
Preventing Microorganism Contamination in Starting Active Materials for Synthesis from Global Regulatory Agencies: Overview for Public Health Implications
 ,
,  , , ,
, , ,  , , ,
, , ,  , , and
, , and
Abstract
1. Introduction
2. Materials and Methods
3. Results
3.1. European Union
3.2. China
3.3. Mexico
3.4. United States
3.5. Canada
3.6. Brazil
3.7. India
3.8. PIC/S
3.9. WHO
4. Discussion
4.1. SAMS Definition and Management
4.2. Microbiological Contamination Control
4.3. Future Perspectives
4.4. Implications for Clinical Practice
4.5. Limitations of the Study
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization (WHO). WHO Guidelines on Good Manufacturing Practices for Active Pharmaceutical Ingredients; World Health Organization (WHO): Geneva, Switzerland, 2010; TRS No. 957; Available online: https://www.who.int/teams/health-product-policy-and-standards/standards-and-specifications/norms-and-standards/gmp (accessed on 25 April 2025).
- European Medicines Agency (EMA). Guideline on the Requirements for Quality Documentation Concerning Biological Investigational Medicinal Products in Clinical Trials; European Medicines Agency (EMA): Amsterdam, The Netherlands, 2022; Available online: https://www.ema.europa.eu/en/requirements-quality-documentation-concerning-biological-investigational-medicinal-products-clinical-trials-scientific-guideline (accessed on 25 April 2025).
- Petrelli, F.; Caraffa, A.; Scuri, S.; Grappasonni, I.; Magrini, E.; Cocchini, A. The Requirements for Manufacturing Highly Active or Sensitising Drugs Comparing Good Manufacturing Practices. Acta Biomed. 2019, 90, 288–299. [Google Scholar] [CrossRef] [PubMed]
- Asif, M.; Usmani, S. Basics of Pharmaceutical Manufacturing and Quality Operations; CRC Press: Boca Raton, FL, USA, 2023. [Google Scholar] [CrossRef]
- U.S. Food and Drug Administration (FDA). Question 3: Media fill contamination. In Questions and Answers on Current Good Manufacturing Practice—Control of Components and Drug Product Containers and Closures; U.S. Food and Drug Administration (FDA): Silver Spring, MD, USA, 2005. Available online: https://www.fda.gov/drugs/guidances-drugs/questions-and-answers-current-good-manufacturing-practice-requirements-control-components-and-drug?utm_ (accessed on 24 June 2025).
- Sandle, T. Pharmaceutical Microbiology: Essentials for Quality Assurance and Quality Control; Elsevier: Amsterdam, The Netherlands, 2015. [Google Scholar] [CrossRef]
- Kanghorban, M.; Farzaneh, A.; Eslami, M. Microbial quality challenges in biopharmaceutical manufacturing: New perspectives. J. Appl. Microbiol. Biotechnol. 2023, 107, 211–225. [Google Scholar]
- Clontz, L. Microbial Limit and Bioburden Tests: Validation Approaches and Global Requirements; CRC Press: Boca Raton, FL, USA, 2009. [Google Scholar] [CrossRef]
- Atzrodt, J.; Derdau, V.; Loewe, C. Late-stage carbon-14 labeling and isotope exchange. JACS Au 2022, 2, 1321–1331. [Google Scholar] [CrossRef]
- Ezeako, C.; Wu, Y.; Zhang, L. Advances in GMP regulatory requirements for pharmaceutical raw materials. Regul. Aff. J. 2024, 29, 45–59. [Google Scholar]
- Ahsan, F.; Rauf, A.; Khan, M. Green synthesis approaches in API manufacturing: A sustainable solution. Green Chem. Lett. Rev. 2020, 13, 295–308. [Google Scholar]
- Gouin, S.; Bory, S.; Morissette, M. Stability and quality control of biopharmaceutical raw materials. J. Biopharm. Sci. 2021, 18, 212–220. [Google Scholar]
- Barhoum, A.; Bechelany, M.; Makhlouf, A.S.H. Handbook of Nanofibers; Springer: Cham, Switzerland, 2019. [Google Scholar] [CrossRef]
- Wu, C.; Chen, H.; Li, Y. Applications of nanomaterials in drug delivery and pharmaceutical development. Adv. Drug Deliv. Rev. 2021, 173, 211–230. [Google Scholar]
- Hauer, B. Embracing nature’s catalysts: Biocatalysis in industrial pharmaceutical production. ACS Catal. 2020, 10, 8418–8427. [Google Scholar] [CrossRef]
- Bornscheuer, U.T.; Hauer, B.; Jaeger, K.E.; Schwaneberg, U. Directed evolution empowered redesign of natural proteins for the sustainable production of chemicals and pharmaceuticals. Angew. Chem. Int. Ed. 2019, 58, 36–40. [Google Scholar] [CrossRef]
- Kalaba, D.; Rusak, J.; Kege, T. Global supply chain risks in pharmaceutical ingredient sourcing. Int. J. Supply Chain Manag. 2024, 13, 102–117. [Google Scholar]
- Decollibus, D.; Romano, M.; Falasca, L. Qualification of suppliers in pharmaceutical manufacturing: A risk-based approach. Pharm. Technol. Eur. 2023, 35, 28–33. [Google Scholar]
- International Council for Harmonisation (ICH). ICH Q10: Pharmaceutical Quality System; International Council for Harmonisation (ICH): Geneva, Switzerland, 2008; Available online: https://www.ich.org/page/quality-guidelines (accessed on 25 April 2025).
- Seoane-Viano, I.; Ochoa-Gonzalez, J.; Trenfield, S.J. Advances in real-time microbial detection technologies in pharmaceutical manufacturing. Trends Biotechnol. 2022, 40, 543–555. [Google Scholar]
- Chaachouay, N.; Zidane, L. Ethnopharmacological properties and microbiological risk of Moroccan medicinal plants. J. Ethnopharmacol. 2024, 312, 116431. [Google Scholar]
- Burlec, A.F.; Hodorogea, R.M.; Petrescu, C. Innovations in sustainable pharmaceutical manufacturing: From raw materials to finished product. Sustain. Health Sci. 2023, 5, 99–112. [Google Scholar]
- Pachnowska, M.; Nowak, M.; Karcz, D. Future directions in pharmaceutical raw material control and risk management. J. Pharm. Innov. 2025, 20, 51–65. [Google Scholar]
- PIC/S–Pharmaceutical Inspection Co-Operation Scheme. PE 009-17: Annex 1–Manufacture of Sterile Medicinal Products; PIC/S: Geneva, Switzerland, 2023. [Google Scholar]
- European Commission. Annex 1 to the EU Guidelines for Good Manufacturing Practice: Manufacture of Sterile Medicinal Products; European Commission: Brussels, Belgium, 2022; Available online: https://health.ec.europa.eu/system/files/2020-02/2020_annex1ps_sterile_medicinal_products_en_0.pdf (accessed on 24 April 2025).
- European Medicines Agency (EMA). Guideline on the Sterilisation of the Medicinal Product, Active Substance, Excipient and Primary Container; European Medicines Agency (EMA): Amsterdam, The Netherlands, 2019; Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-sterilisation-medicinal-product-active-substance-excipient-and-primary-container_en.pdf (accessed on 24 April 2025).
- ECA Academy. Update of the GMP Guideline for Sterile Medicinal Products in China; ECA Academy: Franklin, OH, USA, 2025; Available online: https://www.gmp-compliance.org/gmp-news/update-of-the-gmp-guideline-for-sterile-medicinal-products-in-china (accessed on 24 April 2025).
- COFEPRIS Tracker. Certificado de Buenas Prácticas; COFEPRIS Tracker: Mexico City, Mexico, 2021; Available online: https://www.cofepristracker.org/certificado-buenas-practicas/ (accessed on 24 April 2025).
- NOM-164-SSA1-2015; Good Manufacturing Practices for Drug Substances. Secretaría de Salud, México: México City, México, 2016. Available online: https://www.mexicanlaws.info/mexlaws.com/SALUD/NOM-164-SSA1-2015.htm (accessed on 24 April 2025).
- U.S. Food and Drug Administration (FDA). ICH Q7 Good Manufacturing Practice Guidance for Active Pharmaceutical Ingredients; U.S. Food and Drug Administration (FDA): Silver Spring, MD, USA, 2001. Available online: https://www.fda.gov/media/71518/download (accessed on 25 April 2025).
- U.S. Food and Drug Administration (FDA). ICH Q11 Development and Manufacture of Drug Substances; Food and Drug Administration (FDA): Silver Spring, MD, USA, 2012. Available online: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/q11-development-and-manufacture-drug-substances (accessed on 25 April 2025).
- U.S. Food and Drug Administration (FDA). Sterile Drug Products Produced by Aseptic Processing–Current Good Manufacturing Practice; Food and Drug Administration (FDA): Silver Spring, MD, USA, 2004. Available online: https://www.fda.gov/media/71026/download (accessed on 25 April 2025).
- U.S. Food and Drug Administration (FDA). Compliance Program Guidance Manual 7356.002F.; Food and Drug Administration (FDA): Silver Spring, MD, USA, 2022. Available online: https://www.fda.gov/inspections-compliance-enforcement-and-criminal-investigations/compliance-manuals/compliance-program-manual (accessed on 25 April 2025).
- Health Canada (HC). Good Manufacturing Practices Guidelines for Active Pharmaceutical Ingredient –GUI-0104; Health Canada (HC): Ottawa, ON, Canada, 2022. Available online: https://www.canada.ca/en/health-canada/services/drugs-health-products/compliance-enforcement/information-health-product/drugs/guidelines-active-pharmaceutical-ingredients-0104.html (accessed on 25 April 2025).
- Health Canada (HC). Quality (Chemistry and Manufacturing) Guidance: New Drug Submissions (NDSs) and Abbreviated New Drug Submissions (ANDSs); Health Canada (HC): Ottawa, ON, Canada, 2024. Available online: https://www.canada.ca/en/health-canada/services/drugs-health-products/drug-products/applications-submissions/guidance-documents/quality-new-drug-submissions.html (accessed on 25 April 2025).
- Agência Nacional de Vigilância Sanitária (ANVISA). Resolução RDC No. 301, de 21 de Agosto de 2019; Agência Nacional de Vigilância Sanitária (ANVISA): Brasília, Brazil, 2019. Available online: https://www.in.gov.br/en/web/dou/-/resolucao-rdc-n-301-de-21-de-agosto-de-2019-210546333 (accessed on 25 April 2025).
- Central Drugs Standard Control Organization (CDSCO). Drugs and Cosmetics Act, 1940 and Rules, 1945; Central Drugs Standard Control Organization (CDSCO): New Delhi, India, 2016. Available online: https://cdsco.gov.in/opencms/opencms/en/Acts-Rules (accessed on 25 April 2025).
- Pharmaceutical Inspection Co-operation Scheme (PIC/S). Good Manufacturing Practice Guide–Annex 1: Manufacture of Sterile Medicinal Products; PIC/S: Geneva, Switzerland, 2023; PE 009-17; Available online: https://picscheme.org/en/publications (accessed on 25 April 2025).
- World Health Organization (WHO). WHO Guidelines on Good Manufacturing Practices for Biological Products; World Health Organization (WHO): Geneva, Switzerland, 2016; Available online: https://www.who.int/publications/m/item/trs-1060---annex-3--who-good-manufacturing-practices-for-excipients-used-in-pharmaceutical-products (accessed on 25 April 2025).
- Manousi, N.; Tzanavaras, P.D.; Zacharis, C.K. Determination of bisphosphonate active pharmaceutical ingredients in pharmaceuticals and biological materials: An updated review. J. Pharm. Biomed. Anal. 2022, 219, 114921. [Google Scholar] [CrossRef] [PubMed]
- European Commission. EudraLex-Volume 4-EU Guidelines for Good Manufacturing Practice for Medicinal Products for Human and Veterinary Use. Annex 1: Manufacture of Sterile Medicinal Products; European Commission: Brussels, Belgium, 2023. [Google Scholar]
- Rahbar, N.; Nazernezhad, L.; Asadinezhad, M.; Ramezani, Z.; Kouchak, M. A novel micro-extraction strategy for extraction of bisphosphonates from biological fluids using zirconia nanoparticles coupled with spectrofluorimetry and high performance liquid chromatography. J. Food Drug Anal. 2018, 26, 1303–1311. [Google Scholar] [CrossRef]
- Chen, M.; Liu, K.; Zhong, D.; Chen, X. Trimethylsilyldiazomethane derivatization coupled with solid-phase extraction for the determination of alendronate in human plasma by LC-MS/MS. Anal. Bioanal. Chem. 2012, 402, 791–798. [Google Scholar] [CrossRef]
- Gupta, V.K.; Jain, R.; Sharma, S.; Agarwal, S.; Dwivedi, A. Quantitative determination of alendronate in human urine. Int. J. Electrochem. Sci. 2012, 7, 569–587. [Google Scholar] [CrossRef]
- Chen, Y.; Liu, Y.; Chen, Z.; Chen, M.; Zhu, Y. Simultaneous determination of alendronate, pamidronate, ibandronate and risedronate using ion chromatography with integrated pulsed amperometric detection. Chin. J. Chromatogr. 2013, 30, 414–418. [Google Scholar] [CrossRef]
- Luo, T.; He, S.; Deng, Y.; Zhang, X.; Dong, Y. Establishing tablet dissolution curve through determination of underivatized alendronate sodium by capillary electrophoresis-UV detector. Curr. Pharm. Anal. 2020, 16, 615–622. [Google Scholar] [CrossRef]
- Bertolini, T.; Vicentini, L.; Boschetti, S.; Andreatta, P.; Gatti, R. A novel automated hydrophilic interaction liquid chromatography method using diode-array detector/electrospray ionization tandem mass spectrometry for analysis of sodium risedronate and related degradation products in pharmaceuticals. J. Chromatogr. A 2014, 1365, 131–139. [Google Scholar] [CrossRef] [PubMed]
- U.S. Food and Drug Administration (FDA). Draft Guidance for Industry: Microbiological Quality Considerations in Non-Sterile Drug Manufacturing; Food and Drug Administration (FDA): Silver Spring, MD, USA, 2021.
- Ezzati Nazhad Dolatabadi, J.; Hamishehkar, H.; De La Guardia, M.; Valizadeh, H. A fast and simple spectrofluorometric method for the determination of alendronate sodium in pharmaceuticals. BioImpacts 2014, 4, 39–42. [Google Scholar] [CrossRef] [PubMed]
- Ombelet, S.; Ronat, J.B.; Walsh, T.; Yansouni, C.P.; Cox, J.; Vlieghe, E.; Martiny, D.; Semret, M.; Vandenberg, O.; Jacobs, J.; et al. Clinical bacteriology in low-resource settings: today’s solutions. Lancet Infect. Dis. 2018, 18, e248–e258. [Google Scholar] [CrossRef]
- Natale, A.; Ronat, J.B.; Mazoyer, A.; Rochard, A.; Boillot, B.; Hubert, J.; Baillet, B.; Ducasse, M.; Mantelet, F.; Oueslati, S.; et al. The Mini-Lab: Accessible clinical bacteriology for low-resource settings. Lancet Microbe. 2020, 1, e56–e58. [Google Scholar] [CrossRef]
- Elmalla, S.F.; Mansour, F.R. A simple innovative spectrofluorometric method for the determination of alendronate in bulk and in pharmaceutical tablets. Luminescence 2019, 34, 375–381. [Google Scholar] [CrossRef]
- Institute of Medicine (US) Committee on Accelerating Rare Diseases Research and Orphan Product Development. Development of new therapeutic drugs and biologics for rare diseases. In Rare Diseases and Orphan Products: Accelerating Research and Development; Field, M.J., Boat, T.F., Eds.; National Academies Press (US): Washington, DC, USA, 2010. Available online: https://www.ncbi.nlm.nih.gov/books/NBK56179/ (accessed on 24 April 2025).
- American Society for Microbiology. Reevaluation of Microbial Water Quality: Powerful New Tools for Detection and Risk Assessment: This Report Is Based on a Colloquium Sponsored by the American Academy of Microbiology Held March 3–4, 2000, in Amelia Island, Florida; American Society for Microbiology: Washington, DC, USA, 2000. Available online: http://www.ncbi.nlm.nih.gov/books/NBK562615/ (accessed on 24 April 2025).
- International Council for Harmonisation (ICH). Q7: Good Manufacturing Practice for Active Pharmaceutical Ingredients; International Council for Harmonisation (ICH): Geneva, Switzerland, 2000; Available online: https://database.ich.org/sites/default/files/Q7_Guideline.pdf (accessed on 24 June 2025).
- International Council for Harmonisation (ICH). Q9: Quality Risk Management; International Council for Harmonisation (ICH): Geneva, Switzerland, 2015; Available online: https://www.ema.europa.eu/en/ich-q9-quality-risk-management-scientific-guideline (accessed on 24 June 2025).
- International Council for Harmonisation (ICH). Q10: Pharmaceutical Quality System; International Council for Harmonisation (ICH): Geneva, Switzerland, 2008; Available online: https://database.ich.org/sites/default/files/Q10_Guideline.pdf (accessed on 24 June 2025).
- International Council for Harmonisation (ICH). Q11: Development and Manufacture of Drug Substances; International Council for Harmonisation (ICH): Geneva, Switzerland, 2012; Available online: https://database.ich.org/sites/default/files/Q11%20Guideline.pdf (accessed on 24 June 2025).
- Lezotre, P.L. State of play and review of major cooperation initiatives. In International Cooperation, Convergence and Harmonization of Pharmaceutical Regulations; Elsevier: Amsterdam, The Netherlands, 2014; pp. 7–170. [Google Scholar] [CrossRef]
- Slavich, G.M.; Roos, L.G.; Mengelkoch, S.; Webb, C.A.; Shattuck, E.C.; Moriarity, D.P.; Alley, J.C. Social Safety Theory: Conceptual foundation, underlying mechanisms, and future directions. Health Psychol. Rev. 2023, 17, 5–59. [Google Scholar] [CrossRef]
- Bharate, S.S. Critical analysis of drug product recalls due to nitrosamine impurities. J. Med. Chem. 2021, 64, 2923–2936. [Google Scholar] [CrossRef]
- Chen, H.; Qin, L.; Jiang, C.; Qin, M.; Sun, Y.; Luo, J. Characteristics, risk management and GMP standards of pharmaceutical companies in China. Front. Public Health 2023, 11, 1103555. [Google Scholar] [CrossRef]
- Ferrara, G.; Cangelosi, G.; Morales Palomares, S.; Mancin, S.; Melina, M.; Diamanti, O.; Sguanci, M.; Amendola, A.; Petrelli, F. Optimizing Ultrasound Probe Disinfection for Healthcare-Associated Infection Control: A Comparative Analysis of Disinfectant Efficacy. Microorganisms 2024, 12, 2394. [Google Scholar] [CrossRef]
- Sguanci, M.; Mancin, S.; Morales Palomares, S.; Cangelosi, G.; Parozzi, M.; Piredda, M.; De Marinis, M.G. The Role of Clinical Nurse Specialist and the Safety Management in Operating Theatre during the COVID-19 Pandemic: An Integrative Scoping Review. Perioper. Care Oper. Room Manag. 2024, 37, 100437. [Google Scholar] [CrossRef]
- Assolari, F.; Mancin, S.; Lopane, D.; Dacomi, A.; Coldani, C.; Tomaiuolo, G.; Cattani, D.; Palomares, S.M.; Cangelosi, G.; Mazzoleni, B. Advanced practice nursing in surgery: A scoping review of roles, responsibilities, and educational programs. Int. Nurs. Rev. 2025, 72, e13045. [Google Scholar] [CrossRef] [PubMed]
- Sguanci, M.; Mancin, S.; Gazzelloni, A.; Diamanti, O.; Ferrara, G.; Morales Palomares, S.; Parozzi, M.; Petrelli, F.; Cangelosi, G. The Internet of Things in the Nutritional Management of Patients with Chronic Neurological Cognitive Impairment: A Scoping Review. Healthcare 2024, 13, 23. [Google Scholar] [CrossRef] [PubMed]
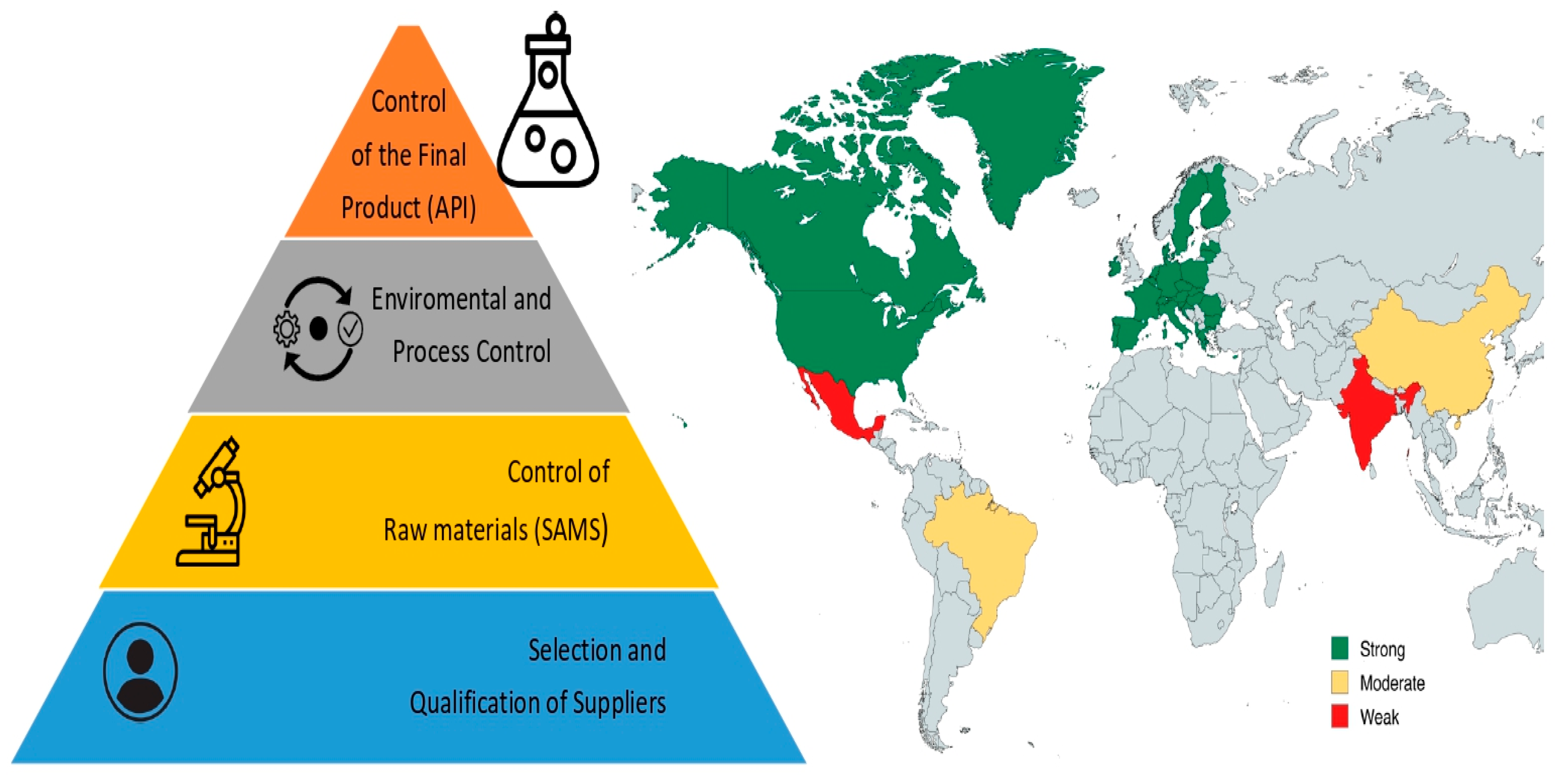
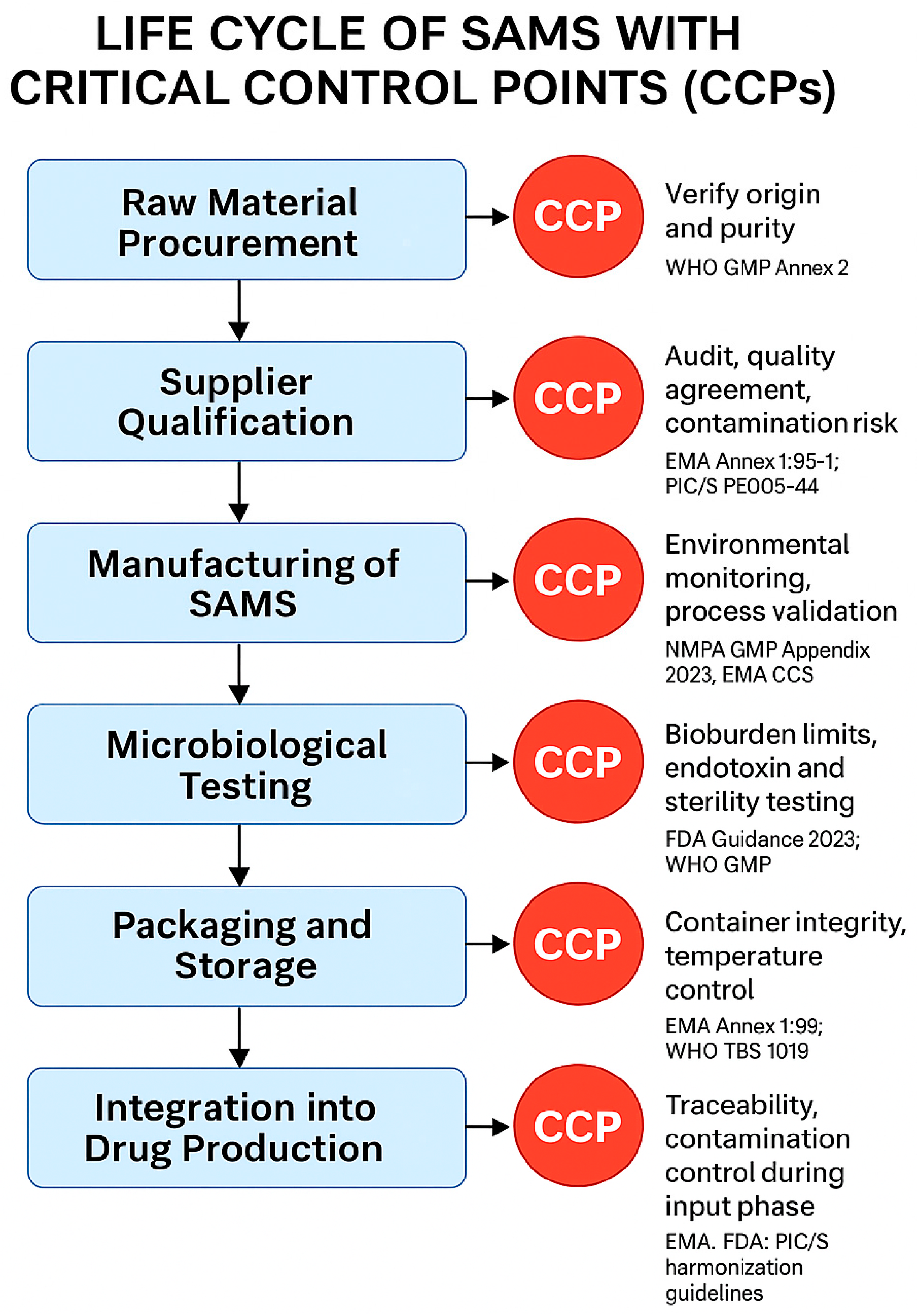

{kind=link}
{kind=link}
| Condition/Institution | EMA (Europe) | NMPA (China) | COFEPRIS (Mexico) | FDA (USA) | Health Canada (Canada) | ANVISA (Brazil) | CDSCO (India) | PIC/S | WHO |
|---|---|---|---|---|---|---|---|---|---|
| Guideline for microbiological control of SAMS | Yes | Yes | No | Yes | Yes | Partial | No | Yes | Yes |
| Measures before SAMS introduction | Yes | Yes | No | Yes | Yes | No | No | Yes | Partial |
| Requirement for risk-based CCS | Yes | Yes | No | Yes | Yes | No | No | Yes | Partial |
| Control of upstream stages (before GMP application) | Yes | Yes * | No | Yes | Yes | No | No | Yes | Encouraged |
| Supplier qualification (microbiological quality) | Yes | Yes | No | Yes | Yes | Partial | No | Yes | Yes |
| Bioburden/sterility validation standards | Yes | Yes | No | Yes | Yes | No | No | Yes | Partial |
| Clear definition of GMP starting point | Yes 1 | Yes | No | Yes | Yes | No | No | Yes | Yes 1 |
| Emphasis on rapid/alternative microbiological methods | Yes | Yes | No | Yes | Partial | No | No | Yes | Partial |
| Mandatory environmental and personnel microbiological monitoring | Yes | Yes | Partial | Yes | Yes | No | No | Yes | Partial |
| Upstream synthetic process description required | N/A | Optional | No | Yes | Yes | Yes | Yes | N/A | N/A |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gravante, F.; Sacchini, F.; Mancin, S.; Lopane, D.; Parozzi, M.; Ferrara, G.; Sguanci, M.; Morales Palomares, S.; Biondini, F.; Marfella, F.; et al. Preventing Microorganism Contamination in Starting Active Materials for Synthesis from Global Regulatory Agencies: Overview for Public Health Implications. Microorganisms 2025, 13, 1595. https://doi.org/10.3390/microorganisms13071595
Gravante F, Sacchini F, Mancin S, Lopane D, Parozzi M, Ferrara G, Sguanci M, Morales Palomares S, Biondini F, Marfella F, et al. Preventing Microorganism Contamination in Starting Active Materials for Synthesis from Global Regulatory Agencies: Overview for Public Health Implications. Microorganisms. 2025; 13(7):1595. https://doi.org/10.3390/microorganisms13071595
Chicago/Turabian StyleGravante, Francesco, Francesco Sacchini, Stefano Mancin, Diego Lopane, Mauro Parozzi, Gaetano Ferrara, Marco Sguanci, Sara Morales Palomares, Federico Biondini, Francesca Marfella, and et al. 2025. "Preventing Microorganism Contamination in Starting Active Materials for Synthesis from Global Regulatory Agencies: Overview for Public Health Implications" Microorganisms 13, no. 7: 1595. https://doi.org/10.3390/microorganisms13071595
APA StyleGravante, F., Sacchini, F., Mancin, S., Lopane, D., Parozzi, M., Ferrara, G., Sguanci, M., Morales Palomares, S., Biondini, F., Marfella, F., Cangelosi, G., Caggianelli, G., & Petrelli, F. (2025). Preventing Microorganism Contamination in Starting Active Materials for Synthesis from Global Regulatory Agencies: Overview for Public Health Implications. Microorganisms, 13(7), 1595. https://doi.org/10.3390/microorganisms13071595