

Mechanisms Operating in the Use of Transition Metal Complexes to Combat Antimicrobial Resistance



Abstract
1. Introduction
2. Microbial Infections
3. Applications of Transition Metal Complexes in Antimicrobial Fields
3.1. Silver Complexes
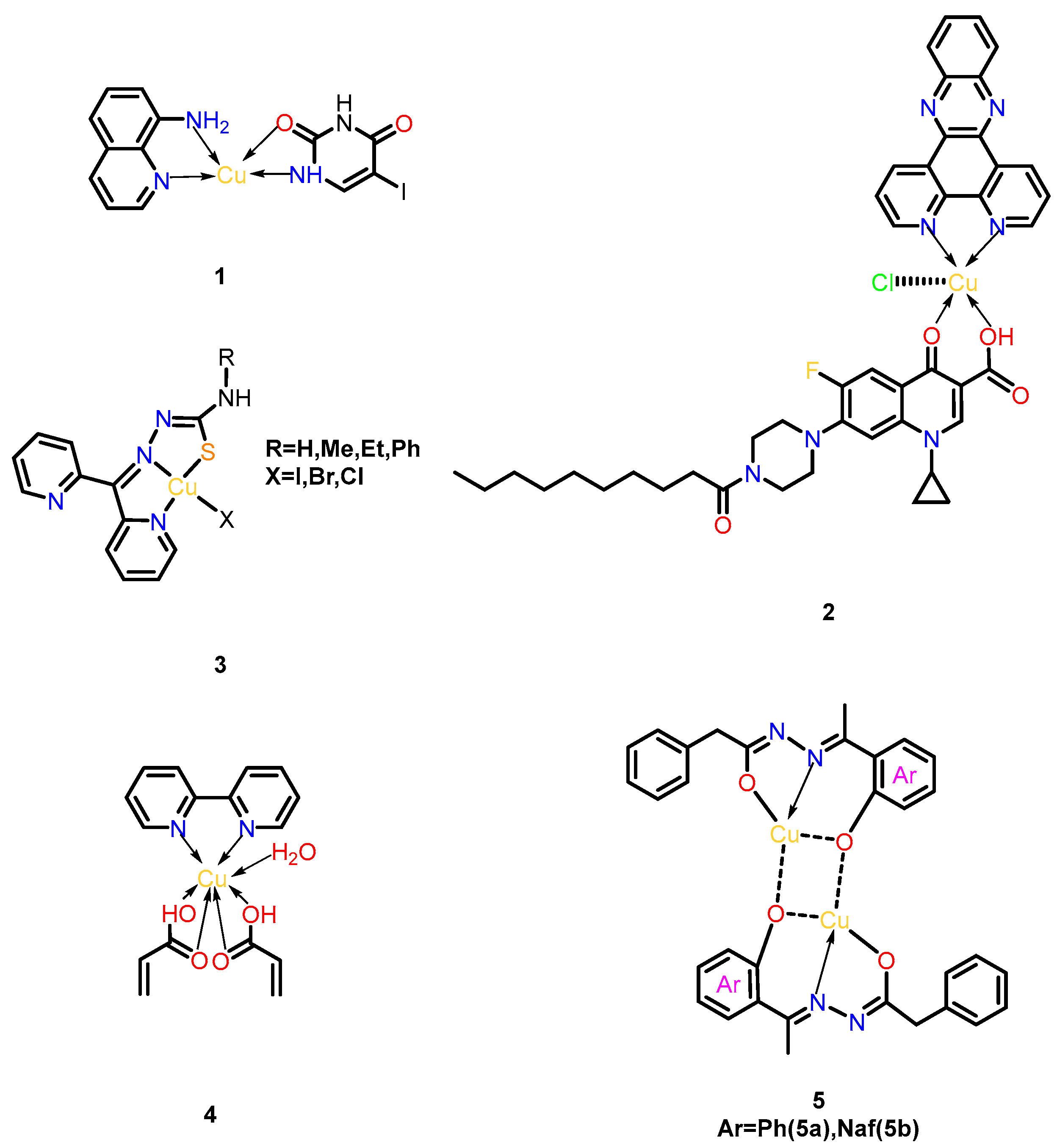
3.2. Copper Complexes
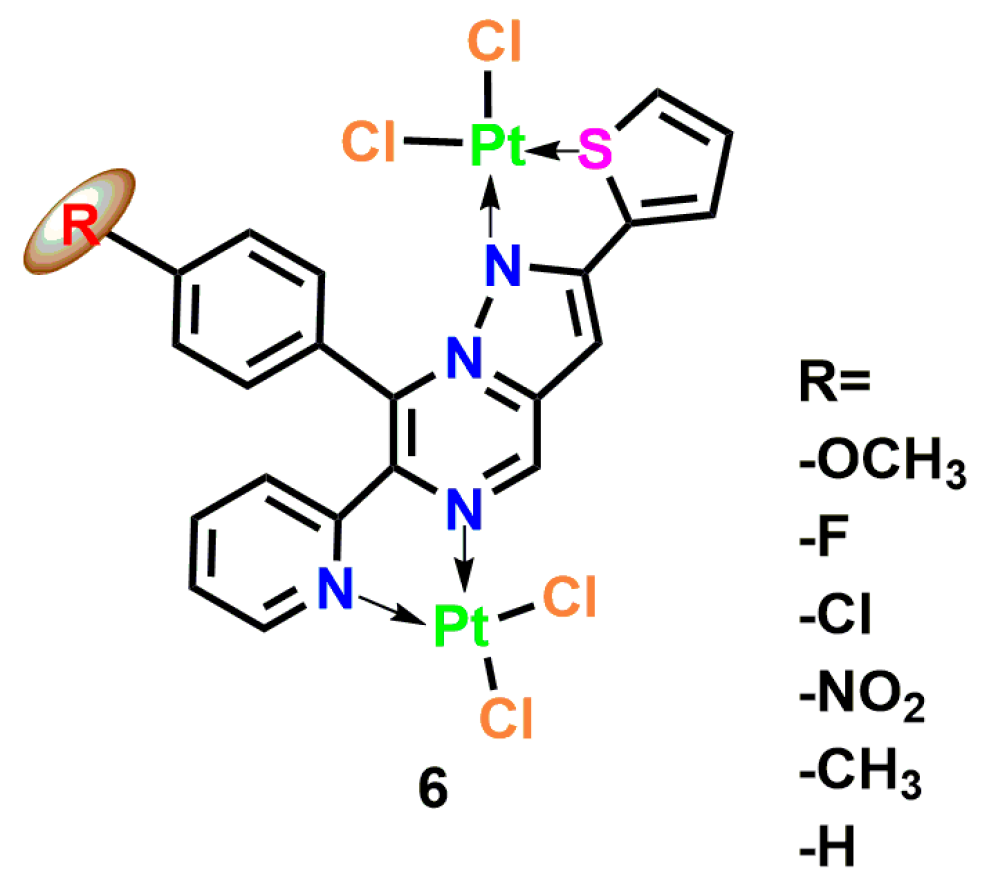
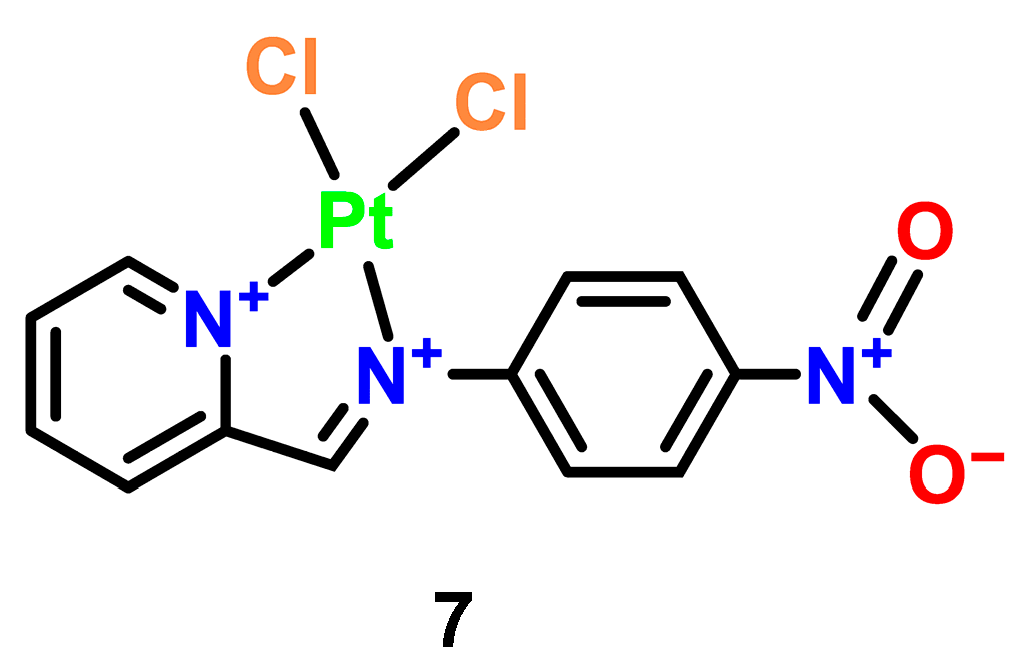
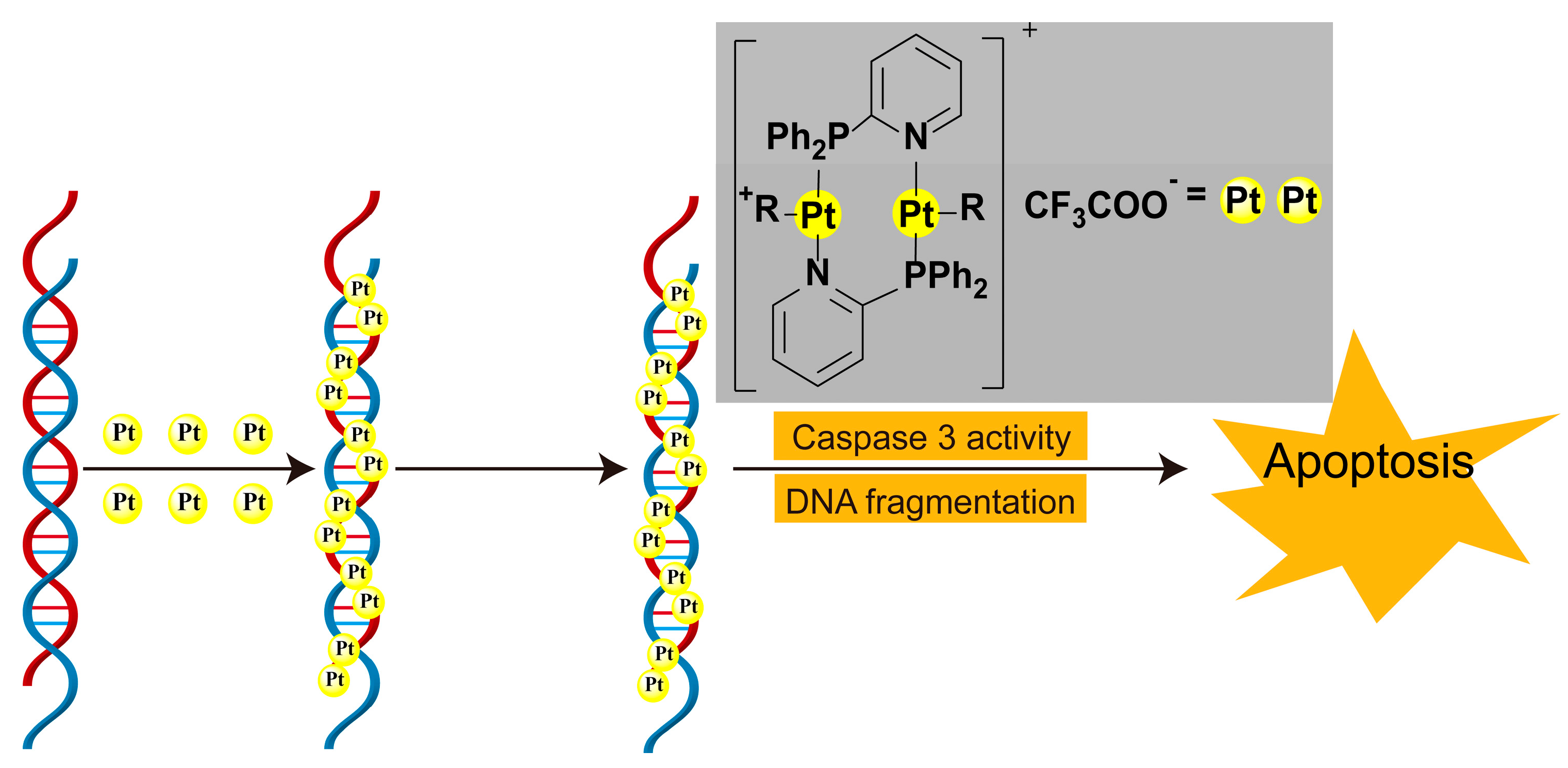
3.3. Platinum Complexes
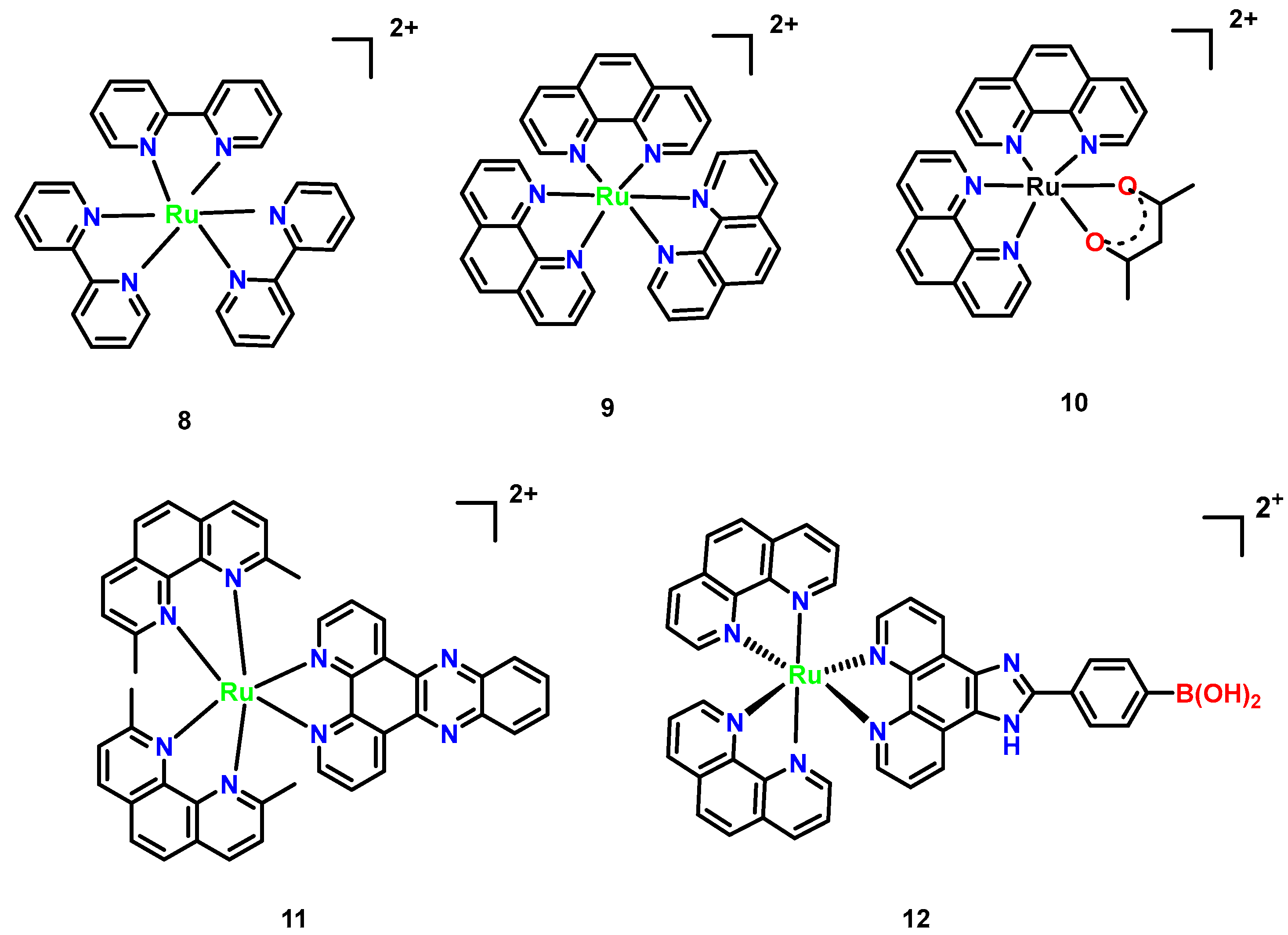
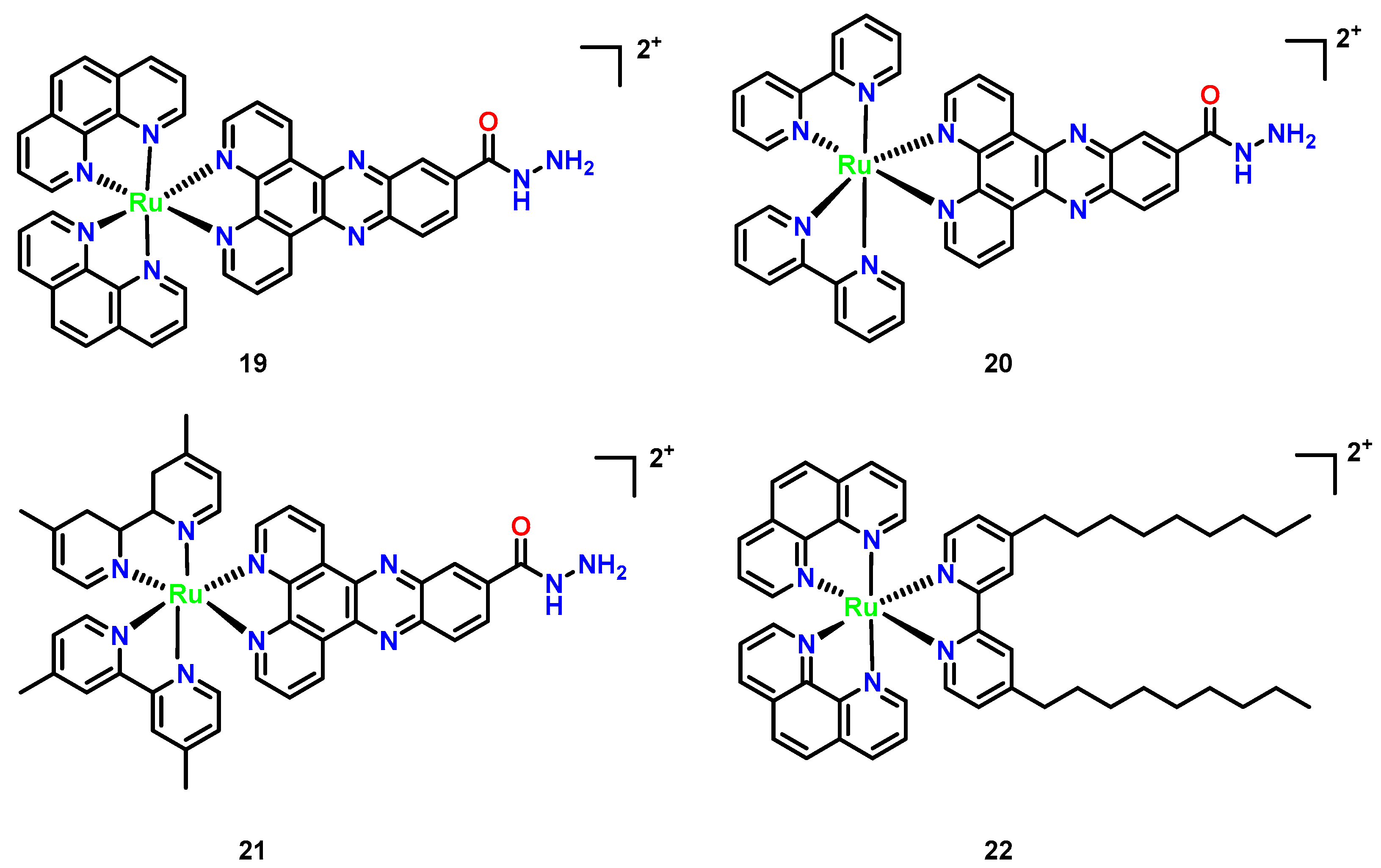
3.4. Ruthenium Complexes
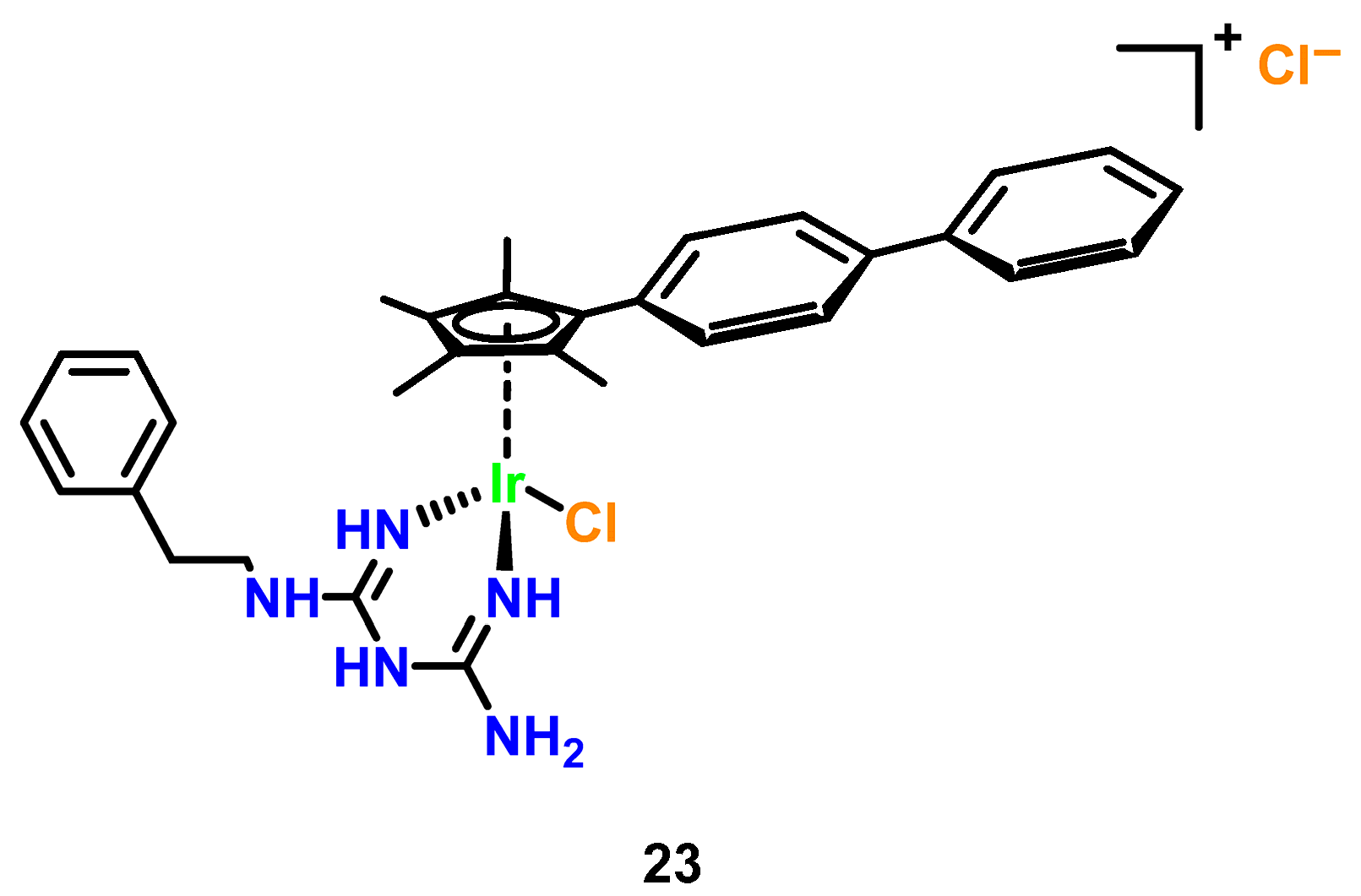
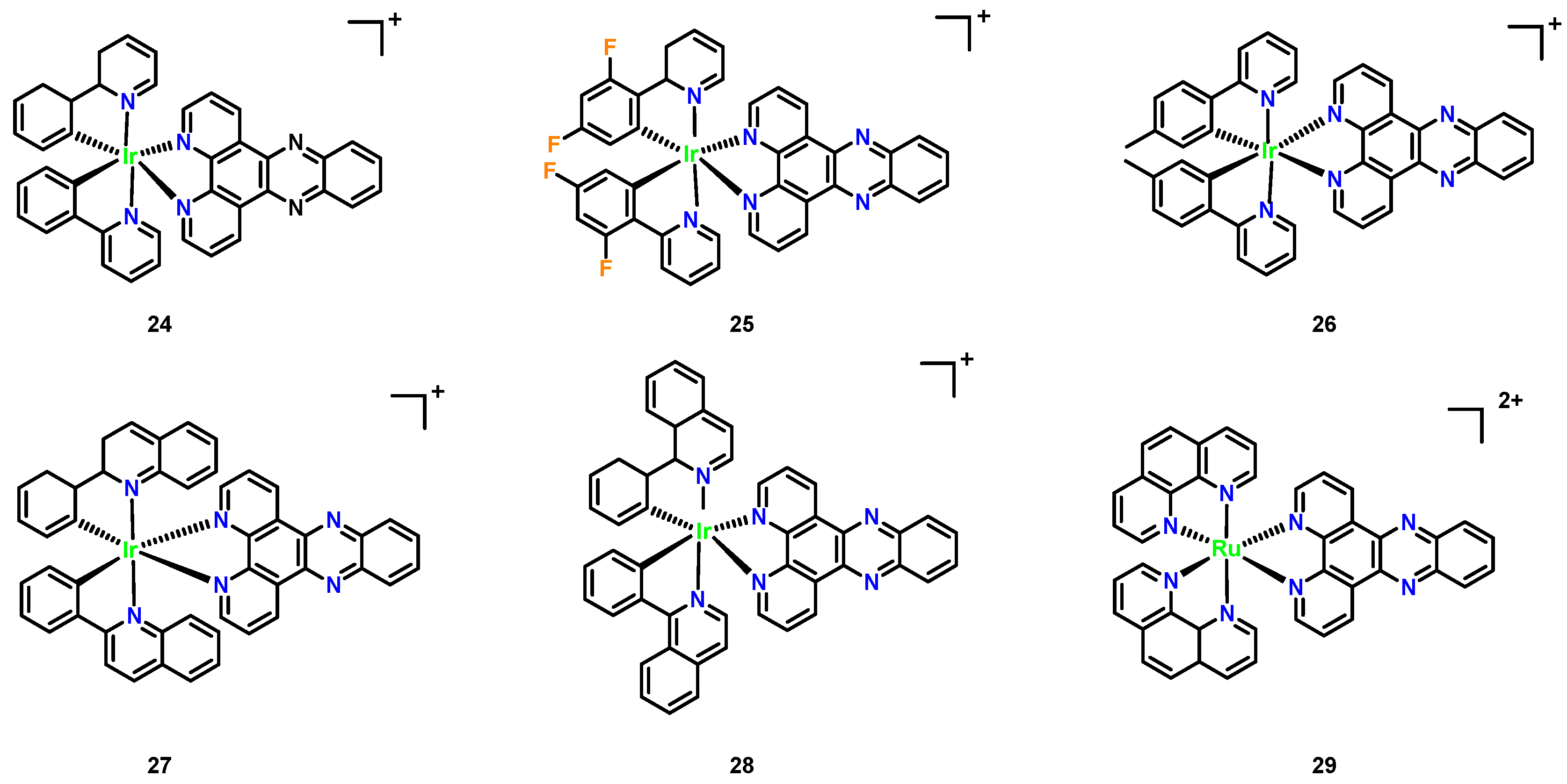
3.5. Iridium Complexes
3.6. Comparative Summary of Antimicrobial Metal Complexes
4. Challenges and Future Perspectives
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ROS | Reactive Oxygen Species |
| MIC | minimum inhibitory concentration |
| MRSA | methicillin-resistant Staphylococcus aureus |
| VRE | vancomycin-resistant Enterococci |
| CRE | carbapenem-resistant Enterobacteriaceae |
| MDR-TB | Multidrug-resistant Mycobacterium tuberculosis |
| XDR | extensively drug-resistant |
| PDR | pandrug-resistant |
| TMDs | transition metal dichalcogenides |
| FDA | U.S. Food and Drug Administration |
| SEM | scanning electron microscopy |
| Co-ADD | Community for Open Antimicrobial Drug Discovery |
| HC50 | hemolytic concentration 50% |
| MDR | multidrug-resistant |
| dppz | dipyrido [3,2-a:2′,3′-c]phenazine |
| pta | 1,3,5-triaza-7-phosphatricyclo [3.3.1.1] decane |
| Cp* | pentamethylcyclopentadienyl |
References
- Mutalik, C.; Lin, I.H.; Krisnawati, D.I.; Khaerunnisa, S.; Khafid, M.; Hsiao, Y.-C.; Kuo, T.-R. Antibacterial pathways in transition metal-based nanocomposites: A mechanistic overview. Int. J. Nanomed. 2022, 17, 6821. [Google Scholar] [CrossRef] [PubMed]
- Frei, A.; Zuegg, J.; Elliott, A.G.; Baker, M.; Braese, S.; Brown, C.; Chen, F.; Dowson, C.G.; Dujardin, G.; Jung, N. Metal complexes as a promising source for new antibiotics. Chem. Sci. 2020, 11, 2627–2639. [Google Scholar] [CrossRef]
- Sharma, S.; Mohler, J.; Mahajan, S.D.; Schwartz, S.A.; Bruggemann, L.; Aalinkeel, R. Microbial biofilm: A review on formation, infection, antibiotic resistance, control measures, and innovative treatment. Microorganisms 2023, 11, 1614. [Google Scholar] [CrossRef] [PubMed]
- Deusenbery, C.; Wang, Y.; Shukla, A. Recent innovations in bacterial infection detection and treatment. ACS Infect. Dis. 2021, 7, 695–720. [Google Scholar] [CrossRef] [PubMed]
- Fang, W.; Wu, J.; Cheng, M.; Zhu, X.; Du, M.; Chen, C.; Liao, W.; Zhi, K.; Pan, W. Diagnosis of invasive fungal infections: Challenges and recent developments. J. Biomed. Sci. 2023, 30, 42. [Google Scholar] [CrossRef]
- Lionakis, M.S.; Drummond, R.A.; Hohl, T.M. Immune responses to human fungal pathogens and therapeutic prospects. Nat. Rev. Immunol. 2023, 23, 433–452. [Google Scholar] [CrossRef]
- O’Neill, J. Review on Antimicrobial Resistance: Tackling Drug-Resistant Infections Globally: Final Report and Recommendations. 2016. Available online: https://www.cabidigitallibrary.org/doi/full/10.5555/20163354200 (accessed on 6 May 2025).
- OneHealthTrust. ResistanceMap: Antibiotic Use. 2023. Available online: https://resistancemap.onehealthtrust.org/AntibioticUse.php (accessed on 6 May 2025).
- Baran, A.; Kwiatkowska, A.; Potocki, L. Antibiotics and bacterial resistance—A short story of an endless arms race. Int. J. Mol. Sci. 2023, 24, 5777. [Google Scholar] [CrossRef]
- Zhang, F.; Cheng, W. The mechanism of bacterial resistance and potential bacteriostatic strategies. Antibiotics 2022, 11, 1215. [Google Scholar] [CrossRef]
- Mlynarczyk-Bonikowska, B.; Kowalewski, C.; Krolak-Ulinska, A.; Marusza, W. Molecular mechanisms of drug resistance in Staphylococcus aureus. Int. J. Mol. Sci. 2022, 23, 8088. [Google Scholar] [CrossRef]
- Ahmed, M.O.; Baptiste, K.E. Vancomycin-resistant enterococci: A review of antimicrobial resistance mechanisms and perspectives of human and animal health. Microb. Drug Resist. 2018, 24, 590–606. [Google Scholar] [CrossRef]
- Ma, J.; Song, X.; Li, M.; Yu, Z.; Cheng, W.; Yu, Z.; Zhang, W.; Zhang, Y.; Shen, A.; Sun, H. Global spread of carbapenem-resistant Enterobacteriaceae: Epidemiological features, resistance mechanisms, detection and therapy. Microbiol. Res. 2023, 266, 127249. [Google Scholar] [CrossRef]
- Singh, V.; Chibale, K. Strategies to combat multi-drug resistance in tuberculosis. Acc. Chem. Res. 2021, 54, 2361–2376. [Google Scholar] [CrossRef] [PubMed]
- Nunes, J.H.B.; Nakahata, D.H.; Corbi, P.P.; de Paiva, R.E.F. Beyond silver sulfadiazine: A dive into more than 50 years of research and development on metal complexes of sulfonamides in medicinal inorganic chemistry. Coord. Chem. Rev. 2023, 490, 215228. [Google Scholar] [CrossRef]
- Wang, H.; Wang, M.; Yang, X.; Xu, X.; Hao, Q.; Yan, A.; Hu, M.; Lobinski, R.; Li, H.; Sun, H. Antimicrobial silver targets glyceraldehyde-3-phosphate dehydrogenase in glycolysis of E. coli. Chem. Sci. 2019, 10, 7193–7199. [Google Scholar] [CrossRef]
- Frei, A. Metal complexes, an untapped source of antibiotic potential? Antibiotics 2020, 9, 90. [Google Scholar] [CrossRef]
- Vincent, M.; Hartemann, P.; Engels-Deutsch, M. Antimicrobial applications of copper. Int. J. Hyg. Environ. Health 2016, 219, 585–591. [Google Scholar] [CrossRef] [PubMed]
- Phopin, K.; Sinthupoom, N.; Treeratanapiboon, L.; Kunwittaya, S.; Prachayasittikul, S.; Ruchirawat, S.; Prachayasittikul, V. Antimalarial and antimicrobial activities of 8-aminoquinoline-uracils metal complexes. Excli J. 2016, 15, 144. [Google Scholar]
- Ude, Z.; Kavanagh, K.; Twamley, B.; Pour, M.; Gathergood, N.; Kellett, A.; Marmion, C.J. A new class of prophylactic metallo-antibiotic possessing potent anti-cancer and anti-microbial properties. Dalton Trans. 2019, 48, 8578–8593. [Google Scholar] [CrossRef]
- Lobana, T.S.; Kaushal, M.; Bala, R.; Nim, L.; Paul, K.; Arora, D.S.; Bhatia, A.; Arora, S.; Jasinski, J.P. Di-2-pyridylketone-N1-substituted thiosemicarbazone derivatives of copper (II): Biosafe antimicrobial potential and high anticancer activity against immortalized L6 rat skeletal muscle cells. J. Inorg. Biochem. 2020, 212, 111205. [Google Scholar] [CrossRef]
- Vasile Scăețeanu, G.; Chifiriuc, M.C.; Bleotu, C.; Kamerzan, C.; Măruţescu, L.; Daniliuc, C.G.; Maxim, C.; Calu, L.; Olar, R.; Badea, M. Synthesis, structural characterization, antimicrobial activity, and in vitro biocompatibility of new unsaturated carboxylate complexes with 2, 2′-bipyridine. Molecules 2018, 23, 157. [Google Scholar] [CrossRef]
- El-Medani, S.M.; Makhlouf, A.A.; Moustafa, H.; Afifi, M.A.; Haukka, M.; Ramadan, R.M. Spectroscopic, crystal structural, theoretical and biological studies of phenylacetohydrazide schiff base derivatives and their copper complexes. J. Mol. Struct. 2020, 1208, 127860. [Google Scholar] [CrossRef]
- Anthony, E.J.; Bolitho, E.M.; Bridgewater, H.E.; Carter, O.W.L.; Donnelly, J.M.; Imberti, C.; Lant, E.C.; Lermyte, F.; Needham, R.J.; Palau, M. Metallodrugs are unique: Opportunities and challenges of discovery and development. Chem. Sci. 2020, 11, 12888–12917. [Google Scholar] [CrossRef] [PubMed]
- Johnstone, T.C.; Suntharalingam, K.; Lippard, S.J. The next generation of platinum drugs: Targeted Pt (II) agents, nanoparticle delivery, and Pt (IV) prodrugs. Chem. Rev. 2016, 116, 3436–3486. [Google Scholar] [CrossRef]
- Lunagariya, M.V.; Thakor, K.P.; Waghela, B.N.; Pathak, C.; Patel, M.N. Design, synthesis, pharmacological evaluation and DNA interaction studies of binuclear Pt (II) complexes with pyrazolo [1,5-a] pyrimidine scaffold. Appl. Organomet. Chem. 2018, 32, e4222. [Google Scholar] [CrossRef]
- Rubino, S.; Pibiri, I.; Minacori, C.; Alduina, R.; Di Stefano, V.; Orecchio, S.; Buscemi, S.; Girasolo, M.A.; Tesoriere, L.; Attanzio, A. Synthesis, structural characterization, anti-proliferative and antimicrobial activity of binuclear and mononuclear Pt (II) complexes with perfluoroalkyl-heterocyclic ligands. Inorganica Chim. Acta 2018, 483, 180–190. [Google Scholar] [CrossRef]
- Olesya, S.; Alexander, P. Antimicrobial activity of mono-and polynuclear platinum and palladium complexes. Foods Raw Mater. 2020, 8, 298–311. [Google Scholar]
- Demberelnyamba, D.; Kim, K.-S.; Choi, S.; Park, S.-Y.; Lee, H.; Kim, C.-J.; Yoo, I.-D. Synthesis and antimicrobial properties of imidazolium and pyrrolidinonium salts. Bioorganic Med. Chem. 2004, 12, 853–857. [Google Scholar] [CrossRef]
- Borowiecki, P.; Milner-Krawczyk, M.; Brzezińska, D.; Wielechowska, M.; Plenkiewicz, J. Synthesis and antimicrobial activity of imidazolium and triazolium chiral ionic liquids. Eur. J. Org. Chem. 2013, 2013, 712–720. [Google Scholar] [CrossRef]
- Birnie, C.R.; Malamud, D.; Schnaare, R.L. Antimicrobial evaluation of N-alkyl betaines and N-alkyl-N, N-dimethylamine oxides with variations in chain length. Antimicrob. Agents Chemother. 2000, 44, 2514–2517. [Google Scholar] [CrossRef]
- Al-Khathami, N.D.; Al-Rashdi, K.S.; Babgi, B.A.; Hussien, M.A.; Arshad, M.N.; Eltayeb, N.E.; Elsilk, S.E.; Lasri, J.; Basaleh, A.S.; Al-Jahdali, M. Spectroscopic and biological properties of platinum complexes derived from 2-pyridyl schiff bases. J. Saudi Chem. Soc. 2019, 23, 903–915. [Google Scholar] [CrossRef]
- Wheate, N.J.; Collins, J.G. Multi-nuclear platinum complexes as anti-cancer drugs. Coord. Chem. Rev. 2003, 241, 133–145. [Google Scholar] [CrossRef]
- Salishcheva, O.V.; Moldagulova, N.E.; Gel’Fman, M.I. Thermodynamic stability and acidic properties of platinum (II) and palladium (II) bromide complexes. Russ. J. Inorg. Chem. 2006, 51, 683–686. [Google Scholar] [CrossRef]
- Ashoo, P.; Yousefi, R.; Nabavizadeh, S.M.; Aseman, M.D.; Paziresh, S.; Ghasemi, A.; Saboury, A.A. Three Pt-Pt complexes with donor-acceptor feature: Anticancer activity, DNA binding studies and molecular docking simulation. Anti-Cancer Agents Med. Chem. 2019, 19, 1762–1774. [Google Scholar] [CrossRef]
- Sousa, S.A.; Leitão, J.H.; Silva, R.A.L.; Belo, D.; Santos, I.C.; Guerreiro, J.F.; Martins, M.; Fontinha, D.; Prudêncio, M.; Almeida, M. On the path to gold: Monoanionic Au bisdithiolate complexes with antimicrobial and antitumor activities. J. Inorg. Biochem. 2020, 202, 110904. [Google Scholar] [CrossRef]
- Claudel, M.; Schwarte, J.V.; Fromm, K.M. New antimicrobial strategies based on metal complexes. Chemistry 2020, 2, 849–899. [Google Scholar] [CrossRef]
- Allardyce, C.S.; Dyson, P.J.; Ellis, D.J.; Salter, P.A.; Scopelliti, R. Synthesis and characterisation of some water soluble ruthenium (II)-arene complexes and an investigation of their antibiotic and antiviral properties. J. Organomet. Chem. 2003, 668, 35–42. [Google Scholar] [CrossRef]
- Mansour, A.M.; Radacki, K. Protein binding affinity of biologically active thiourea based half-sandwich Ru (II) cymene complexes. Polyhedron 2020, 175, 114175. [Google Scholar] [CrossRef]
- Kljun, J.; Scott, A.J.; Lanišnik Rižner, T.; Keiser, J.; Turel, I. Synthesis and biological evaluation of organoruthenium complexes with azole antifungal agents. First crystal structure of a tioconazole metal complex. Organometallics 2014, 33, 1594–1601. [Google Scholar] [CrossRef]
- Li, F.; Collins, J.G.; Keene, F.R. Ruthenium complexes as antimicrobial agents. Chem. Soc. Rev. 2015, 44, 2529–2542. [Google Scholar] [CrossRef]
- Sun, D.; Zhang, W.; Lv, M.; Yang, E.; Zhao, Q.; Wang, W. Antibacterial activity of ruthenium (II) polypyridyl complex manipulated by membrane permeability and cell morphology. Bioorganic Med. Chem. Lett. 2015, 25, 2068–2073. [Google Scholar] [CrossRef]
- Pandrala, M.; Li, F.; Feterl, M.; Mulyana, Y.; Warner, J.M.; Wallace, L.; Keene, F.R.; Collins, J.G. Chlorido-containing ruthenium (II) and iridium (III) complexes as antimicrobial agents. Dalton Trans. 2013, 42, 4686–4694. [Google Scholar] [CrossRef] [PubMed]
- Kumar, Y.P.; Devi, C.S.; Srishailam, A.; Deepika, N.; Kumar, V.R.; Reddy, P.V.; Nagasuryaprasad, K.; Singh, S.S.; Nagababu, P.; Satyanarayana, S. Studies on photocleavage, DNA binding, cytotoxicity, and docking studies of ruthenium (II) mixed ligand complexes. J. Fluoresc. 2016, 26, 2119–2132. [Google Scholar] [CrossRef] [PubMed]
- Fu, C.; Lv, Q.; Fan, J.; Wu, S.; Lei, M.; Zhang, X.; Li, X.; Zhou, W.; Yu, Y.; Ren, W. Discovery of polypyridyl iridium (III) complexes as potent agents against resistant Candida albicans. Eur. J. Med. Chem. 2022, 233, 114250. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Fu, C.; Zhao, Z.; Fu, A. Targeted theranostic of cryptococcal encephalitis by a novel polypyridyl ruthenium complex. Mol. Pharm. 2019, 17, 145–154. [Google Scholar] [CrossRef]
- Chen, F.; Moat, J.; McFeely, D.; Clarkson, G.; Hands-Portman, I.J.; Furner-Pardoe, J.P.; Harrison, F.; Dowson, C.G.; Sadler, P.J. Biguanide iridium (III) complexes with potent antimicrobial activity. J. Med. Chem. 2018, 61, 7330–7344. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Organism | MIC (μg/mL) | ||
|---|---|---|---|
| 26 | Fluconazole | 29 | |
| C. albicans SC5314 | 1 | 1 | 64 |
| C. albicans G5 | 2 | 32 | >64 |
| C. albicans Caci17 | 8 | >64 | >64 |
| C. parapsilosis ATCC2019 | 2 | 2 | 8 |
| C. krusei ATCC6258 | 8 | 32 | 64 |
| C. neoformans H99 | 4 | 4 | 4 |
| C. neoformans 5-FC | 4 | 2 | 4 |
| C. gattii R265 | 2 | 4 | >64 |
| HC50 | 64 | - | >256 |
| Metal | Key Mechanisms | MIC Range (μg/mL) | Model Pathogens | Major Limitations |
|---|---|---|---|---|
| Silver | ROS generation, membrane disruption, | 0.5–168 | P. aeruginosa, MRSA | Mammalian cytotoxicity, efflux pump induction |
| Copper | DNA gyrase inhibition, Fenton-like ROS | 0.5–32 | S. aureus, E. coil | Narrow therapeutic window, oxidation instability |
| Platinum | DNA crosslinking, helical distortion | 2–16 | MDR-TB, CRE | Nephrotoxicity, poor solubility |
| Ruthenium | DNA/RNA damage, membrane penetration | 1.6–16.2 | C. neoformans, MRSA | Hemolytic effects, Gram-negative selectivity |
| Iridium | DNA intercalation, biofilm disruption | 0.125–8 | VRE, C. albicans | Efflux-mediated resistance (Pseudomonas) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, S.; Wang, M.; Liu, Z.; Fu, C. Mechanisms Operating in the Use of Transition Metal Complexes to Combat Antimicrobial Resistance. Microorganisms 2025, 13, 1570. https://doi.org/10.3390/microorganisms13071570
Wu S, Wang M, Liu Z, Fu C. Mechanisms Operating in the Use of Transition Metal Complexes to Combat Antimicrobial Resistance. Microorganisms. 2025; 13(7):1570. https://doi.org/10.3390/microorganisms13071570
Chicago/Turabian StyleWu, Shiming, Meishu Wang, Ziyi Liu, and Chen Fu. 2025. "Mechanisms Operating in the Use of Transition Metal Complexes to Combat Antimicrobial Resistance" Microorganisms 13, no. 7: 1570. https://doi.org/10.3390/microorganisms13071570
APA StyleWu, S., Wang, M., Liu, Z., & Fu, C. (2025). Mechanisms Operating in the Use of Transition Metal Complexes to Combat Antimicrobial Resistance. Microorganisms, 13(7), 1570. https://doi.org/10.3390/microorganisms13071570