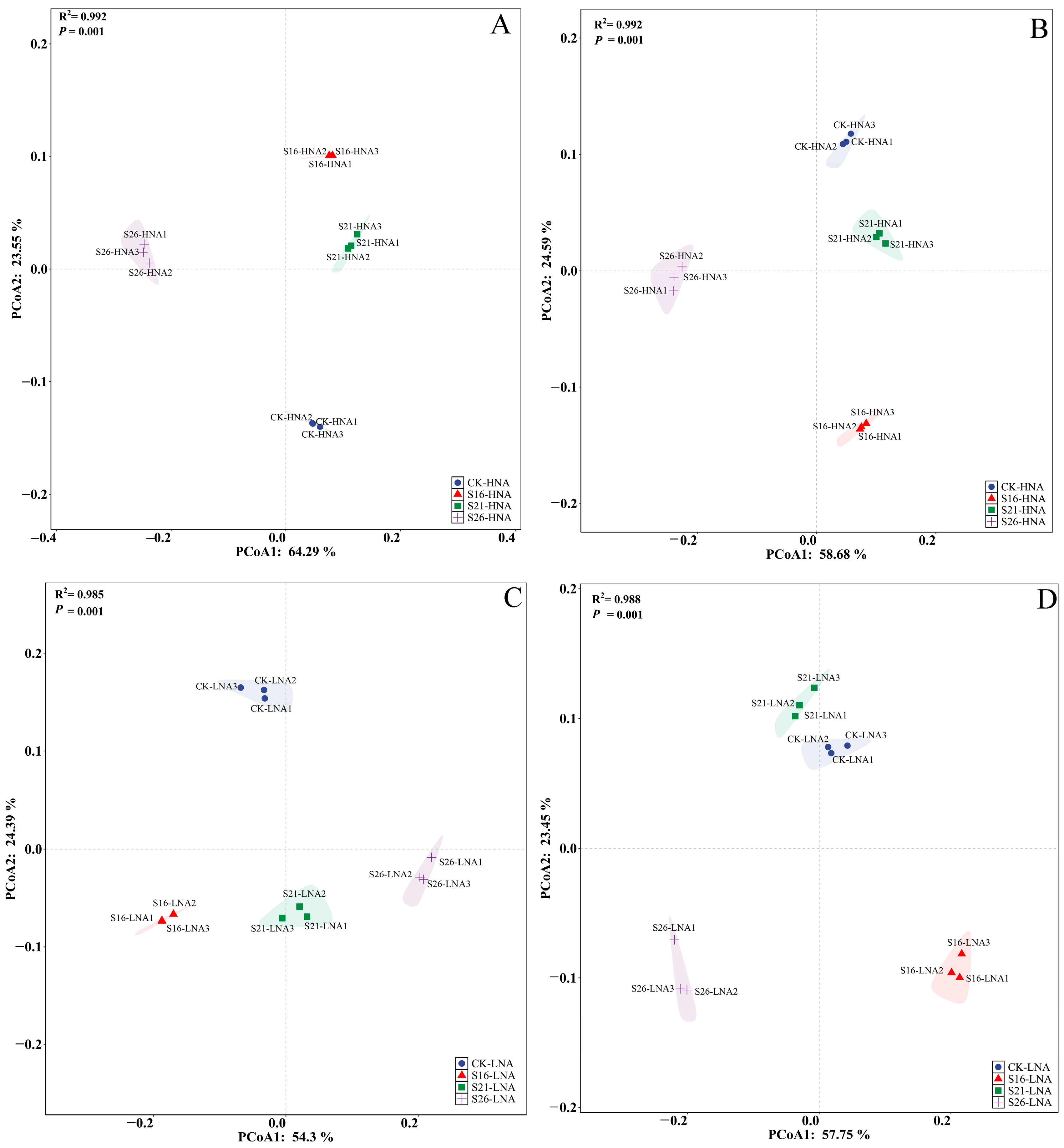
3.4. Effects of Salinity on the Bacterial Community Hosting ARGs and VFGs in the Marine Environment
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sun, J.; Xie, Y.-G.; Zhou, H.; Chen, L.; Qu, Y.-N.; Cheng, H.; Chen, Z.; Hua, Z.-S.; Wang, Y. Distribution Patterns and Ecological Risks of Antibiotic Resistance Genes in the Yap Trench. Water Res. 2025, 281, 123589. [Google Scholar] [CrossRef]
- Kim, H.; Kim, M.; Kim, S.; Lee, Y.M.; Shin, S.C. Characterization of Antimicrobial Resistance Genes and Virulence Factor Genes in an Arctic Permafrost Region Revealed by Metagenomics. Environ. Pollut. 2022, 294, 118634. [Google Scholar] [CrossRef]
- Xu, N.; Qiu, D.; Zhang, Z.; Wang, Y.; Chen, B.; Zhang, Q.; Wang, T.; Hong, W.; Zhou, N.-Y.; Penuelas, J.; et al. A Global Atlas of Marine Antibiotic Resistance Genes and Their Expression. Water Res. 2023, 244, 120488. [Google Scholar] [CrossRef] [PubMed]
- Farrell, M.L.; Chueiri, A.; O’Connor, L.; Duane, S.; Maguire, M.; Miliotis, G.; Cormican, M.; Hooban, B.; Leonard, A.; Gaze, W.H.; et al. Assessing the Impact of Recreational Water Use on Carriage of Antimicrobial Resistant Organisms. Sci. Total Environ. 2023, 888, 164201. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Zhang, Y.; Wu, J.; Wang, J.; Zhang, C.; Lin, Y. Occurrence and Spatial Distribution of Antibiotic Resistance Genes in the Bohai Sea and Yellow Sea Areas, China. Environ. Pollut. 2019, 252, 450–460. [Google Scholar] [CrossRef] [PubMed]
- Mao, G.; Ji, M.; Jiao, N.; Su, J.; Zhang, Z.; Liu, K.; Chen, Y.; Liu, Y. Monsoon Affects the Distribution of Antibiotic Resistome in Tibetan Glaciers. Environ. Pollut. 2023, 317, 120809. [Google Scholar] [CrossRef]
- Zheng, B.; Liu, W.; Xu, H.; Li, J.; Jiang, X. Occurrence and Distribution of Antimicrobial Resistance Genes in the Soil of an Industrial Park in China: A Metagenomics Survey. Environ. Pollut. 2021, 273, 116467. [Google Scholar] [CrossRef]
- Pan, Y.; Zeng, J.; Li, L.; Yang, J.; Tang, Z.; Xiong, W.; Li, Y.; Chen, S.; Zeng, Z. Coexistence of Antibiotic Resistance Genes and Virulence Factors Deciphered by Large-Scale Complete Genome Analysis. Msystems 2020, 5, 1–15. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, T.L.; Xiang, Q.; Fu, C.X.; Qiao, M.; Ding, L.J.; Zhu, D. Selective Enrichment of Virulence Factor Genes in the Plastisphere under Antibiotic and Heavy Metal Pressures. J. Hazard. Mater. 2024, 465, 133319. [Google Scholar] [CrossRef]
- Xue, X.; Su, X.; Zhou, L.; Ji, J.; Qin, Z.; Liu, J.; Li, K.; Wang, H.; Wang, Z. Antibiotic-Induced Recruitment of Specific Algae-Associated Microbiome Enhances the Adaptability of Chlorella Vulgaris to Antibiotic Stress and Incidence of Antibiotic Resistance. Environ. Sci. Technol. 2023, 57, 13336–13345. [Google Scholar] [CrossRef]
- Li, N.; Fan, X.Y.; Li, X. Unveiling the characteristics of free-living and particle-associated antibiotic resistance genes associated with bacterial communities along different processes in a full-scale drinking water treatment plant. J. Hazard. Mater. 2024, 476, 135194. [Google Scholar] [CrossRef]
- Zhu, Y.G.; Zhao, Y.; Li, B.; Huang, C.L.; Zhang, S.Y.; Yu, S.; Chen, Y.S.; Zhang, T.; Gillings, M.R.; Su, J.Q. Continental-Scale Pollution of Estuaries with Antibiotic Resistance Genes. Nat. Microbiol. 2017, 2, 16270. [Google Scholar] [CrossRef]
- Veldhuis, M.J.W.; Cucci, T.L.; Sieracki, M.E. Cellular DNA Content of Marine Phytoplankton Using Two New Fluorochromes: Taxonomic and Ecological Implications. J. Phycol. 1997, 33, 527–541. [Google Scholar] [CrossRef]
- Gasol, J.M.; Li Zweifel, U.; Peters, F.; Fuhrman, J.A.; Hagström, Å. Significance of Size and Nucleic Acid Content Heterogeneity as Measured by Flow Cytometry in Natural Planktonic Bacteria. Appl. Environ. Microbiol. 1999, 65, 4475–4483. [Google Scholar] [CrossRef]
- Lebaron, P.; Servais, P.; Baudoux, A.C.; Bourrain, M.; Courties, C.; Parthuisot, N. Variations of Bacterial-Specific Activity with Cell Size and Nucleic Acid Content Assessed by Flow Cytometry. Aquat. Microb. Ecol. 2002, 28, 131–140. [Google Scholar] [CrossRef]
- Wang, Y.; Hammes, F.; Boon, N.; Chami, M.; Egli, T. Isolation and Characterization of Low Nucleic Acid (LNA)-Content Bacteria. ISME J. 2009, 3, 889–902. [Google Scholar] [CrossRef]
- Hu, W.; Zhang, H.; Lin, X.; Liu, R.; Bartlam, M.; Wang, Y. Characteristics, Biodiversity, and Cultivation Strategy of Low Nucleic Acid Content Bacteria. Front. Microb. 2022, 13, 900669. [Google Scholar] [CrossRef]
- Mao, G.; Ji, M.; Xu, B.; Liu, Y.; Jiao, N. Variation of High and Low Nucleic Acid-Content Bacteria in Tibetan Ice Cores and Their Relationship to Black Carbon. Front. Microb. 2022, 13, 84432. [Google Scholar] [CrossRef]
- Proctor, C.R.; Besmer, M.D.; Langenegger, T.; Beck, K.; Walser, J.C.; Ackermann, M.; Bürgmann, H.; Hammes, F. Phylogenetic Clustering of Small Low Nucleic Acid-Content Bacteria across Diverse Freshwater Ecosystems. ISME J. 2018, 12, 1344–1359. [Google Scholar] [CrossRef]
- Santos, M.; Oliveira, H.; Pereira, J.L.; Pereira, M.J.; Gonçalves, F.J.M.; Vidal, T. Flow Cytometry Analysis of Low/High DNA Content (LNA/HNA) Bacteria as Bioindicator of Water Quality Evaluation. Ecol. Indic. 2019, 103, 774–781. [Google Scholar] [CrossRef]
- Song, Y.; Wang, Y.; Mao, G.; Gao, G.; Wang, Y. Impact of Planktonic Low Nucleic Acid-Content Bacteria to Bacterial Community Structure and Associated Ecological Functions in a Shallow Lake. Sci. Total Environ. 2019, 658, 868–878. [Google Scholar] [CrossRef] [PubMed]
- Zheng, N.; Hu, W.; Liu, Y.; Li, Z.; Jiang, Y.; Bartlam, M.; Wang, Y. Phycospheric Bacteria Limits the Effect of Nitrogen and Phosphorus Imbalance on Diatom Bloom. Sci. Total Environ. 2024, 935, 173477. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Zheng, N.; Zhang, Y.; Li, S.; Bartlam, M.; Wang, Y. Metagenomics Analysis Reveals Effects of Salinity Fluctuation on Diversity and Ecological Functions of High and Low Nucleic Acid Content Bacteria. Sci. Total Environ. 2024, 933, 173186. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Zheng, N.; Zhang, Y.; Bartlam, M.; Wang, Y. Spatiotemporal Dynamics of High and Low Nucleic Acid-Content Bacterial Communities in Chinese Coastal Seawater: Assembly Process, Co-Occurrence Relationship and the Ecological Functions. Front. Microb. 2023, 14, 1219655. [Google Scholar] [CrossRef]
- Ma, L.; Li, B.; Zhang, T. New Insights into Antibiotic Resistome in Drinking Water and Management Perspectives: A Metagenomic Based Study of Small-Sized Microbes. Water Res. 2019, 152, 191–201. [Google Scholar] [CrossRef]
- Nakai, R.; Abe, T.; Takeyama, H.; Naganuma, T. Metagenomic Analysis of 0.2-Μm-Passable Microorganisms in Deep-Sea Hydrothermal Fluid. Mar. Biotechnol. 2011, 13, 900–908. [Google Scholar] [CrossRef]
- Ali Alharbi, S. Isolation of Ultrasmall (Filterable) Bacteria from Patients Suffering from ME, and Patients and Staff of a Paediatric Hospital. Saudi J. Biol. Sci. 2020, 27, 1566–1568. [Google Scholar] [CrossRef]
- He, X.; McLean, J.S.; Edlund, A.; Yooseph, S.; Hall, A.P.; Liu, S.Y.; Dorrestein, P.C.; Esquenazi, E.; Hunter, R.C.; Cheng, G.; et al. Cultivation of a Human-Associated TM7 Phylotype Reveals a Reduced Genome and Epibiotic Parasitic Lifestyle. Pro. Natl. Acad. Sci. USA 2015, 112, 244–249. [Google Scholar] [CrossRef]
- Zheng, N.; Hu, W.; Zhou, X.; Liu, Y.; Bartlam, M.; Wang, Y. Influence of Phycospheric Bacterioplankton Disruption or Removal on Algae Growth and Survival. Environ. Res. 2023, 237, 117060. [Google Scholar] [CrossRef]
- Zhang, Y.-J.; Hu, H.-W.; Yan, H.; Wang, J.-T.; Lam, S.K.; Chen, Q.-L.; Chen, D.; He, J.-Z. Salinity as a Predominant Factor Modulating the Distribution Patterns of Antibiotic Resistance Genes in Ocean and River Beach Soils. Sci. Total Environ. 2019, 668, 193–203. [Google Scholar] [CrossRef]
- Dželalija, M.; Fredotović, Ž.; Udiković-Kolić, N.; Kalinić, H.; Jozić, S.; Šamanić, I.; Ordulj, M.; Maravić, A. Large-Scale Biogeographical Shifts of Abundance of Antibiotic Resistance Genes and Marine Bacterial Communities as Their Carriers along a Trophic Gradient. Int. J. Mol. Sci. 2024, 25, 654. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Xiang, J.; Xie, Y.; Yu, K.; Gin, K.Y.H.; Zhang, B.; He, Y. Dynamic Distribution and Driving Mechanisms of Antibiotic Resistance Genes in a Human-Intensive Watershed. Water Res. 2022, 222, 118841. [Google Scholar] [CrossRef] [PubMed]
- Lew, S.; Glińska-Lewczuk, K.; Burandt, P.; Kulesza, K.; Kobus, S.; Obolewski, K. Salinity as a Determinant Structuring Microbial Communities in Coastal Lakes. Int. J. Environ. Res. Public Health 2022, 19, 4592. [Google Scholar] [CrossRef] [PubMed]
- Shindoh, S.; Kadoya, A.; Kanechi, R.; Watanabe, K.; Suzuki, S. Marine Bacteria Harbor the Sulfonamide Resistance Gene Sul4 without Mobile Genetic Elements. Front. Microbiol. 2023, 14, 1230548. [Google Scholar] [CrossRef]
- Cuadrat, R.R.C.; Sorokina, M.; Andrade, B.G.; Goris, T.; Dávila, A.M.R. Global Ocean Resistome Revealed: Exploring Antibiotic Resistance Gene Abundance and Distribution in TARA Oceans Samples. GigaScience 2020, 9, giaa046. [Google Scholar] [CrossRef]
- Yang, P.; Hao, S.; Han, M.; Xu, J.; Yu, S.; Chen, C.; Zhang, H.; Ning, K. Analysis of Antibiotic Resistance Genes Reveals Their Important Roles in Influencing the Community Structure of Ocean Microbiome. Sci. Total Environ. 2022, 823, 153731. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An Ultra-Fast All-in-One FASTQ Preprocessor. Bioinformatics 2018, 38, i884–i890. [Google Scholar] [CrossRef]
- Li, D.; Liu, C.M.; Luo, R.; Kunihiko, S.; Tak-Wah, L. MEGAHIT: An Ultra-Fast Single-Node Solution for Large and Complex Metagenomics Assembly via Succinct de Bruijn Graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef]
- Tang, S.; Borodovsky, M. Ab Initio Gene Identification in Metagenomic Sequences. Nucleic Acids Res. 2010, 38, e132. [Google Scholar] [CrossRef]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for Clustering the next-Generation Sequencing Data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S. Fast Gapped-Read Alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and Sensitive Protein Alignment Using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef] [PubMed]
- Alcock, B.P.; Raphenya, A.R.; Lau, T.T.Y.; Tsang, K.K.; Bouchard, M.; Edalatmand, A.; Huynh, W.; Nguyen, A.-L.V.; Cheng, A.A.; Liu, S.; et al. CARD 2020: Antibiotic Resistome Surveillance with the Comprehensive Antibiotic Resistance Database. Nucleic Acids Res. 2019, 48, D517. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Zheng, D.; Jin, Q.; Chen, L.; Yang, J. VFDB 2019: A Comparative Pathogenomic Platform with an Interactive Web Interface. Nucleic Acids Res. 2019, 47, D687–D692. [Google Scholar] [CrossRef]
- Bray, N.L.; Pimentel, H.; Melsted, P.; Pachter, L. Near-Optimal Probabilistic RNA-Seq Quantification. Nat. Biotechnol. 2016, 34, 525–527. [Google Scholar] [CrossRef]
- Wang, Y.F.; Liu, Y.J.; Fu, Y.M.; Xu, J.Y.; Zhang, T.L.; Cui, H.L.; Qiao, M.; Rillig, M.C.; Zhu, Y.G.; Zhu, D. Microplastic diversity increases the abundance of antibiotic resistance genes in soil. Nat. Commun. 2024, 15, 9788. [Google Scholar] [CrossRef]
- Liu, Q.; Jia, J.; Hu, H.; Li, X.; Zhao, Y.; Wu, C. Nitrogen and Phosphorus Limitations Promoted Bacterial Nitrate Metabolism and Propagation of Antibiotic Resistome in the Phycosphere of Auxenochlorella Pyrenoidosa. J. Hazard. Mater 2024, 468, 133786. [Google Scholar] [CrossRef]
- Ma, B.; Wang, Y.; Ye, S.; Liu, S.; Stirling, E.; Gilbert, J.A.; Faust, K.; Knight, R.; Jansson, J.K.; Cardona, C.; et al. Earth Microbial Co-Occurrence Network Reveals Interconnection Pattern across Microbiomes. Microbiome 2020, 8, 1–12. [Google Scholar] [CrossRef]
- Zhang, Z.; Wu, C.; Di, Y.; Zhang, J.; Chai, B.; Zhou, S. Identification and Relative Contributions of Environmental Driving Factors for Abundant and Rare Bacterial Taxa to Thermal Stratification Evolution. Environ. Res. 2023, 232, 116424. [Google Scholar] [CrossRef]
- Niu, Z.-G.; Zhang, K.; Zhang, Y. Occurrence and Distribution of Antibiotic Resistance Genes in the Coastal Area of the Bohai Bay, China. Mar. Pollut. Bull. 2016, 107, 245–250. [Google Scholar] [CrossRef]
- Hatosy, S.M.; Martiny, A.C. The Ocean as a Global Reservoir of Antibiotic Resistance Genes. Appl. Environ. Microbiol. 2015, 81, 7593–7599. [Google Scholar] [CrossRef]
- Wang, X.; Lin, Y.; Li, S.; Wang, J.; Li, X.; Zhang, D.; Duan, D.; Shao, Z. Metagenomic analysis reveals the composition and sources of antibiotic resistance genes in coastal water ecosystems of the yellow sea and yangtze river delta. Environ. Pollut. 2025, 371, 125923. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, Q.; Wang, T.; Xu, N.; Lu, T.; Hong, W.; Penuelas, J.; Gillings, M.; Wang, M.; Gao, W.; et al. Assessment of Global Health Risk of Antibiotic Resistance Genes. Nat. Commun. 2022, 13, 1553. [Google Scholar] [CrossRef]
- Peng, F. Urbanization Drives Riverine Bacterial Antibiotic Resistome More than Taxonomic Community at Watershed Scale. Environ. Int. 2020, 137, 105524. [Google Scholar] [CrossRef]
- Pei, R.; Kim, S.C.; Carlson, K.H.; Pruden, A. Effect of River Landscape on the Sediment Concentrations of Antibiotics and Corresponding Antibiotic Resistance Genes (ARG). Water Res. 2006, 40, 2427–2435. [Google Scholar] [CrossRef]
- Liang, J.; Mao, G.; Yin, X.; Ma, L.; Liu, L.; Bai, Y.; Zhang, T.; Qu, J. Identification and Quantification of Bacterial Genomes Carrying Antibiotic Resistance Genes and Virulence Factor Genes for Aquatic Microbiological Risk Assessment. Water Res. 2020, 168, 115160. [Google Scholar] [CrossRef]
- Zhang, B.; Xia, Y.; Wen, X.; Wang, X.; Yang, Y.; Zhou, J.; Zhang, Y. The Composition and Spatial Patterns of Bacterial Virulence Factors and Antibiotic Resistance Genes in 19 Wastewater Treatment Plants. PLoS ONE 2016, 11, e0167422. [Google Scholar] [CrossRef]
- Beceiro, A.; Tomás, M.; Bou, G. Antimicrobial Resistance and Virulence: A Successful or Deleterious Association in the Bacterial World? Clin. Microbiol. Rev. 2013, 26, 185–230. [Google Scholar] [CrossRef]
- Livermore, D.M. Bacterial Resistance: Origins, Epidemiology, and Impact. Clin. Infect. Dis. 2003, 36, S11–S23. [Google Scholar] [CrossRef]
- Penadés, J.R.; Chen, J.; Quiles-Puchalt, N.; Carpena, N.; Novick, R.P. Bacteriophage-Mediated Spread of Bacterial Virulence Genes. Curr. Opin. Microbiol. 2015, 23, 171–178. [Google Scholar] [CrossRef]
- Meng, M.; Li, Y.; Yao, H. Plasmid-Mediated Transfer of Antibiotic Resistance Genes in Soil. Antibiotics 2022, 11, 525. [Google Scholar] [CrossRef]
- Jebri, S.; Rahmani, F.; Hmaied, F. Bacteriophages as Antibiotic Resistance Genes Carriers in Agro-food Systems. J. Appl. Microbiol. 2021, 130, 688–698. [Google Scholar] [CrossRef]
- McMahon, M.A.S.; Xu, J.; Moore, J.E.; Blair, I.S.; McDowell, D.A. Environmental Stress and Antibiotic Resistance in Food-Related Pathogens. Appl. Environ. Microbiol. 2007, 73, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Qi, L.; Li, W.; Hu, B.X.; Dai, Z. Bacterial Community Variations with Salinity in the Saltwater-Intruded Estuarine Aquifer. Sci. Total Environ. 2021, 755, 142423. [Google Scholar] [CrossRef] [PubMed]
- Qiao, L.; Liu, X.; Zhang, S.; Zhang, L.; Li, X.; Hu, X.; Zhao, Q.; Wang, Q.; Yu, C. Distribution of the Microbial Community and Antibiotic Resistance Genes in Farmland Surrounding Gold Tailings: A Metagenomics Approach. Sci. Total Environ. 2021, 779, 146502. [Google Scholar] [CrossRef] [PubMed]
- Søborg, D.A.; Hendriksen, N.B.; Kroer, N. Occurrence and Expression of Bacterial Human Virulence Gene Homologues in Natural Soil Bacteria. FEMS Microbiol. Ecol. 2014, 90, 520–532. [Google Scholar] [CrossRef]
- Van Goethem, M.W.; Pierneef, R.; Bezuidt, O.K.I.; Van De Peer, Y.; Cowan, D.A.; Makhalanyane, T.P. A Reservoir of ‘Historical’ Antibiotic Resistance Genes in Remote Pristine Antarctic Soils. Microbiome 2018, 6, 40. [Google Scholar] [CrossRef]
- Uyaguari-Díaz, M.I.; Croxen, M.A.; Luo, Z.; Cronin, K.I.; Chan, M.; Baticados, W.N.; Nesbitt, M.J.; Li, S.; Miller, K.M.; Dooley, D.; et al. Human Activity Determines the Presence of Integron-Associated and Antibiotic Resistance Genes in Southwestern British Columbia. Front. Microbiol. 2018, 9, 852. [Google Scholar] [CrossRef]
- Han, M.; Zhang, L.; Zhang, N.; Mao, Y.; Peng, Z.; Huang, B.; Zhang, Y.; Wang, Z. Antibiotic Resistome in a Large Urban-Lake Drinking Water Source in Middle China: Dissemination Mechanisms and Risk Assessment. J. Hazard. Mater. 2022, 424, 127745. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hu, W.; Zhou, X.; Liu, Y.; Zhang, Y.; Wang, Y. Effects of Salinity Fluctuation on Antimicrobial Resistance and Virulence Factor Genes of Low and High Nucleic Acid-Content Bacteria in a Marine Environment. Microorganisms 2025, 13, 1710. https://doi.org/10.3390/microorganisms13071710
Hu W, Zhou X, Liu Y, Zhang Y, Wang Y. Effects of Salinity Fluctuation on Antimicrobial Resistance and Virulence Factor Genes of Low and High Nucleic Acid-Content Bacteria in a Marine Environment. Microorganisms. 2025; 13(7):1710. https://doi.org/10.3390/microorganisms13071710
Chicago/Turabian StyleHu, Wei, Xinzhu Zhou, Yu Liu, Yadi Zhang, and Yingying Wang. 2025. "Effects of Salinity Fluctuation on Antimicrobial Resistance and Virulence Factor Genes of Low and High Nucleic Acid-Content Bacteria in a Marine Environment" Microorganisms 13, no. 7: 1710. https://doi.org/10.3390/microorganisms13071710
APA StyleHu, W., Zhou, X., Liu, Y., Zhang, Y., & Wang, Y. (2025). Effects of Salinity Fluctuation on Antimicrobial Resistance and Virulence Factor Genes of Low and High Nucleic Acid-Content Bacteria in a Marine Environment. Microorganisms, 13(7), 1710. https://doi.org/10.3390/microorganisms13071710