
Preliminary Insights into the Gut Microbiota of Captive Tigers in Republic of Korea: Influence of Geographic and Individual Variation
 ,
,  , , and
, , and
Abstract
1. Introduction
2. Results
2.1. Alpha Diversity Outcomes and Interpretations
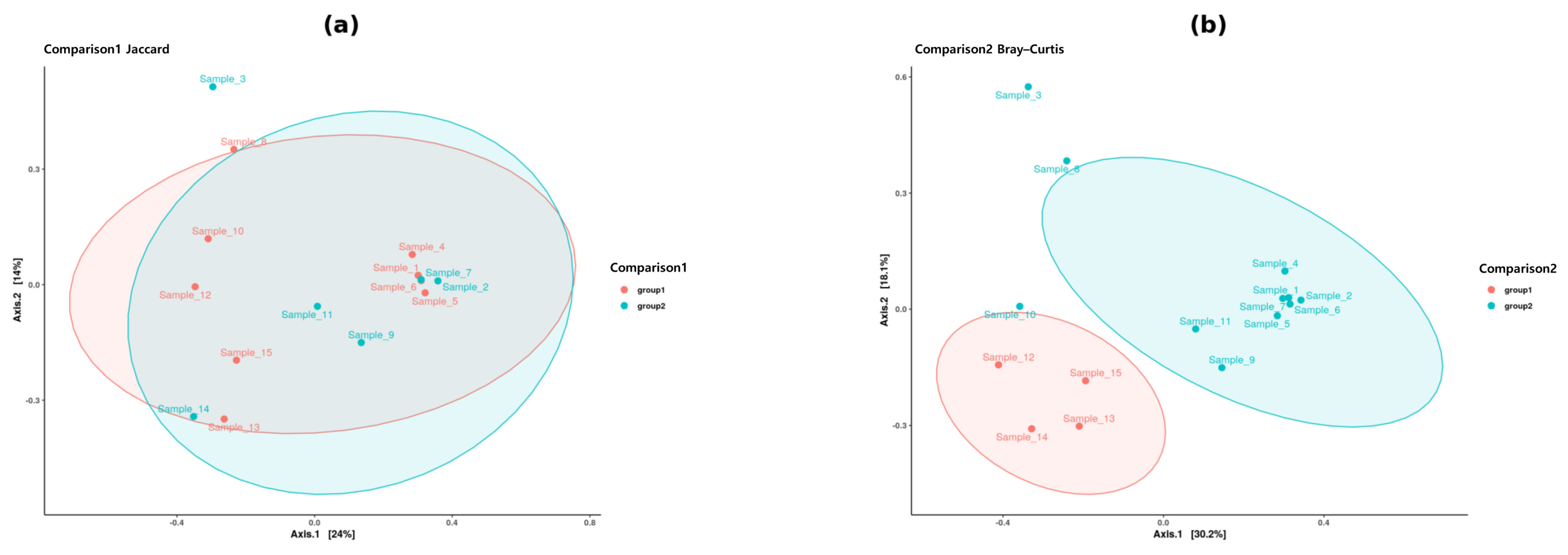
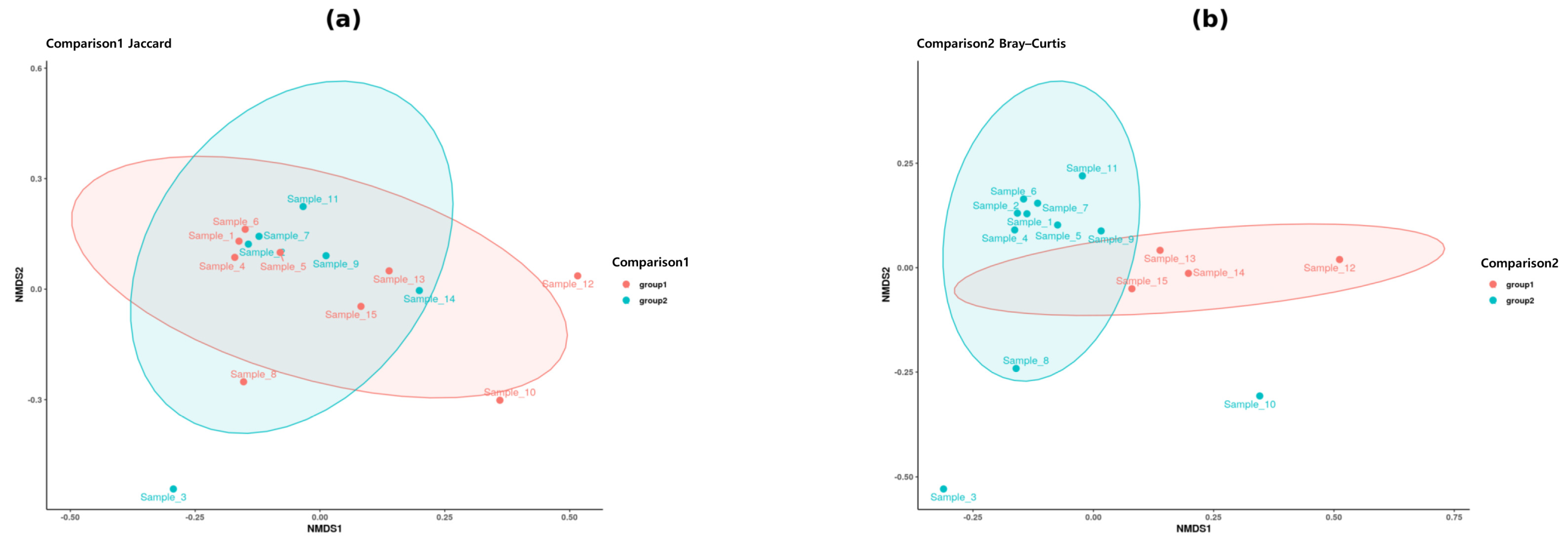
2.2. Beta Diversity Outcomes and Interpretations
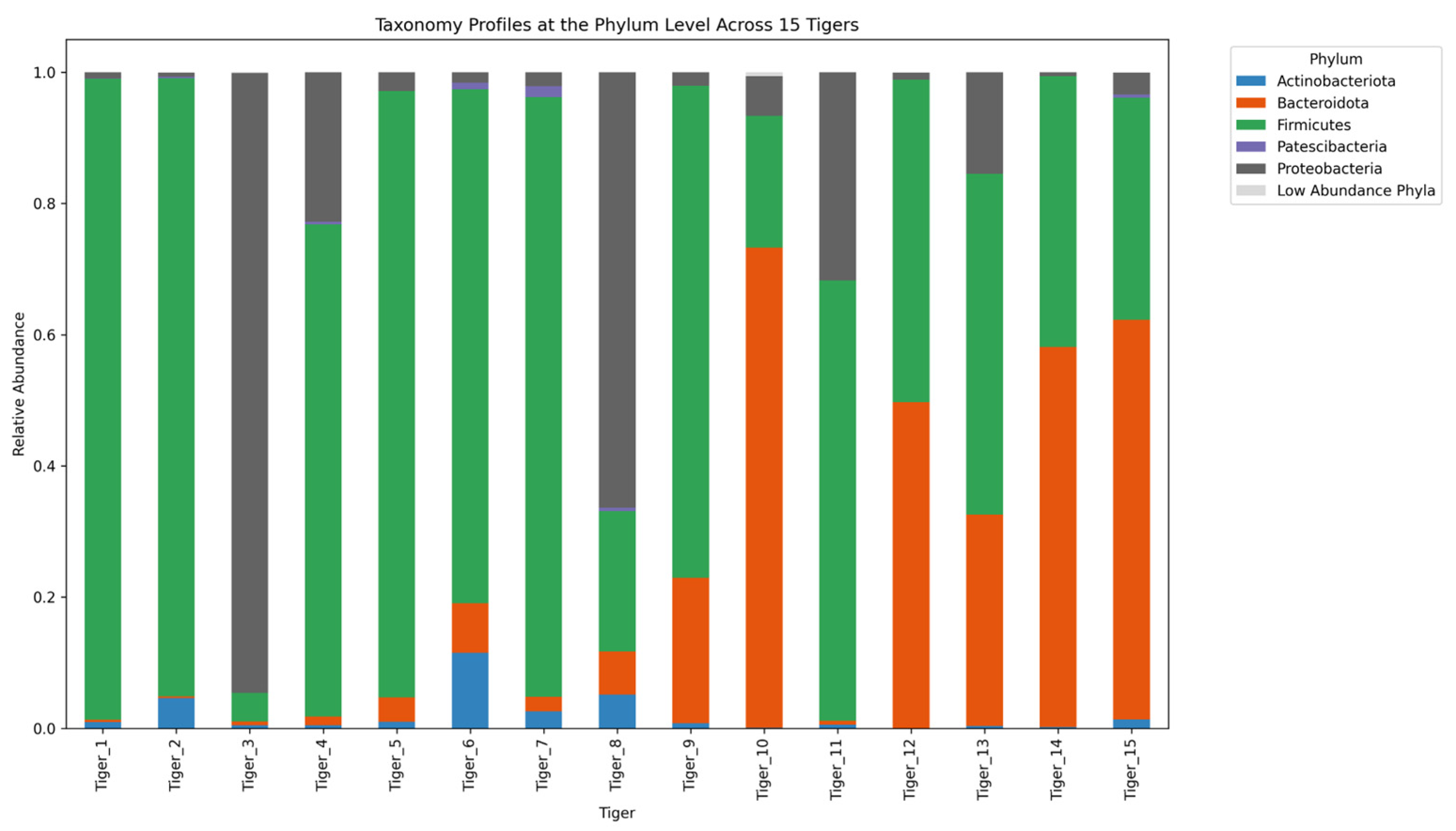
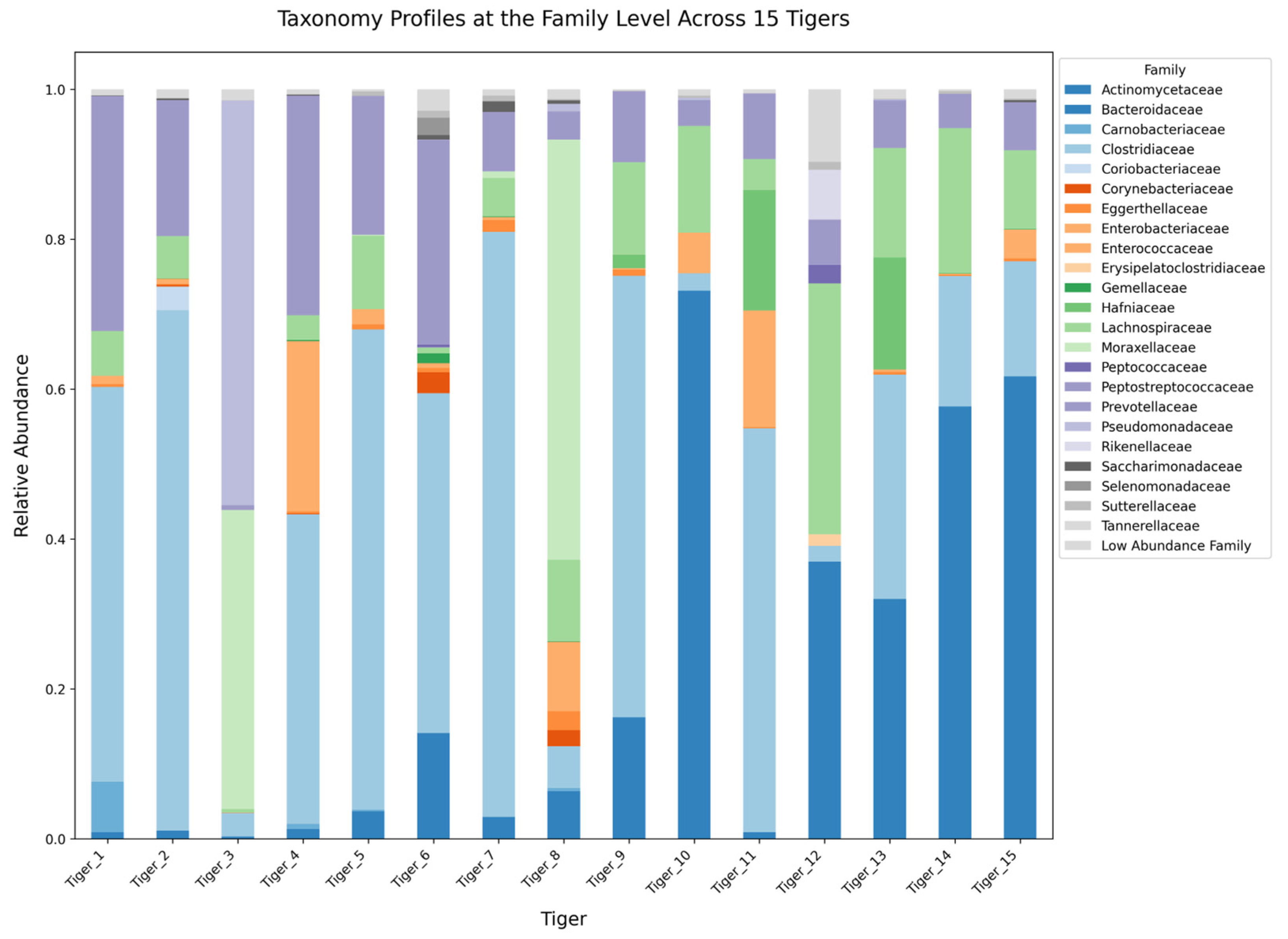
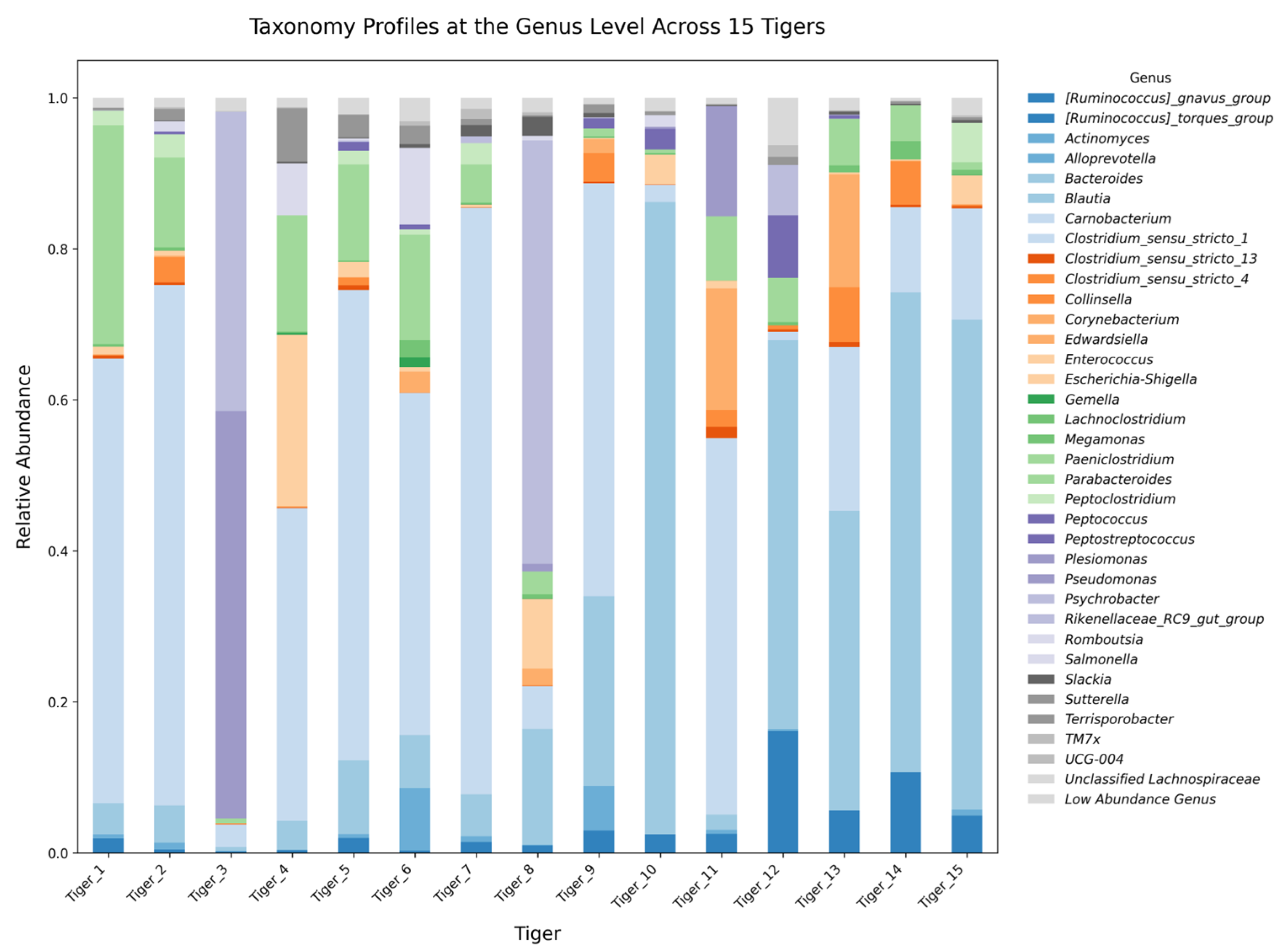
2.3. Taxonomic Composition
3. Discussion
4. Materials and Methods
4.1. Ethical Approval
4.2. Sample Collection and Health Status
4.3. Experimental Grouping for Microbiome Analysis
4.4. DNA Extraction and 16S rRNA Gene Sequencing
4.5. Library Preparation and Sequencing
4.6. Quality Control and Preprocessing
4.7. Alpha Diversity Analysis
4.8. Beta Diversity Analysis
4.9. Taxonomic Composition Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| SCFA | short-chain fatty acids |
| Faith PD | Faith’s Phylogenetic Diversity |
| PCoA | Principal Coordinate Analysis |
| NMDS | Nonmetric multidimensional scaling |
| PERMANOVA | Permutational Multivariate Analysis of Variance |
References
- Lozupone, C.A.; Stombaugh, J.I.; Gordon, J.I.; Jansson, J.K.; Knight, R. Diversity, stability and resilience of the human gut microbiota. Nature 2012, 489, 220–230. [Google Scholar] [CrossRef] [PubMed]
- McKenzie, V.J.; Kueneman, J.G.; Harris, R.N. Probiotics as a tool for disease mitigation in wildlife: Insights from food production and medicine. Ann. New York Acad. Sci. 2018, 1429, 18–30. [Google Scholar] [CrossRef] [PubMed]
- Redford, K.H.; Segre, J.A.; Salafsky, N.; del Rio, C.M.; McAloose, D. Conservation and the microbiome. Conserv. Biol. 2012, 26, 195–197. [Google Scholar] [CrossRef] [PubMed]
- Goodrich, J.; Wibisono, H.; Miquelle, D.; Lynam, A.J.; Sanderson, E.; Chapman, S.; Gray, T.N.E.; Chanchani, P.; Harihar, A. Panthera tigris: The IUCN Red List of Threatened Species 2022. Available online: https://www.iucnredlist.org/species/15955/214862019 (accessed on 2 February 2025).
- Deng, P.; Swanson, K.S. Gut microbiota of humans, dogs and cats: Current knowledge and future opportunities and challenges. Br. J. Nutr. 2015, 113 (Suppl. S1), S6–S17. [Google Scholar] [CrossRef]
- Handl, S.; Dowd, S.E.; Garcia-Mazcorro, J.F.; Steiner, J.M.; Suchodolski, J.S. Massive parallel 16S rRNA gene pyrosequencing reveals highly diverse fecal bacterial and fungal communities in healthy dogs and cats. FEMS Microbiol. Ecol. 2011, 76, 301–310. [Google Scholar] [CrossRef]
- Thompson, L.R.; Sanders, J.G.; McDonald, D.; Amir, A.; Ladau, J.; Locey, K.J.; Prill, R.J.; Tripathi, A.; Gibbons, S.M.; Ackermann, G.; et al. A communal catalogue reveals Earth’s multiscale microbial diversity. Nature 2017, 551, 457–463. [Google Scholar] [CrossRef]
- Becker, A.A.; Hesta, M.; Hollants, J.; Janssens, G.P.; Huys, G. Phylogenetic analysis of faecal microbiota from captive cheetahs reveals underrepresentation of Bacteroidetes and Bifidobacteriaceae. BMC Microbiol. 2014, 14, 43. [Google Scholar] [CrossRef]
- Ning, Y.; Qi, J.; Dobbins, M.T.; Liang, X.; Wang, J.; Chen, S.; Ma, J.; Jiang, G. Comparative analysis of microbial community structure and function in the gut of wild and captive Amur tiger. Front. Microbiol. 2020, 11, 1665. [Google Scholar] [CrossRef]
- Zhu, L.; Wu, Q.; Dai, J.; Zhang, S.; Wei, F. Evidence of cellulose metabolism by the giant panda gut microbiome. Proc. Natl. Acad. Sci. USA 2011, 108, 17714–17719. [Google Scholar] [CrossRef]
- Zhao, G.; Nyman, M.; Jönsson, J.A. Rapid determination of short-chain fatty acids in colonic contents and faeces of humans and rats by acidified water-extraction and direct-injection gas chromatography. Biomed. Chromatogr. 2006, 20, 674–682. [Google Scholar] [CrossRef]
- Koh, A.; De Vadder, F.; Kovatcheva-Datchary, P.; Bäckhed, F. From dietary fiber to host physiology: Short-chain fatty acids as key bacterial metabolites. Cell 2016, 165, 1332–1345. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.-J.; Lee, S.; Kim, B.; Kwak, D.; Kim, T.; Seo, M.-G. Gut microbiome diversity and composition in captive Siberian tigers (Panthera tigris altaica): The influence of diet, health status, and captivity on microbial communities. Microorganisms 2024, 12, 2165. [Google Scholar] [CrossRef] [PubMed]
- Karmacharya, D.; Manandhar, P.; Manandhar, S.; Sherchan, A.M.; Sharma, A.N.; Joshi, J.; Bista, M.; Bajracharya, S.; Awasthi, N.P.; Sharma, N.; et al. Gut microbiota and their putative metabolic functions in fragmented Bengal tiger population of Nepal. PLoS ONE 2019, 14, e0221868. [Google Scholar] [CrossRef]
- Gani, M.; Mohd-Ridwan, A.R.; Mohd-Kulaimi, N.A.; Khairulmunir, M.; Abdul-Latiff, M.A.B.; Md-Zain, B.M. Comparative analysis of gut microbiomes in captive tigers across ex situ facilities in peninsular Malaysia. Microbiology 2024, 93, 904–915. [Google Scholar] [CrossRef]
- He, F.; Liu, D.; Zhang, L.; Zhai, J.; Ma, Y.; Xu, Y.; Jiang, G.; Rong, K.; Ma, J. Metagenomic analysis of captive Amur tiger faecal microbiome. BMC Vet. Res. 2018, 14, 379. [Google Scholar] [CrossRef]
- Khairulmunir, M.; Gani, M.; Mohd-Ridwan, A.R.; Karuppannan, K.V.; Abdul-Latiff, M.A.B.; Md-Zain, B.M. Alteration of the gut microbial composition of critically endangered Malayan tigers (Panthera tigris jacksoni) in captivity during enrichment phase. Mol. Biol. Rep. 2024, 51, 742. [Google Scholar] [CrossRef]
- Zhou, L.; Xu, X.; Zhang, Z.; Zhang, X.; Lin, K.; Luo, H.; Huang, C.; Lin, X.; Zhang, C.; Qing, Y.; et al. Seasonal and environmental influences on the gut microbiota of south China tigers (Panthera tigris amoyensis). Animals 2025, 15, 1471. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- DeSantis, T.Z.; Hugenholtz, P.; Larsen, N.; Rojas, M.; Brodie, E.L.; Keller, K.; Huber, T.; Dalevi, D.; Hu, P.; Andersen, G.L. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 2006, 72, 5069–5072. [Google Scholar] [CrossRef]
- Anderson, M.J. A new method for non-parametric multivariate analysis of variance. Austral Ecol. 2001, 26, 32–46. [Google Scholar] [CrossRef]
- Lin, H.; Peddada, S.D. Analysis of compositions of microbiomes with bias correction. Nat. Commun. 2020, 11, 3514. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Comparison | Distance Metric | PERMANOVA p-Value | Significance |
|---|---|---|---|
| Comparison 1 (Female vs. Male) | Bray–Curtis | >0.05 | Not significant |
| Comparison 1 (Female vs. Male) | Weighted UniFrac | >0.05 | Not significant |
| Comparison 2 (Gwangju vs. Seoul) | Bray–Curtis | <0.05 | Significant |
| Comparison 2 (Gwangju vs. Seoul) | Weighted UniFrac | <0.05 | Significant |
| Tiger ID | Sample Location | Subspecies | Name | Sample Collection Date | Birth Year | Sex | Diet | Health Status |
|---|---|---|---|---|---|---|---|---|
| 1 | Seoul Grand Park | Siberian Tiger | Penza | November 2023 | 2010 | Female | Chicken: 3–5 kg, Beef: 2–3 kg, Rabbit (once a week) | Healthy |
| 2 | Seoul Grand Park | Siberian Tiger | Seonho | November 2023 | 2013 | Male | Chicken: 3–5 kg, Beef: 2–3 kg, Rabbit (once a week) | Born together with #6 |
| 3 | Seoul Grand Park | Siberian Tiger | Rostov | November 2023 | 2010 | Male | Chicken: 3–5 kg, Beef: 2–3 kg, Rabbit (once a week) | Hypothyroidism |
| 4 | Seoul Grand Park | Siberian Tiger | Sarang | November 2023 | 2022 | Female | Chicken: 3–5 kg, Beef: 2–3 kg, Rabbit (once a week) | Born together with #5 |
| 5 | Seoul Grand Park | Siberian Tiger | Haerang | November 2023 | 2022 | Female | Chicken: 3–5 kg, Beef: 2–3 kg, Rabbit (once a week) | Born together with #4 |
| 6 | Seoul Grand Park | Siberian Tiger | Miho | November 2023 | 2013 | Female | Chicken: 3–5 kg, Beef: 2–3 kg, Rabbit (once a week) | Born together with #2 |
| 7 | Seoul Grand Park | Siberian Tiger | Joseph | November 2023 | 2011 | Male | Chicken: 3–5 kg, Beef: 2–3 kg, Rabbit (once a week) | Healthy |
| 8 | Seoul Grand Park | Siberian Tiger | Geumgang | November 2023 | 2018 | Female | Chicken: 3–5 kg, Beef: 2–3 kg, Rabbit (once a week) | Born together with #9, #11 |
| 9 | Seoul Grand Park | Siberian Tiger | Baekdu | December 2023 | 2018 | Male | Chicken: (3–5 kg, Beef: 2–3 kg | Healthy |
| 10 | Seoul Grand Park | Siberian Tiger | Beautiful | December 2023 | 2005 | Female | Chicken: 3–5 kg, Beef: 2–3 kg, Rabbit (once a week) | Chronic renal failure |
| 11 | Seoul Grand Park | Siberian Tiger | Taebaek | December 2023 | 2018 | Male | Chicken: 3–5 kg, Beef: 2–3 kg, Rabbit (once a week) | Born together with #8, #9 |
| 12 | Uchi Zoo | Bengal Tiger | Love | December 2023 | 2009 | Female | Chicken: 3–4.5 kg | Mammary tumor |
| 13 | Uchi Zoo | Bengal Tiger | Ho Soon | December 2023 | 2012 | Female | Chicken: 3–4.5 kg | Healthy |
| 14 | Uchi Zoo | Bengal Tiger | Parker | December 2023 | 2011 | Male | Chicken: 3–4.5 kg | Healthy |
| 15 | Uchi Zoo | Siberian Tiger | Homin | December 2023 | 2007 | Female | Chicken: 3–4.5 kg | Healthy |
| Comparison | Group A (n = ) | Group B (n = ) | Criteria |
|---|---|---|---|
| Comparison 1 | Samples 1, 4, 5, 6, 8, 10, 12, 13, and 15 | Samples 2, 3, 7, 9, 11, and 14 | Female vs. Male |
| Comparison 2 | Samples 12, 13, 14, and 15 | Samples 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, and 11 | Gwangju (Uchi Zoo) vs. Seoul Grand Park |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, B.; Lee, S.; Lee, Y.-J.; Kang, Y.-M.; Rhee, M.H.; Kwak, D.; Yeo, Y.-G.; Kang, J.W.; Kim, T.; Seo, M.-G. Preliminary Insights into the Gut Microbiota of Captive Tigers in Republic of Korea: Influence of Geographic and Individual Variation. Microorganisms 2025, 13, 1427. https://doi.org/10.3390/microorganisms13061427
Kim B, Lee S, Lee Y-J, Kang Y-M, Rhee MH, Kwak D, Yeo Y-G, Kang JW, Kim T, Seo M-G. Preliminary Insights into the Gut Microbiota of Captive Tigers in Republic of Korea: Influence of Geographic and Individual Variation. Microorganisms. 2025; 13(6):1427. https://doi.org/10.3390/microorganisms13061427
Chicago/Turabian StyleKim, Beoul, Saebom Lee, You-Jeong Lee, Yong-Myung Kang, Man Hee Rhee, Dongmi Kwak, Yong-Gu Yeo, Ju Won Kang, Taehwan Kim, and Min-Goo Seo. 2025. "Preliminary Insights into the Gut Microbiota of Captive Tigers in Republic of Korea: Influence of Geographic and Individual Variation" Microorganisms 13, no. 6: 1427. https://doi.org/10.3390/microorganisms13061427
APA StyleKim, B., Lee, S., Lee, Y.-J., Kang, Y.-M., Rhee, M. H., Kwak, D., Yeo, Y.-G., Kang, J. W., Kim, T., & Seo, M.-G. (2025). Preliminary Insights into the Gut Microbiota of Captive Tigers in Republic of Korea: Influence of Geographic and Individual Variation. Microorganisms, 13(6), 1427. https://doi.org/10.3390/microorganisms13061427