Abstract
Consumption of contaminated drinking water is known to cause waterborne diseases such as diarrhoea, dysentery, typhoid, and hepatitis. This study applied whole-genome sequencing (WGS) to detect, identify, compare, and profile diarrhoeagenic pathogens (Vibrio cholerae, Shiga toxin-producing Escherichia coli, and Escherichia coli O157:H7) from 3168 water samples and 135 faecal samples (human and animal). Culture-based methods, MALDI-TOF mass spectrometry, and PCR were employed prior to WGS for identification of pathogens. Culture-based results revealed high presumptive prevalence of STEC (40.2%), V. cholerae (37.1%), and E. coli O157:H7 (22.7%). The MALDI-TOF confirmed 555 isolates with V. cholerae identified as Vibrio albensis. Shiga toxin-producing Escherichia coli (STEC) was more prevalent in wastewater (60%), treated water (54.1%), and groundwater (36.8%). PCR detected 46.4% of virulence genes from the water isolates and 66% of virulence genes from the STEC stool isolates. WGS also revealed STEC (92.9%) as the most prevalent species and found common virulence (e.g., hcp1/tssD1 and hlyE) and resistance (e.g., acrA and baeR) genes in all three types of samples. Five resistance and thirteen virulence genes overlapped among treated water and stool isolates. These findings highlight the diarrhoeagenic pathogens’ public health risk in water sources and underscore the need for better water quality monitoring and treatment standards.
1. Introduction
Tracking and monitoring the spread of diarrhoeagenic pathogens is crucial for effective public health interventions, especially in rural communities where access to healthcare resources may be limited. Diarrhoeal diseases continue to pose a substantial burden on global health, contributing to high morbidity and mortality rates, particularly among vulnerable populations [1]. Waterborne pathogens pose significant risks to public health worldwide, causing a range of diseases and outbreaks [2]. Identifying and characterising these pathogens is crucial for effective surveillance, outbreak response, and the implementation of appropriate control measures. Rural communities, in particular, face unique challenges when it comes to the surveillance and management of diarrhoeal diseases [3]. Limited access to healthcare facilities, inadequate sanitation infrastructure, and poor water quality contribute to increased susceptibility to diarrhoeagenic pathogens.
Consumption of contaminated drinking water can cause waterborne diseases such as diarrhoea, dysentery, typhoid, and hepatitis A [4]. These waterborne diseases are caused by bacteria such as Vibrio cholerae, Shigella, and Salmonella spp. and protozoan parasites such as Giardia spp. and Cryptosporidium spp., among others. For this study, two bacteria were selected, namely Vibrio cholerae and Escherichia coli. Several studies have identified and detected the virulome and resistome of these bacteria using different methods, including WGS [5,6,7,8,9]. For E. coli, this study focused on Shiga toxin-producing E. coli (STEC), including E. coli O157:H7, which has been reported to cause human diseases and outbreaks and has been found in healthy cattle, sheep, and goats [10,11,12,13]. By elucidating the genomic characteristics and transmission patterns of these pathogens, we can inform public health strategies and interventions aimed at reducing the burden of diarrhoeal diseases in vulnerable communities and clinical guidance on suitable treatment regimens.
Traditional methods for pathogen detection and profiling often rely on culture-based techniques and targeted testing, which have limitations in terms of sensitivity, specificity, and the ability to capture the genetic diversity of pathogens [14,15]. In recent years, the advent of next-generation sequencing technologies, particularly whole-genome sequencing (WGS), has revolutionised the field of pathogen identification and characterisation. In addition, WGS allows for the comprehensive analysis of the entire genetic material of microorganisms, providing high-resolution insights into their genome composition, serotype, antimicrobial resistance, and virulence factors [12]. By leveraging the power of WGS, researchers can gain a deeper understanding and comprehensively analyse diarrhoeagenic pathogens circulating in rural communities.
In this study, we applied the culture-dependent method for these two selected bacterial pathogens (V. cholerae and E. coli) to diverse water sources, although a significant number of studies have generally described the conventional method as time-consuming and laborious. Thereafter, PCR and WGS were used to provide a broad knowledge of the microorganisms studied and offered some advantages, such as the detection of multiple genes simultaneously and cost-effectivity. Moreover, molecular methods meet the requirements for the reliable analysis of pathogenic bacteria. These include high specificity, high sensitivity, good reproducibility, and automation.
The objective of this study was to advance our understanding of the genome characteristics and the distribution of waterborne pathogens among various water sources used by communities in rural areas. Through the identification and comparison of pathogen genomes, we can gain insights into the transmission dynamics, variations in virulence factor genes (VF genes), and antimicrobial resistance (AMR) profiles of these organisms. Such information is vital for improving water quality management, implementing targeted interventions, and preventing waterborne disease outbreaks. Overall, the use of whole-genome sequencing (WGS), the most widely used form of next-generation sequencing (NGS), for waterborne pathogen surveillance represents a powerful tool for public health and environmental monitoring. By enhancing our ability to identify, compare, and profile pathogens from diverse water sources, this study contributes to the development of more effective strategies for preventing and controlling waterborne diseases, ultimately safeguarding public health and promoting water safety.
2. Materials and Methods
2.1. Ethical Consideration and Study Areas
This study was approved by the Tshwane University of Technology (TUT) through its Faculty Committee for Research Ethics (FCRE 2019/09/011 (FCPS 03) (SCI)). The purpose of the investigation was explained to the Vhembe District Municipality in order to access all water sources and permission was granted by the municipal committee. Permission was also obtained from the tribal authorities or chief in selected villages, and consent was obtained from the selected households at the beginning of the project.
2.2. Sample Collection
Surface water, groundwater, treated drinking water, wastewater and stool samples were collected from different sources in three local municipalities, namely the Thulamela Local Municipality, Collins Chabane Local Municipality, and Makhado Local Municipality of the Vhembe District Municipality. A random sampling approach was employed to select sampling points in terms of water source type and location. The matrices included surface water (R and D), groundwater (S, B, and DUG), treated water (TWPC), stored water (HC), wastewater (SP), and stool samples (SS) of humans and animals (SS) from areas surrounding water sources and from pit latrines to track and compare the pathogens detected in faeces with those found in water samples (Table S1). After collection, all samples were stored in an insulated cooler box with ice packs and transported to the Microbiology Laboratory at Tshwane University of Technology (TUT), where analyses were performed within 24 h.
2.3. Enrichment and Isolation of Diarrhoeagenic Pathogens
To isolate Shiga toxin-producing Escherichia coli (STEC), Oxoid™ Tryptone Soya Broth (Thermo Fisher Scientific in Johannesburg, South Africa) was prepared according to the manufacturer’s instructions. Prior to water sample collection, dilutions were made using autoclaved distilled water; the dilution volumes are shown in Table S2. Approximately 1 mL of water samples and wastewater effluent was added to 9 mL of the prepared Tryptone Soya Broth (TSB). Human and animal stool samples (approximately 3 g) were mixed with 9 mL of brain heart infusion broth for growth enrichment (Thermo Fisher Scientific in Johannesburg, South Africa). The mixture was then incubated at 37 °C for 18–24 h. Subsequently, 100 µL of the incubated broth was streaked onto plates containing CHROMagar™ O157 for E. coli O157:H7 isolation and CHROMagar™ STEC for non-O157 STEC isolation, both obtained from MediaMage, Johannesburg, South Africa. The plates were incubated at 37 °C for 18–24 h. Distinctive pink to mauve colonies on the plates represented E. coli O157:H7 and non-O157 STEC. For Vibrio cholerae isolation, 100 mL of a water sample was filtered using a 0.45 µm membrane filter (Merck, Darmstadt, Germany). The filters, along with 1 mL of wastewater and 1 mL of prepared stool solution, were separately placed in test tubes containing double-strength alkaline peptone water (pH 8.5) (Merck, Darmstadt, Germany and Merck, Johannesburg, South Africa). The test tube was vigorously shaken and incubated for 6–8 h at 37 °C. Afterwards, 100 µL of the overnight culture was streaked onto plates containing CHROMagar™ Vibrio supplemented with 0.95% alkaline peptone water (APW). The plates were incubated aerobically for 24 h at 37 °C. Blue colonies observed on the media indicated the presence of V. cholerae. Positive controls for STEC (ATCC 43888) and V. cholerae (ATCC 14035) were obtained from the TUT Microbiology Laboratory. All colonies (pink to mauve colonies for STEC and E. coli O157:H7 and blue colonies for V. cholerae) were isolated and purified for further molecular analysis.
2.4. Bacterial Identification
2.4.1. Mass Spectrometry Technology (MALDI-TOF)
The identification of bacterial isolates involved the use of matrix-assisted laser desorption ionisation time-of-flight (MALDI-TOF) mass spectrometry technology [16]. This analysis was performed at the MALDI-TOF diagnostic service located at the Department of Microbiology and Plant Pathology, University of Pretoria, South Africa, using the MALDI-TOF Mass Spectrometer (Bruker Daltonics GmbH & Co., Bremen, Germany). This method was performed following the procedure described by Ramaite et al. [17]. Positive isolates identified through MALDI-TOF MS were selected for further molecular analysis.
2.4.2. Conventional PCR for Identification of Target Pathogens
For molecular study, it is important to mention that only one bacterial isolate per positive samples was taken into consideration for a specific target bacteria.
- a.
- DNA extraction
Of the 555 (17%) presumptive positive samples determined by culturing, n = 223 (40.2%) were STEC, n = 126 (227%) were E. coli O157:H7, and n = 206 (37.1%) were V. cholerae. The MALDI-TOF MS results confirmed that most of these isolates were STEC [24% (n = 36.8)], followed by E. coli O157:H7 [15.7% (n = 87)] and V. cholerae [13.7% (n = 76). The preserved bacterial isolates were thawed and subsequently centrifuged at 13,000× g for 1 min. The ZymoBIOMICS™ DNA Miniprep Kit from Zymo Research (Inqaba Biotechnical Industries, Pretoria, South Africa) was utilised to extract the total genomic DNA from the bacterial pellets, following the instructions provided by the manufacturer. To determine the quantity and quality of the extracted DNA, a NanoDropTM 2000 spectrophotometer (Thermo Scientific in Johannesburg, South Africa) was employed. The genomic DNA (gDNA) suspension obtained was stored at −80 °C until further analysis, either through polymerase chain reaction (PCR) or whole-genome sequencing (WGS).
- b.
- DNA Amplification by conventional multiplex PCR
To detect E. coli and V. cholerae, all bacterial isolates in this study underwent further analysis using conventional multiplex polymerase chain reaction (PCR) amplification. Each multiplex PCR (mPCR) reaction was conducted in a total volume of 25 μL. This volume included the following reagents: 5 μL of template DNA, 0.5 μL of forward primer (10 M), 0.5 μL of reverse primer (10 M), 12.5 μL of NEB Taq 2X master mix, and 6.5 μL of nuclease-free water, which were obtained from Inqaba Biotechnical Industries, Pretoria, South Africa. The amplification process was carried out using a MiniAmp™ Plus Thermal Cycler (Thermo Fisher Scientific, Johannesburg, South Africa). For the detection of Shiga toxin-producing Escherichia coli (STEC), a multiplex PCR assay targeting four (4) important virulence-associated genes, namely stx1, stx2, eae, and hlyA, was performed. The identification of E. coli O157:H7 involved the detection of three (3) virulence-associated genes: rfbO157, fliCH7, and eaeA. Vibrio cholerae was detected using three (3) virulence-associated genes: ctxAB, tcpA, and ompW. Detailed descriptions of the primers and annealing temperatures are given in Table 1. Following PCR amplification, the products were separated using electrophoresis on a 1% agarose gel prepared in 1× TAE buffer, as described by Ramaite et al. [17].

Table 1.
Primer sequences for the detection of STEC, E. coli O157:H7, and V. cholera.
2.4.3. Library Preparation and Sequencing
Library preparation and sequencing of single isolate procedures were carried out at the Agricultural Research Council Biotechnology Platform in Pretoria, South Africa, utilising the Illumina HiSeq® 2500 platform. Initially, the quantity and quality of the DNA were assessed using the Qubit™ dsDNA BR assay kit from Invitrogen, Thermo Fisher Scientific, Waltham, MA, USA. Approximately 2 µg of isolated genomic DNA from each single isolate was employed for constructing Illumina paired-end libraries using the Illumina Nextera XT DNA library preparation method. The generated libraries underwent validation through quality control checks to ensure their suitability for sequencing. Barcodes were assigned to all qualified libraries, and sequencing was performed in accordance with the manufacturer’s instructions. This study employed whole-genome sequencing (WGS) rather than metagenomic sequencing to obtain high-resolution genetic profiles for single bacterial isolates. The aim of the sequencing process was to achieve an average genome coverage of 30× or greater for all isolates. The resulting sequence data were obtained in FASTQ format files and subsequently subjected to bioinformatic analysis.
2.4.4. Bioinformatic Analysis
To assess the quality of the raw sequence reads, FastQC v.0.11.9 [21] was utilised for quality control, including the removal of low-quality sequences. Trimmomatic (v0.36) [22] was employed to trim the reads by removing adapter sequences and low-quality regions. The UCHIME algorithm [23] was applied to eliminate any human DNA contamination. Initially, the Kraken 2 v2.1.0 software program was used to identify the most abundant bacteria in the raw reads. Subsequently, the assembled scaffolds underwent analysis with autoMLST to determine the most closely related species. A species was considered identifiable if the average nucleotide identity (ANI) was equal to or greater than 0.96. In cases where the ANI fell below 0.96, the closest species was indicated as either E. coli or V. cholerae. The raw BLAST® outputs were further filtered based on three (3) criteria to improve result reliability: (i) a minimum length of 100 base pairs (length = qEnd to qStart); (ii) a percentage pairwise sequence identity (PIDE) greater than 70; and (iii) a query coverage exceeding 70. De novo assembly of the raw reads was performed using SPAdes De Novo Assembler Software v.1.03 [24]. The quality of the assembly was assessed using QUAST version 5.3.0 [25]. The assembled sequences were submitted to the NCBI Sequence Read Archive with BioProject accession number PRJNA964706. Gene recognition was conducted using Prodigal v.2.6.3 [26]. Functional annotation was conducted to identify antimicrobial resistance (AMR) determinants and virulence factors (VFs) by querying the CARD database v.1.2.1 and VFDB database [27], respectively. An AMR and VF determinant was considered part of the core resistome if it was present in all the assessed matrices.
2.5. Statistical Analysis
The prevalence disparity among the matrices surface water (R and D), groundwater (S, B, and DUG), treated water (TWPC), stored water (HC), wastewater (SP), and stool samples (SS) was evaluated using the chi-square (χ2) test. To determine the relative abundance of antimicrobial resistance genes (ARGs) and virulence factors (VFs) or virulence factor (VF) genes, the results were presented as heat maps. Graphical visualisations were created using Excel (2019) and the Venny (version 2.1) tool online.
3. Results
3.1. Prevalence of Potential Diarrhoeagenic Pathogens by Culture-Based Methods
A total of 3303 samples were collected from various sources comprising surface water, groundwater, treated water, stored water, wastewater, and stool samples. The detailed sample collection data for dry and wet seasons are given in Table S1. The culture-based results indicate varying levels of potential STEC, E. coli O157:H7, and V. cholerae contamination in various water sources and stool samples (Table 2). Surface water had comparatively high contamination with 23.5% and 22.8% detection of potential STEC and V. cholerae, respectively. Groundwater and treated water had low rates of contamination with potential STEC in 3.8% and 2.7% of samples, respectively, and potential V. cholerae was isolated from 1.4% and 2.3%, demonstrating relatively good microbial quality. Stored water, on the other hand, depicted increased contamination with potential STEC at 5.9% and V. cholerae at 5.4%, probably resulting from recontamination following treatment. Potential E. coli O157:H7 was found to be more prevalent in wastewater, at 12.5% (n = 13), among all water samples, with the highest prevalence of 30.4% in stool samples. Potential STEC and V. cholerae were isolated in 8.7% and 17.3% wastewater samples, confirming them as critical pathogen transmission sources. Stool samples also captured the highest prevalence, with potential STEC detected in 32.6% and V. cholerae in 33.3%, confirming human infection and potential disease transmission. Overall, the results put surface water, wastewater, and stored water into the spotlight as major contamination hotspots, while treated and groundwater sources recorded comparatively lower microbial dangers.
Table 2.
Culture-based detection of STEC, E. coli O157:H7, and V. cholerae in water and stool samples.
3.2. Isolates Confirmed as Diarrhoeagenic Pathogens
Of the 555 presumptive positive samples determined by culturing methods, n = 349 (62.9%) for E. coli and (n = 206 (37.1%) for V. cholerae, the MALDI-TOF MS Biotyper® results confirmed that 223 (40.2%) isolates were STEC, 126 (27%) isolates were E. coli O157:H7, and 206 (37.1%) isolates of V. cholerae were identified as V. albensis. The MALDI-TOF results also revealed high variations in the identification of targeted pathogens in various water sources and stool samples (Table 3). The highest presence of STEC was observed in wastewater (60%), followed by treated water (54.1%) and groundwater (36.8%). Stored water showed a moderate contamination rate of 42.6%, which is higher than groundwater, pointing to recontamination during storage. Escherichia coli O157:H7 showed the overall lowest prevalence, ranging from 0.7–2.8% in water samples and at 1.1% in stool samples. Vibrio cholerae occurred in lower rates across all sample types, with the highest in treated water (21.6%) and groundwater (14.7%). Wastewater and stool samples had 17.5% and 16.2% V. cholerae detection rates, respectively, suggesting human exposure and faecal shedding of the pathogen. This study also revealed that among the isolates categorised as presumptive E. coli, several other bacterial species, such as Plesiomonas shigelloides (1.4%; n = 5) and Proteus mirabilis (0.9%; n = 3), were identified and the remaining 14.3% (n = 50) could not be identified. For presumptive V. cholerae, the remaining isolates were mostly Aeromonas spp. (18.45%; n = 38), Pseudomonas aeruginosa (9.71%; n = 20), Pseudomonas mendocina (8.74%; n = 18), Morganella morganii (7.3%; n = 15), and Enterobacter cloacae (5.8%; n = 12) and the remaining 13.1% (n = 27) could not be identified.
Table 3.
Identification of E. coli and V. cholerae by MALDI-TOF in water and stool samples.
3.3. Abundance of Virulence-Associated Genes Identified from Various Matrices by PCR
Overall, among the isolates that were confirmed as E. coli by MALDI-TOF MS, the PCR results revealed that 6.4% (n = 147) and 26.2% (n = 83) of isolates were from water samples and 66% (n = 33) and 24% (n = 12) were from stool samples, which harboured one or more virulence-associated genes specific to STEC and E. coli O157:H7, respectively. Additionally, 14.8% (n = 47) of isolates from water samples and 36% (n = 18) of isolates from stool samples possessed one or more virulence-associated genes of V. cholerae. Overall, STEC was detected in 49% of all samples, with the highest prevalence in stool samples (66%) followed by surface water (50%), wastewater (50%), and stored water samples (48.1%). For E. coli O157H:7, 26.2% (n = 83) originated from water samples and 24.0% (n = 12) from stool samples. Vibrio cholerae was found in 17.7% of total samples, with the highest prevalence in stool samples (36.0%), implying active V. cholerae infection. Surface water (22.2%) and wastewater (17.5%) also showed significant contamination. Detection in treated water was 17.1% and in groundwater was 12.1%. None of the spring water samples tested positive for V. cholerae (Table S3 in Supplementary Data). Notably, a total of 34 isolates (9.1%) did not harbour any of the target genes (stx1, stx2, eae, hlyA, rfbO157, fliCH7, eaeA, ctxA, tcpA, and ompW) for either STEC, E. coli O157:H7, or V. cholerae.
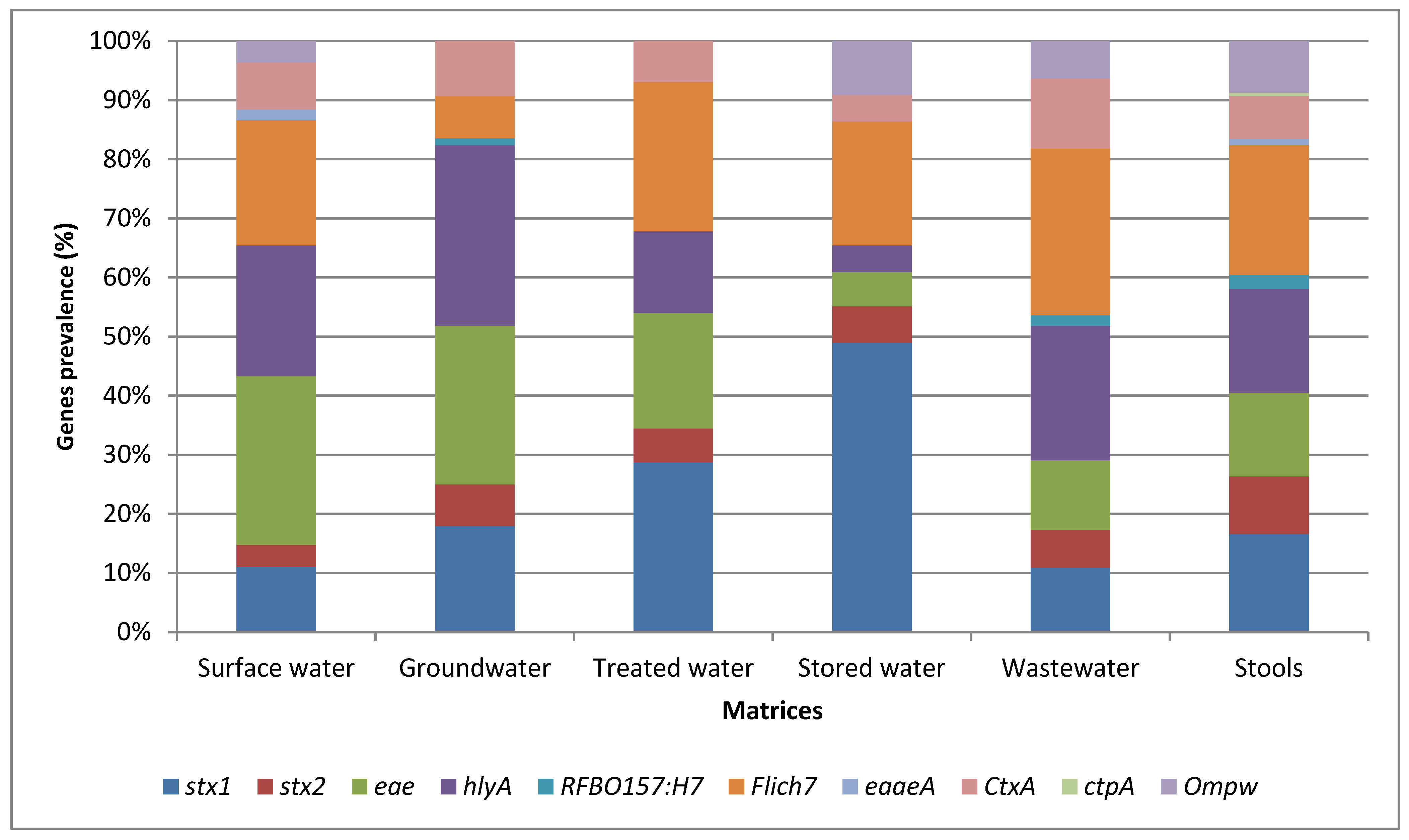
Figure 1 illustrates the relative abundance of the virulence-associated genes identified in the various matrices assessed by conventional PCR. Among all the target genes, the stx1 gene detected in STEC exhibited the highest prevalence of 72.1% from stored water. The stx2 gene was found to have the highest prevalence of 40% in STEC isolated from stool samples. The highest prevalence of fliCH7 isolated in E. coli O157:H7 was detected from stool samples [n = 45 (90%)], while the highest prevalence of hlyA in STEC isolated in water samples was detected at 62.5% from groundwater samples. Furthermore, the eae gene exhibited the highest prevalence of 77.5% in STEC isolates from surface water samples. The least frequently detected virulence-associated genes in the isolates from the various matrices were tcpA and eaeA (carried by V. cholerae) and rfbO157 (carried by E. coli O157:H7) (Figure 1).
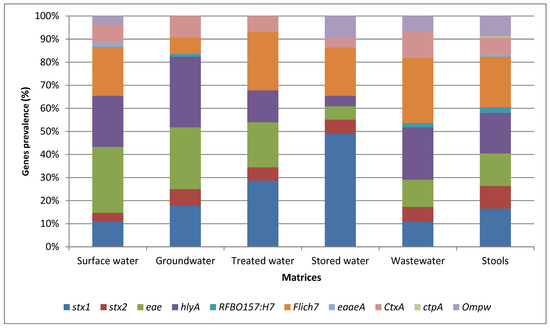
Figure 1.
Relative abundance of the most abundant virulence-associated genes identified in the various matrices assessed by conventional PCR.
3.4. Bioinformatic Analysis
Conventional PCR results revealed that isolates from each source were positive and were showing similar patterns of genes. These isolates were regarded as containing similar organisms; consequently, their DNA was pooled to make one sample. A total of 35 pooled samples were subjected to whole-genome sequencing (WGS) using the HiSeq 2500 platform. Of these, there were 74.3% (n = 26) of STEC, 5.7% (n = 2) of E. coli O157:H7, and 20% (n = 7) of V. cholerae. Vibrio cholerae isolates yielded very low reads due to insufficient sequencing depth. For E. coli, the raw sequence data obtained from water samples and stool samples were collected and pre-processed to remove any noise, ensuring the quality and the reliability of the subsequent analysis. Sequencing resulted in the generation of over 7.13 gigabytes (Gb) of unzipped data for functional annotations. The raw reads used in this study have been deposited in the NCBI (accession number PRJNA1123913). On average, each sample yielded approximately 1,635,181 raw reads. The summary statistics provided include sample ID, number of contigs, total length of each base pair (bp), number of sequences, N50, guanine–cytosine (GC) content (%), CDS, and GenBank accession number (provided in Table 4. The resulting genome assemblies of E. coli ranged between 1,255,779 and 3,968,282 bp in size. Hand-dug wells had the highest number of contigs (approximately 3588 contigs); the lowest number of contigs was observed in septic tank wastewater (1429 contigs). The N50 values for all assemblies ranged from 705 bp to 1224 bp. All 28 samples of E. coli genomes sequenced in the present study contained an average GC content of 52%. The genome coverage for all samples was >30×. Table 5 gives a summary of the genome assemblies of E. coli.
Table 4.
Summary statistics comparing the genome assemblies of the Escherichia coli population in various water types sampled.
Table 5.
PCR detection of E. coli and V. cholerae virulence genes in water and stool samples.
3.4.1. Identification
The AutoMLST results revealed that the most abundant bacterial genera were E. coli [100% (n = 28)], where 92.9% (n = 26) were STEC isolates and 7.1% (n = 2) were E. coli O157:H7 isolates. However, we were not able to distinguish between STEC and E. coli O157:H7 due to lower coverage (<30×), which had led to incomplete O/H antigen gene detection. For V. cholerae, only one (1) out of seven (7) sequences was closely related to V. cholerae, with an ANI = 0.96. Of the 26 STEC sequences, most of them were from the groundwater (n = 8) and stored water (n = 6), followed by stool samples (n = 4), wastewater (n = 4), surface water (n = 3), and treated water (n = 1). The two sequences for E. coli O157:H7 were each from surface water and a stool sample.
3.4.2. Most Abundant Virulence Factors (VFs)
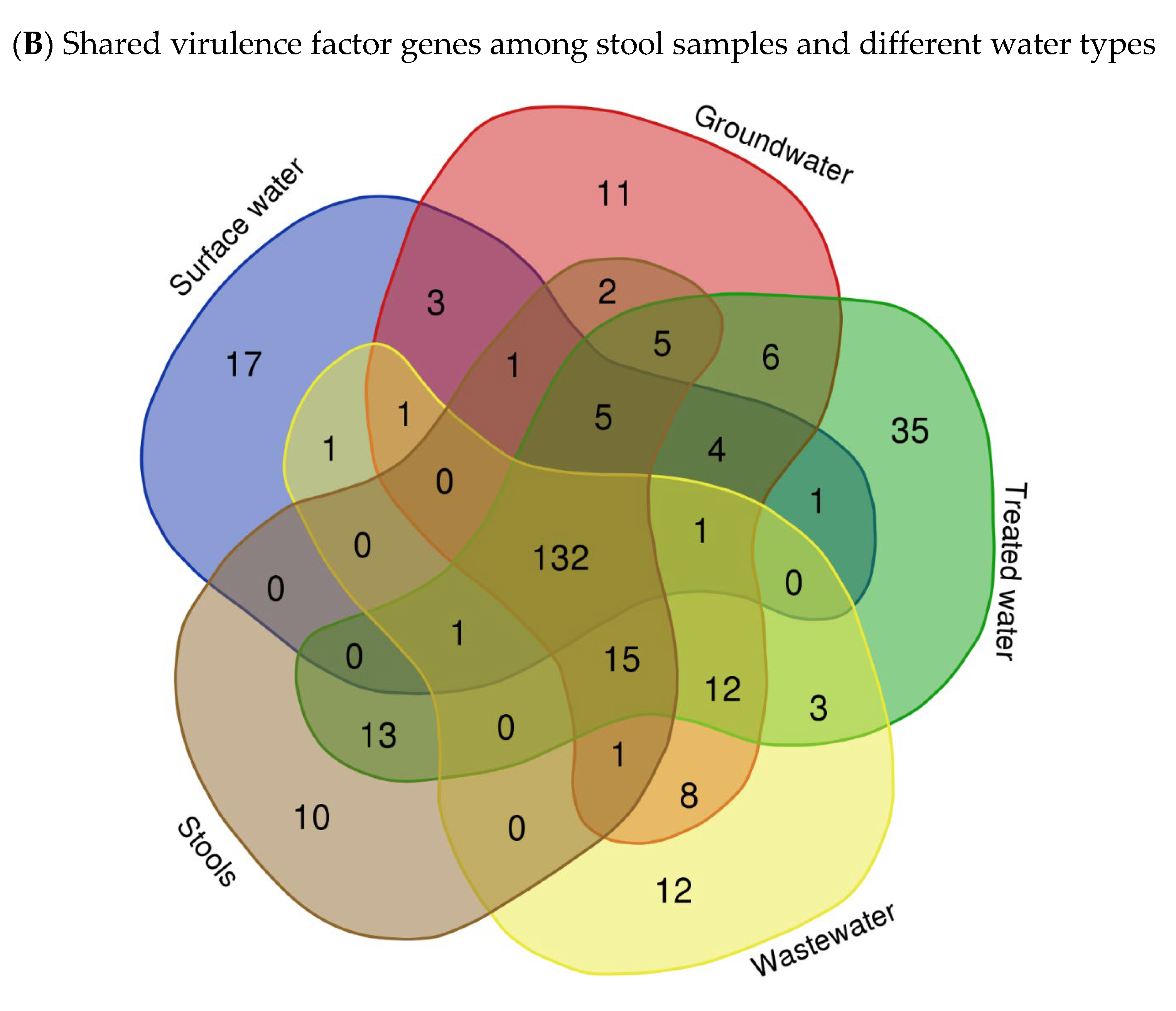
The virulome of E. coli isolates was analysed, indicating variations in the virulence factor genes (VF genes). A total of 153 VF genes across all the samples were identified using virulome analysis. Among the different sample types, the septic tank wastewater (SP) exhibited the highest number of VF genes, with a count of 118, followed by stool samples (SS) with 116 VF genes and the household water storage containers (HC) with 115 VF genes, while the spring water (S) samples exhibited the lowest number of VF genes at 18. In Figure 2A, which illustrates the relative abundance of VF genes in the E. coli isolates in the various types of water, it can be observed that upaG and ehaG (shown as upaG/ehaG on the heat map) were consistently the most frequently identified VF genes in the majority of the E. coli isolates, occurring 92 times, followed by the ehaA, fepA, and cheA VF genes, which were detected with counts of 52, 50, and 47, respectively. In contrast, gspM was the least identified VF gene, detected in only four (4) E. coli isolates. Furthermore, the hcp1/tssD1, hlyE, mrkA, and mrkB genes encoding virulence factors were each detected five (5) times in very few E. coli isolates. The most frequent VF genes identified, like upaG/ehaG, are associated with adhesion mechanisms, while ehaA is implicated in autotransporter-mediated pathogenesis and fepA encodes a siderophore receptor implicated in iron acquisition [12]. Several VF genes known to contribute to the virulence and pathogenicity of E. coli leading to diarrhoeal diseases were detected, including cfaC, cfaD/cfaE, csgA, eaeH, espX4, fepA, fimD, ibeC, and ompA genes. Moreover, the findings indicated that a total of 132 VF genes were shared between the stool samples (SSs) and all the different types of water sources analysed, including surface water (R and D), treated (drinking) water (TWPC and HC), groundwater (S and B), and wastewater (SP) (Figure 2B). Stool samples shared 13 VF genes with the drinking water in households, but only two (2) VF genes with groundwater. Wastewater and groundwater shared eight (8) VF genes. Fifteen VF genes were simultaneously identified in stools, wastewater, and drinking water in households. Around 12 VF genes were detected in wastewater, groundwater, and drinking water in households. Groundwater and drinking water shared six (6) VF genes. However, no shared VF genes were detected among E. coli in surface water, groundwater, stools, and wastewater.

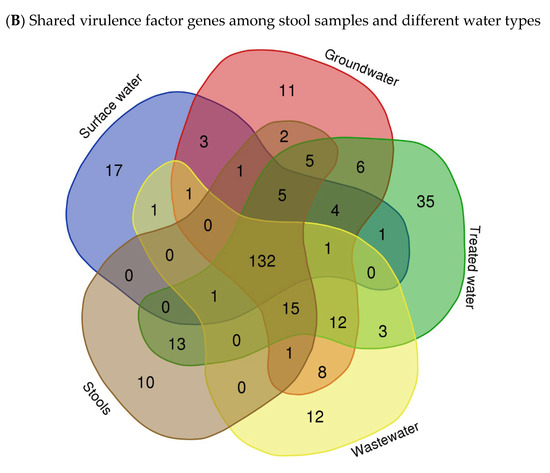
Figure 2.
Heat map showing the abundance of VF genes of E. coli isolated in various types of water (A) (with colour gradient ranges from pale orange (low or zero abundance) to intermediate shades (moderate abundance) to dark/deep orange (high abundance)) and Venn diagram showing the number of shared VF genes among stool samples and different types of water (B).
3.4.3. Most Abundant Antibiotic Resistance Genes (ARGs)
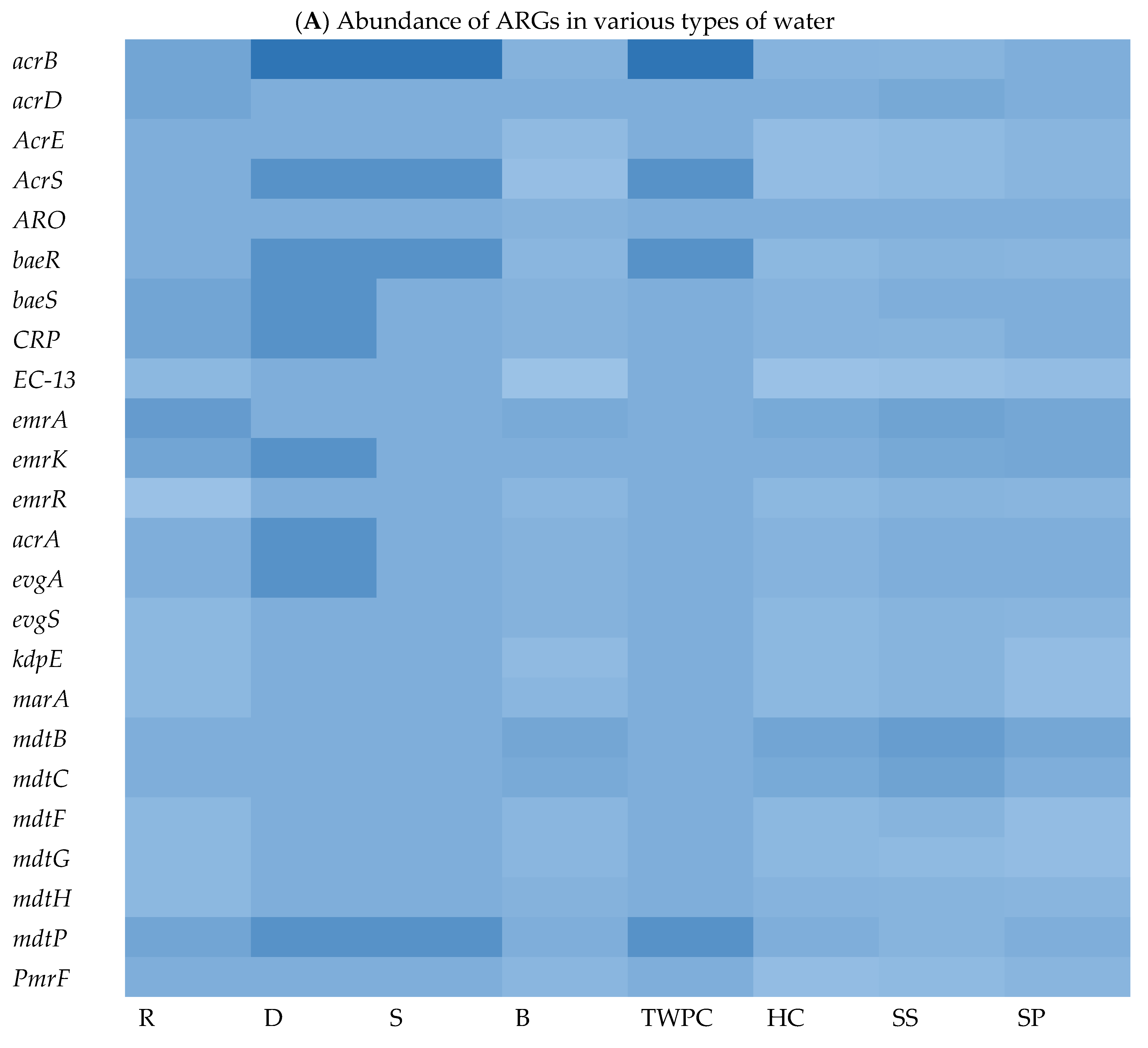
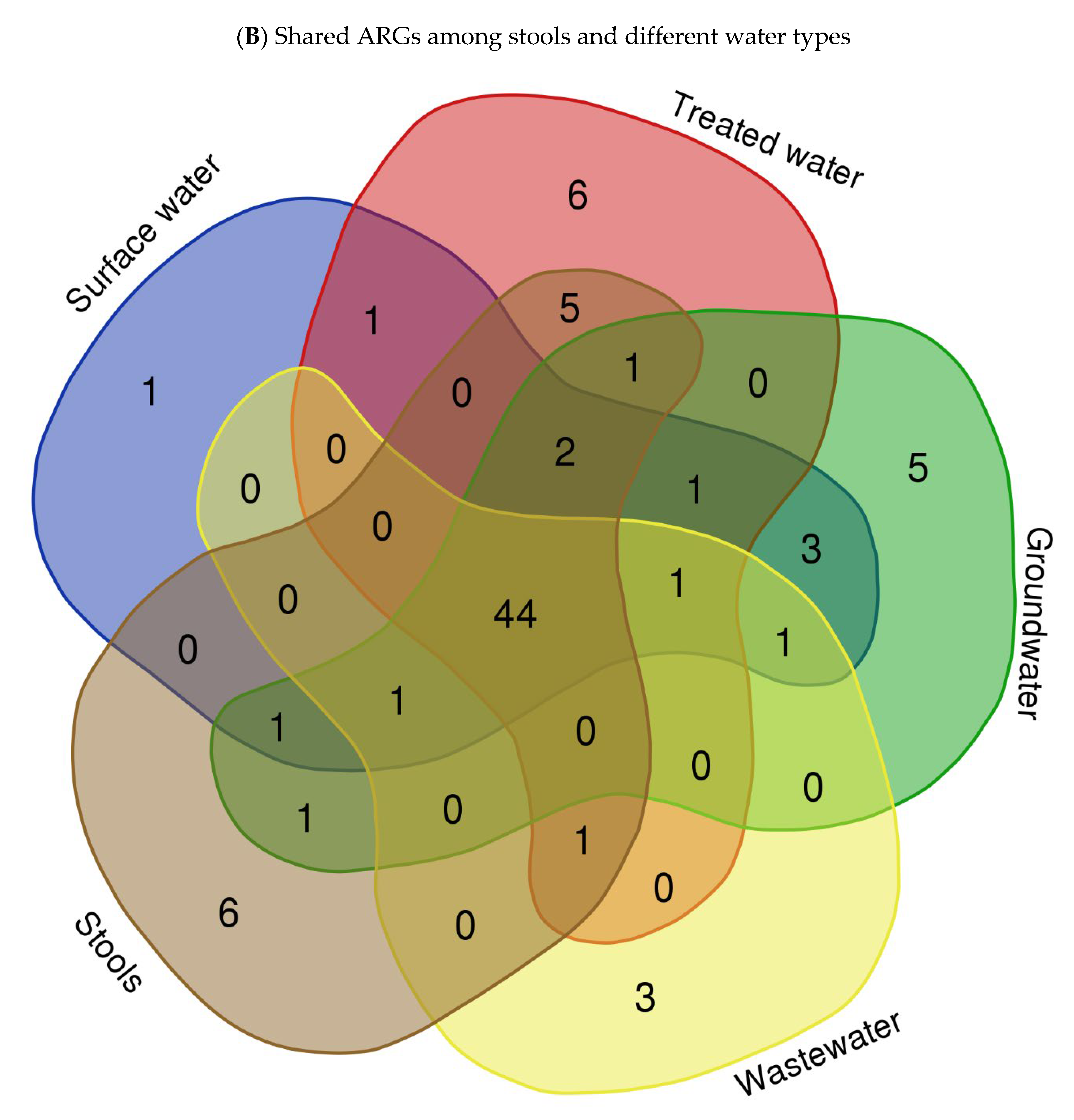
To explore the ARG content of each matrix, WGS sequences were queried against the Comprehensive Antibiotic Resistance Database (CARD) using BLASTx. The results of this analysis revealed a wide range of ARGs found in the different assessed matrices. We identified an average of 29, 178, 131, 29, 101, 188, 39, and 232 ARGs in isolates from springs (S), stool samples (SS), septic tank wastewater (SP), tap water at the point of use in the community (TWPC), rivers (R), household containers (HC), dams (D), and boreholes (B), respectively. Figure 3A shows the heat map of relative abundance of ARG profiles per assessed samples of the most abundant ARGs across all matrices sampled. In terms of ARG abundance, the highest abundance was observed for mdtB (n = 36), followed by emrA (n = 35), detected across all water sources. The mdtC, emrK, and acrB resistance genes were detected 32 times each in all matrices. The S, D, and TWPC samples were found to have the highest number of the acrB gene, followed by the AcrS, baeR, and mdtP genes. The dam (D) samples also showed a high number of emrK, acrA, and evgA genes. Among the identified ARGs, one of the most common ARGs (acrA) was detected in this study. The most frequent ARGs identified, like mdtB, emrA, and acrB, are principally related to multidrug efflux systems, which are implicated in resistance via the active exportation of antibiotics from the bacterial cell [5]. The core resistome was defined as shared ARGs found in all assessed matrices (Figure 3B). The core resistome was found to be 44 ARGs. Three (3) ARGs were shared between groundwater and surface water, while one (1) ARG was shared between surface water and drinking water at the household level and two (2) ARGs were shared among surface water, groundwater, wastewater, and household drinking water.
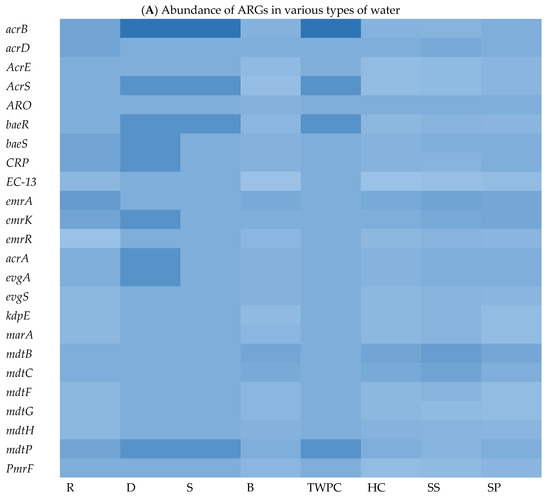
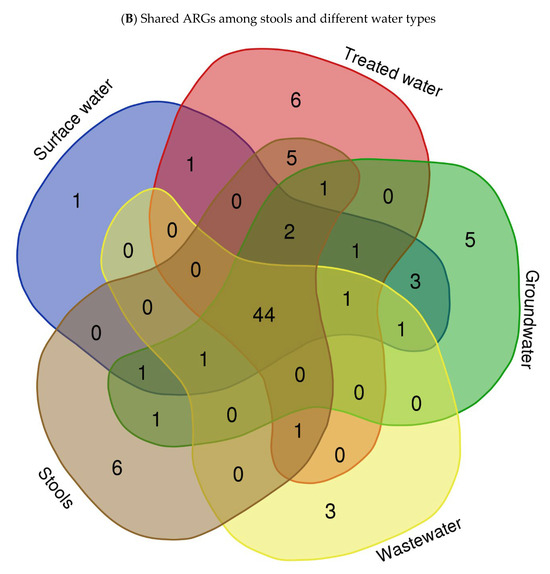
Figure 3.
Heat map showing abundance of ARGs in different types of water (A) (with colour scale ranges from pale blue (low or zero abundance) to intermediate shades (moderate abundance) to dark/deep blue (high abundance))and Venn diagram showing shared ARGs among different stool samples and different types of water (B).
4. Discussion
The present study focused on the detection and identification of E. coli and V. cholerae strains. It compared the results of culturing, MALDI-TOF MS, and PCR-based detection of the main virulence-associated genes with WGS-derived findings of the virulome and resistome. These analyses were conducted in various types of water and stool samples. For pathogenic STEC, E. coli O157:H7, and V. cholerae isolates, culture-based methods revealed that the highest prevalence was found in stool samples with a prevalence rate of 32.6%, 30.4%, and 33.3%, respectively (Figure 1). This is to be expected, as E. coli is a ubiquitous human intestinal flora and is commonly excreted in stool, particularly with enteric infections. This finding is aligned with the fact that cholera is a diarrhoeal disease primarily transmitted through the faecal–oral route [28]. A study by Gwimbi et al. [29] also found a high prevalence of E. coli in samples from open wells and streams compared to unprotected springs. Surface water also showed a relatively high prevalence of presumptive pathogenic STEC (23.5%) and V. cholerae (22.8%). In Kenya, Sila [30] also reported that V. cholerae was mostly detected at high prevalence in Kauthulini River (36%) and Athi River (21%). These findings indicate that rivers may be contaminated with faecal matter, possibly from human or animal sources. Contaminated river water can pose a health risk to individuals who use the water source for drinking, bathing, or other purposes [31].
Springs, which are usually groundwater sources, can also be affected by contamination from nearby surface water or faulty sanitation systems. Groundwater and tap water at the point of use in the community had the lowest prevalence of presumptive pathogenic STEC, with rates of 3.8% and 2.7%, respectively. Similarly, for Vibrio cholerae isolates, groundwater had the lowest prevalence of isolates (1.4%), followed by tap water at the point of use in the community (2.3%) (Table 2). Boreholes are deep wells that extract water from underground water sources, and hence they typically have lower levels of contamination [32]. Similar results were obtained by Obanor et al. [33], where borehole water samples were found to have the lowest prevalence of E. coli. These findings are expected since these microbes are common inhabitants of the intestinal tracts of animals and can be shed in faeces, especially during episodes of gastrointestinal infections. It is in accordance with the fact that cholera is a diarrhoeal disease primarily transmitted through the faecal–oral route [28]. Similarly, Sila [30] also found that V. cholerae were mainly detected at high prevalence in Kenya, with 36% in Kauthulini River and 21% in Athi River. Consequently, the target bacterial isolates were subjected to MALDI-TOF MS Biotyper® analysis. The high prevalence in stool samples suggested a significant level of faecal contamination.
The MALDI-TOF MS results revealed that more of the isolates were confirmed as STEC (36.8%, n = 204) than E. coli O157:H7 (15.7%, n = 87). For V. cholerae, 13.7% (n = 76) of the isolates were identified as V. albensis, a species closely related to Vibrio cholerae. These findings support the assumption that the presumptive E. coli and V. cholerae (identified as V. albensis) isolates in this study were indeed representative of pathogenic strains. Vibrio albensis is a non-O1 serovar V. cholerae, a luminescent bacterium that shares more than 70% of its DNA sequences with V. cholerae [34]. However, it is important to note that further characterisation and testing would be required to determine specific pathotypes within the STEC, E. coli O157:H7, and V. cholerae isolates. A small percentage of pathogenic STEC, E. coli O157:H7, and V. cholerae isolates were identified as other species: 1.4% (n = 5) as Plesiomonas shigelloides, 0.9% (n = 3) as Proteus mirabilis, Aeromonas spp. at 18.45% (n = 38), Pseudomonas aeruginosa at 9.71% (n = 20), Pseudomonas mendocina at 8.74 % (n = 18), Morganella morganii at 7.3% (n = 15), and Enterobacter cloacae at 5.8% (n = 12). These results suggest the presence of multiple bacterial species in water sources that may have pathogenic potential; for example, Plesiomonas shigelloides is known to cause diarrhoeal disease; Proteus mirabilis and Morganella morganii cause urinary tract infections; some species of Aeromonas cause gastrointestinal infections; and Pseudomonas aeruginosa cause urinary tract infections and respiratory tract infections [35]. Several isolates could not be identified; this is an indication of the presence of bacterial species that were not covered by the analysis or of potentially novel or unknown organisms. This is one of the general limitations of MALDI-TOF, as its database may not capture all the bacterial species, particularly emerging or rare ones [17]. Further investigation would be needed to determine the identity and significance of these unidentifiable isolates.
In this study, among the matrices tested, PCR results also showed that stool samples exhibited the highest prevalence of STEC, with 66% (n = 33) of samples testing positive (Table 5). In contrast, the highest prevalence of Vibrio cholerae being detected in hand-dug wells (Table S3 in Supplementary Data) indicates that hand-dug wells may serve as a reservoir for V. cholerae. Hand-dug wells are vulnerable to contamination, particularly if they are in areas with inadequate sanitation practices or in proximity to faecal sources [36]. The detection of V. cholerae in hand-dug wells raises concerns about the potential for cholera transmission if contaminated water from these wells is consumed without prior treatment. None of the tested springs showed the presence of virulence-associated genes of Vibrio cholerae (Table S3 in Supplementary Data). This finding suggested that the spring water tested in this study did not harbour pathogenic strains of Vibrio cholerae. Springs are typically considered a relatively safe source of drinking water, as groundwater is generally protected from surface contamination [37]. However, it is worth noting that the absence of virulence-associated genes does not completely rule out the presence of non-pathogenic or environmental strains of Vibrio cholerae in the springs. The presence of these virulence-associated genes indicates the need for proper sanitation and water treatment practices to prevent the transmission of waterborne diseases.
This study also found that a total of 34 isolates (9.1%) did not contain any of the target genes for either STEC, E. coli O157:H7, or V. cholerae when using conventional PCR. This suggested that these isolates were free from the specific pathogenic strains of these bacteria targeted in this study. Nevertheless, it is important to note that the absence of the target genes does not guarantee the absence of other potential pathogens or indicators of faecal contamination in those samples. Additionally, the absence of virulence-associated genes of V. cholerae in the tested spring water samples (Table S3 in Supplementary Data) is encouraging, indicating that these springs may pose a lower risk of Vibrio cholerae infection. However, it is crucial to regularly monitor and assess the quality of water sources to ensure public health and prevent waterborne illnesses.
The findings also showed the highest prevalence of the stx1 gene in STEC was isolated from stored water [(n = 119 (72.1%)]; this gene is carried by STEC (Figure 1). This finding suggested a potential risk of contamination in household storage containers, which might be related to improper cleaning and maintenance practices, allowing for the survival and growth of pathogenic E. coli strains. The fliCH7 gene encodes the H7 flagellar antigen, which is commonly linked to certain pathogenic E. coli strains [38]. The hlyA gene, associated with the production of haemolysin [39], exhibited the highest prevalence in STEC isolated from spring (S) samples [n = 10 (71.4%)]. This suggested that springs might be a source of STEC strains capable of producing haemolysin, which can cause damage to host cells and potentially contribute to the pathogenicity of E. coli infections. The eae gene, which is associated with the intimin protein involved in attaching and effacing lesions [40], exhibited the highest prevalence in STEC isolated from surface water [n = 8 (77.5%)]. The highest prevalence of the fliCH7 gene was observed in E. coli O157:H7 isolated from stool samples [n = 45 (90%)]. The high prevalence of the fliCH7 gene in stool samples may indicate the presence of pathogenic E. coli O157:H7 strains in the human intestinal tract and the potential for faecal–oral transmission. This finding demonstrates the potential presence of E. coli strains with attaching and effacing capabilities in hand-dug wells, which may pose a risk of infection if water is consumed without prior treatment.
Conversely, the tcpA gene carried by V. cholerae and the eaeA and rfbO157 genes carried by E. coli O157:H7 were the least frequently detected in the isolates from the various matrices (Figure 1). The low prevalence of these genes suggested a lower overall risk of infection with these specific pathogenic strains in the tested samples. Overall, these results provide insight into the distribution and prevalence of specific virulence-associated genes of pathogenic STEC, E. coli O157:H7, and V. cholerae strains in different matrices. Understanding the presence of these genes in various sources is valuable for assessing the potential health risks associated with water and environmental contamination and can aid in implementing appropriate control measures to mitigate these risks.
The average number of raw reads generated per sample was 1,635,181. It is worth noting that the genome coverage for all samples was reported to be greater than 30×, indicating that each position in the genome was covered by sequencing reads at least 30 times on average. Adequate genome coverage is crucial for the accurate assembly and analysis of genomic data, as it ensures a sufficient depth of sequencing to capture the genetic information present in the sample [41]. Hence, STEC emerged as the most frequently identified species in the water samples, also previously examined in the study area [42].
Whole-genome sequencing (WGS) and virulome analysis of E. coli isolated from the various types of water indicated variations in the virulence factor genes (VF genes) among the various water types. Virulome analysis identified a total of 153 VF genes across all the selected samples. The different sample types showed different numbers of VF genes. In Figure 3A, which shows the relative abundance of virulence factor genes in different water sources, the adhesion- and invasion-related genes, such as the upaG and the ehaG trimeric autotransporter adhesins (TAAs), were consistently the most frequently identified genes in most of the E. coli isolates, occurring 92 times. These TAAs have been characterised previously by Totsika et al. [43]. According to these authors, ehaG from enterohaemorrhagic Escherichia coli (EHEC) O157:H7 and upaG from uropathogenic E. coli (UPEC) were found to share similar properties for pathogenesis, but these TAAs also showed differences. While ehaG mediates binding to colorectal epithelial cells, upaG mediates binding to bladder epithelial cells. Moreover, the availability of genome sequencing through WGS has significantly increased the amount of information generated in laboratory testing using conventional methods. By sequencing the entire genome, researchers can uncover a broader range of virulence factors and genetic markers associated with diarrhoeal diseases [44]. In this case, the study identified specific VF genes, including hcp1/tssD1, hlyE, mrkA, and mrkB, which are known to be associated with diarrhoeal diseases.
The presence of these VF genes in various water sources provides additional evidence that these genes are widespread and have a significant impact on the pathogenicity of E. coli. The VF genes shared between stool samples and different water sources, as demonstrated in this study, have also been observed in other studies. A study by Ateba and Mbewe [45] explored the occurrence of virulence factor genes in E. coli isolates from various sources, including water and stools, and found a significant overlap between their virulence factor gene profiles. This implies a potential exchange of pathogenic strains and genes between animals and environmental reservoirs. It is important to note that the absence of VF gene sharing among surface water, groundwater, stools, and wastewater, as reported in this study, may suggest distinct sources and routes of contamination. These findings emphasise the importance of understanding the sources and transmission dynamics of pathogenic strains in water environments to mitigate the risks associated with waterborne diseases. However, human faecal contamination remains a major concern since it introduces enteric pathogens into water and increases the degree of exposure to infectious strains. These findings have implications for water quality management, public health protection, and the development of targeted interventions to reduce the risk of waterborne infections.
One of the objectives of this study was to explore the ARG content in each matrix and to identify the abundance and shared ARGs among them. The WGS analysis revealed a wide range of ARGs across isolates from different matrices (Figure 3). On average, this study identified 29, 178, 131, 29, 101, 188, 39, and 232 ARGs in the matrices S, SS, SP, TWPC, R, HC, D, and B, respectively. This reveals variations in the ARG content among the different water sources and sample types. The most abundant ARG was found to be mdtB, detected 36 times, followed by emrA, detected 35 times across all water sources. The prevalence of mdtB and emrA genes in these matrices implies that they are widely distributed and may play a significant role in antibiotic resistance within the bacterial populations present in the analysed water sources. These genes are associated with efflux pump systems, which are mechanisms that bacteria use to actively remove antibiotics from the cell, reducing their effectiveness [46].
It is important to record that the prevalence and significance of ARGs can vary depending on bacterial species, geographical locations, and other factors. This means that bacteria harbouring these ARGs can resist the effects of multiple classes of antibiotics, making it challenging to treat infections caused by these resistant strains. In this study, acrA, acrD, acrE, acrS, baeR, and baeS genes were widely distributed among the target E. coli strains in different matrices sampled and their presence is often associated with multidrug resistance. Understanding the prevalence and mechanisms of antibiotic resistance genes is crucial for the development of effective strategies to combat bacterial infections [47]. This knowledge can help in the selection of appropriate antibiotics and the development of new drugs or alternative treatment approaches to combat antibiotic-resistant bacteria.
This study also investigated the core resistome, which refers to the shared ARGs found in isolates from all assessed matrices. The core resistome was found to consist of 44 ARGs in isolates from various water matrices. Furthermore, the WGS analysis identified ARGs shared between different water sources. For example, three (3) ARGs were shared between isolates from groundwater and surface water (Figure 3B). Additionally, surface water, groundwater, wastewater, and household drinking water shared two ARGs (Figure 3B). These results provide insight into the diversity and abundance of ARGs in different water sources and highlight the presence of shared antibiotic resistance genes among these matrices. The findings are consistent with previous studies that have also reported variations in the presence of ARGs in different types of water. For instance, a study by Yang et al. [48] investigated ARGs in different water sources, from river water to tap water, and found differences in the abundance and composition of antibiotic resistance genes among the samples.
These data clearly demonstrate the value of WGS as a powerful tool that goes beyond traditional epidemiological methods, enabling a comprehensive and thorough detection of VF genes and ARGs of the studied pathogen such E. coli. In addition, in this study, it was shown that one ARG was shared between surface water and drinking water in households; the persistence of ARGs in HH drinking water highlights potential concerns regarding the spread of antibiotic resistance from one source to another (Figure 3B). It suggests that even if the water passes through treatment processes, which aim to remove harmful microorganisms, ARGs may remain present and pose a risk to public health. Understanding the ARG content and the distribution patterns of antibiotic resistance genes in water sources is crucial for addressing the issue of antibiotic resistance and developing strategies for water quality management and public health protection. It allows for targeted interventions to mitigate the spread of antibiotic resistance and reduce the potential risks associated with the presence of ARGs in the environment.
5. Conclusions
In conclusion, lack of or inadequate access to clean water and poor sanitation facilities in rural communities can lead to the occurrence of waterborne diseases. Pathogenic microorganisms are responsible for waterborne infections, which can be transferred from one water source to another if appropriate water treatment is not practised. This study’s findings highlight a high level of detection of Shiga toxin-producing Escherichia coli, E. coli O157:H7, and Vibrio cholerae in water and stool samples, emphasizing the potential health risks of water contamination. Although culture-based techniques and MALDI-TOF mass spectrometry are used for the initial identification, WGS provides more detailed information regarding the genetic makeup of the pathogens, such as the availability of antimicrobial resistance (AMR) genes and virulence factors (VFs). The existence of virulence genes such as hcp1/tssD1, hlyE, mrkA, and mrkB and antibiotic resistance determinants such as acrA, acrD, acrE, acrS, baeR, and baeS indicates the capacity for more aggressive disease and drug-related problems. The similarity of VF genes and ARGs across different matrices, i.e., stool and treated water samples, works to reinforce the potential survival of the pathogens and possible transmission through the water route. These findings support the need for ongoing surveillance, more effective water treatment methods, and strict public health practices to prevent waterborne disease outbreaks.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/microorganisms13061373/s1, Table S1: Total number of samples collected during wet and dry season; Table S2: Water sample volume used for membrane filtration; Table S3: The prevalence of STEC, E. coli O157:H7 and V. cholerae isolates per matrix; Figure S1: Shiga-toxin Escherichia coli Gel 1 Note: lane M-gene ruler, 1-WS9, 2-WS41, 3-WS27, 4-WS27, 5-DS14, 6-SS1, 7-WS42, 8-DS7, 9-DS43, 10-WS6, 11-WS4, 12-DS5, 13-DS20, 14-DS51; Figure S2: Escherichia coli O157h:7 Gel 1. Note: lane M-gene ruler, 1-WO8, 2-DO7, 3-DO4, 4-D014, 5-DO15, 6-DO12, 7-DO1, 8-WO7, 9-WO6, 10-WO18, 11-WO23, 12-DO11, 13-DO2, 14-DO51; Figure S3: Vibrio cholerae Gel 1. Note: lane M-gene ruler, 1-VW43, 2-WV14, 3-VW12, 4-VW11, 5-WV16, 6-WV13, 7-WV52, 8-VW48, 9-WV91, 10-VW17, 11-WV54; Figure S4: Vibrio cholerae Gel 2. Note: lane M-gene ruler, 12-WV14, 13-WV42, 14-VW21, 15-WV34, 16-WV28, 17-WV40, 18-WV22, 19-WV4, 20-WV2, 21-WV30,22-WV23, 23-WV19, 24-VW8, 25-WV51, 26-WV39.
Author Contributions
M.N.B.M.: Conceptualization, Methodology, Formal analysis, Investigation, Writing—original draft—review, Visualization, Project administration, and Supervision. A.M.: Study design, Methodology, Formal analysis, Investigation, Data curation, and Writing—original draft. All authors have read and agreed to the published version of the manuscript.
Funding
The National Research Foundation and the Department of Science and Technology funded our research under the South African Research Chairs Initiative (SARChI) in Water Quality and Wastewater Management (UID87310). Further funding was provided by the Tshwane University of Technology. Arinao Murei acknowledges that a National Research Foundation (NRF) (UID138785) scholarship also provided financial support.
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki, and approved by the Faculty Committee for Research Ethics of Tshwane University of Technology (TUT) ((FCRE 2019/09/011 (FCPS 03) (SCI)).
Informed Consent Statement
Not applicable.
Data Availability Statement
The original contributions presented in this study are included in the article/Supplementary Materials. Further inquiries can be directed to the corresponding authors.
Acknowledgments
The authors would like to acknowledge the assistance of MSc students in TUT Water Science and Technologies for data collection, as well as the editor.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Omarova, A.; Tussupova, K.; Berndtsson, R.; Kalishev, M.; Sharapatova, K. Protozoan parasites in drinking water: A system approach for improved water, sanitation and hygiene in developing countries. Int. J. Environ. Res. Public. Health 2018, 15, 495. [Google Scholar] [CrossRef] [PubMed]
- WHO. Guidelines for Drinking Water Quality: Fourth Edition, Incoporating the First and Second Addenda; WHO: Geneva, Switzerland, 2022. [Google Scholar]
- Edokpayi, J.N.; Rogawski, E.T.; Kahler, D.M.; Hill, C.L.; Reynolds, C.; Nyathi, E.; Smith, J.A.; Odiyo, J.O.; Samie, A.; Bessong, P.; et al. Challenges to sustainable safe drinking water: A case study of water quality and use across seasons in rural communities in Limpopo Province, South Africa. Water 2018, 10, 159. [Google Scholar] [CrossRef] [PubMed]
- Amatobi, D.A.; Agunwamba, J.C. Improved quantitative microbial risk assessment (QMRA) for drinking water sources in developing countries. Appl. Water Sci. 2022, 12, 49. [Google Scholar] [CrossRef]
- Karama, M.; Mainga, A.O.; Cenci-Goga, B.T.; Malahlela, M.; El-Ashram, S.; Kalake, A. Molecular profiling and antimicrobial resistance of Shiga toxin-producing Escherichia coli O26, O45, O103, O121, O145 and O157 isolates from cattle on cow-calf operations in South Africa. Sci. Rep. 2019, 9, 11930. [Google Scholar] [CrossRef]
- Liu, Y.; Li, H.; Chen, X.; Tong, P.; Zhang, Y.; Zhu, M.; Su, Z.; Yao, G.; Li, G.; Cai, W. Characterization of Shiga toxin-producing Escherichia coli isolated from cattle and sheep in Xinjiang Province, China, using whole-genome sequencing. Transbound. Emerg. Dis. 2022, 69, 413–422. [Google Scholar] [CrossRef]
- Greig, D.R.; Hickey, T.J.; Boxall, M.D.; Begum, H.; Gentle, A.; Jenkins, C.; Chattaway, M.A. A real-time multiplex PCR for the identification and typing of Vibrio cholerae, Diagn. Microbiol. Infect. Dis. 2018, 90, 171–176. [Google Scholar] [CrossRef]
- Pasquali, F.; Palma, F.; Trevisani, M.; Parisi, A.; Lucchi, A.; De Cesare, A.; Manfreda, G. Whole genome sequencing based typing and characterisation of Shiga-toxin producing Escherichia coli strains belonging to O157 and O26 serotypes and isolated in dairy farms. Ital. J. Food Saf. 2018, 7, 181–188. [Google Scholar] [CrossRef]
- Bénard, A.H.; Guenou, E.; Fookes, M.; Ateudjieu, J.; Kasambara, W.; Siever, M.; Debes, A.K. Whole genome sequence of Vibrio cholerae directly from dried spotted filter paper. PLoS Negl. Trop. Dis. 2019, 13, e0007330. [Google Scholar] [CrossRef]
- Delannoy, S.; Mariani-Kurkdjian, P.; Webb, H.E.; Bonacorsi, S.; Fach, P. The mobilome; A major contributor to Escherichia coli stx2-Positive O26: H11 strains intra-serotype diversity. Front. Microbiol. 2017, 8, 1625. [Google Scholar] [CrossRef]
- Amézquita-López, B.A.; Soto-Beltrán, M.; Lee, B.G.; Yambao, J.C.; Quiñones, B. Isolation, genotyping and antimicrobial resistance of Shiga toxin-producing Escherichia coli. J. Microbiol. Immunol. Infect. 2018, 51, 425–434. [Google Scholar] [CrossRef]
- Rumore, J.; Tschetter, L.; Kearney, A.; Kandar, R.; McCormick, R.; Walker, M.; Peterson, C.L.; Reimer, A.; Nadon, C. Evaluation of whole-genome sequencing for outbreak detection of Verotoxigenic Escherichia coli O157:H7 from the Canadian perspective. BMC Genom. 2018, 19, 870. [Google Scholar] [CrossRef] [PubMed]
- Shridhar, P.B.; Worley, J.N.; Gao, X.; Yang, X.; Noll, L.W.; Shi, X.; Bai, J.; Meng, J.; Nagaraja, T.G. Analysis of virulence potential of Escherichia coli O145 isolated from cattle feces and hide samples based on whole genome sequencing. PLoS ONE 2019, 14, e0225057. [Google Scholar] [CrossRef] [PubMed]
- Alhamlan, F.S.; Al-Qahtani, A.A.; Al-Ahdal, M.N. Recommended advanced techniques for waterborne pathogen detection in developing countries. J. Infect. Dev. Ctries. 2015, 9, 128–135. [Google Scholar] [CrossRef] [PubMed]
- García-Aljaro, C.; Blanch, A.R.; Campos, C.; Jofre, J.; Lucena, F. Pathogens, faecal indicators and human-specific microbial source-tracking markers in sewage. J. Appl. Microbiol. 2019, 126, 701–717. [Google Scholar] [CrossRef]
- Cuénod, A.; Foucault, F.; Pflüger, V.; Egli, A. Factors associated with MALDI-TOF mass spectral quality of species identification in clinical routine diagnostics. Front. Cell. Infect. Microbiol. 2021, 11, 1–15. [Google Scholar] [CrossRef]
- Ramaite, K.; Ekwanzala, M.D.; Dewar, J.B.; Momba, M.N.B. Human-associated methicillin-resistant Staphylococcus aureus Clonal Complex 80 isolated from cattle and aquatic environments. Antibiotics 2021, 10, 1038. [Google Scholar] [CrossRef]
- Bannon, J.; Melebari, M.; Jordao, C., Jr.; Leon-Velarde, C.G.; Warriner, K. Incidence of Top 6 Shiga Toxigenic Escherichia Coli within Two Ontario Beef Processing Facilities: Challenges in Screening and Confirmation Testing. AIMS Microbiol. 2016, 2, 278–291. [Google Scholar] [CrossRef]
- Fayemi, O.E.; Akanni, G.B.; Elegbeleye, J.A.; Aboaba, O.O.; Njage, P.M. Prevalence, characterization and antibiotic resistance of Shiga toxigenic Escherichia coli serogroups isolated from fresh beef and locally processed ready-to-eat meat products in Lagos, Nigeria. Food Microbiol. 2021, 347, 109191. [Google Scholar] [CrossRef]
- Mehrabadi, J.F.; Morsali, P.; Nejad, H.R.; Fooladi, A.A.I. Detection of toxigenic Vibrio cholerae with new multiplex PCR. J. Infect. Public. Health 2012, 5, 263–267. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 16 April 2023).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Edgar, R.C.; Haas, B.J.; Clemente, J.C.; Quince, C.; Knight, R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 2011, 27, 2194–2200. [Google Scholar] [CrossRef] [PubMed]
- Prjibelski, A.; Antipov, D.; Meleshko, D.; Lapidus, A.; Korobeynikov, A. Using SPAdes De Novo Assembler. Curr. Protoc. Bioinform. 2020, 70, e102. [Google Scholar] [CrossRef] [PubMed]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef]
- Hyatt, D.; Chen, G.-L.; LoCascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Inform. 2010, 11, 119. [Google Scholar] [CrossRef]
- Liu, B.; Zheng, D.; Jin, Q.; Chen, L.; Yang, J. VFDB 2019: A comparative pathogenomic platform with an interactive web interface. Nucleic Acids Res. 2019, 47, D687–D692. [Google Scholar] [CrossRef]
- Javaid, M.; Qasim, H.; Zia, H.Z.; Bashir, M.A.; Samiullah, K.; Hashem, M.; Morsy, K.; Dajem, S.B.; Muhammad, T.; Shaheen, M.; et al. Bacteriological composition of groundwater and its role in human health. J. King Saud Univ. Sci. 2022, 34, 102128. [Google Scholar] [CrossRef]
- Gwimbi, P.; George, M.; Ramphalile, M. Bacterial contamination of drinking water sources in rural villages of Mohale Basin, Lesotho: Exposures through neighbourhood sanitation and hygiene practices. Environ. Health Prev. Med. 2019, 24, 33. [Google Scholar] [CrossRef]
- Sila, O.N.A. Physico-chemical and bacteriological quality of water sources in rural settings, a case study of Kenya, Africa. Sci. Afr. 2019, 2, e00018. [Google Scholar] [CrossRef]
- Pang, X.; Qiu, Y.; Gao, T.; Zurawell, R.; Neumann, N.F.; Craik, S.; Lee, B.E. Prevalence, levels and seasonal variations of human enteric viruses in six major rivers in Alberta, Canada. Water Res. 2019, 153, 349–356. [Google Scholar] [CrossRef]
- Genter, F.; Putri, G.L.; Pratama, M.A.; Priadi, C.; Willetts, J.; Foster, T. Microbial contamination of groundwater self-supply in urban Indonesia: Assessment of sanitary and socio-economic risk factors. Water Resour. Res. 2022, 58, e2021WR031843. [Google Scholar] [CrossRef]
- Obanor, O.; Afegbua, S.L.; Ameh, J.B. Sanitary status and water quality of some drinking water sources and antibiogram of Shiga toxin-producing Escherichia coli O157:H7 isolated from Shika, Zaria, Nigeria. Int. J. Environ. Health Res. 2022, 33, 1604–1616. [Google Scholar] [CrossRef]
- Hada, H.S.; Stemmler, J.; Grossbard, M.L.; West, P.A.; Potrikus, C.J.; Hastings, J.W. Characterization of non-O1 serovar Vibrio cholerae (Vibrio albensis). Syst. Appl. Microbiol. 1985, 6, 203–209. [Google Scholar] [CrossRef]
- Gronthoud, F.A. (Ed.) Practical Clinical Microbiology and Infectious Diseases: A Hands-On Guide; CRC Press: Boca Raton, FL, USA, 2020; p. 484. [Google Scholar]
- Kurwadkar, S. Occurrence and distribution of organic and inorganic pollutants in groundwater. Water Environ. Res. 2019, 91, 1001–1008. [Google Scholar] [CrossRef]
- Hakim, A.L.; Saputra, D.D.; Tanika, L.; Kusumawati, I.A.; Sari, R.R.; Andreotti, F.; Bagbohouna, M.; Abdurrahim, A.Y.; Wamucii, C.; Lagneaux, E.G.; et al. Protected spring and sacred forest institutions at the instrumental—relational value interface. Curr. Opin. Environ. Sustain. 2023, 62, 101292. [Google Scholar] [CrossRef]
- Tayh, G.; Boubaker, S.M.; Khedher, R.B.; Jbeli, M.; Chehida, F.B.; Mamlouk, A.; Messadi, L. Prevalence, virulence genes, and antimicrobial profiles of Escherichia coli O157:H7 isolated from healthy cattle in Tunisia. J. Infect. Dev. Ctries 2022, 16, 1308–1316. [Google Scholar] [CrossRef]
- Nhu, N.T.K.; Phan, M.D.; Forde, B.M.; Murthy, A.M.; Peters, K.M.; Day, C.J.; Schembri, M.A. Complex multilevel control of hemolysin production by uropathogenic Escherichia coli. mBio 2019, 10, e02248-19. [Google Scholar] [CrossRef]
- Awad, W.S.; El-Sayed, A.A.; Mohammed, F.F.; Bakry, N.M.; Abdou, N.E.M.; Kamel, M.S. Molecular characterisation of pathogenic Escherichia coli isolated from diarrheic and in-contact cattle and buffalo calves. Trop. Anim. Health Prod. 2020, 52, 3173–3185. [Google Scholar] [CrossRef]
- Lou, R.N.; Jacobs, A.; Wilder, A.P.; Therkildsen, N.O. A beginner’s guide to low-coverage whole genome sequencing for population genomics. Mol. Ecol. 2021, 30, 5966–5993. [Google Scholar] [CrossRef]
- Traoré, A.N.; Mulaudzi, K.; Chari, G.J.; Foord, S.H.; Mudau, L.S.; Barnard, T.G.; Potgieter, N. The impact of human activities on microbial quality of rivers in the Vhembe District, South Africa. Int. J. Environ. Res. Public Health 2016, 13, 817. [Google Scholar] [CrossRef]
- Totsika, M.; Wells, T.J.; Beloin, C.; Valle, J.; Allsopp, L.P.; King, N.P.; Ghigo, J.M.; Schembri, M.A. Molecular characterization of the EhaG and UpaG trimeric autotransporter proteins from pathogenic Escherichia coli. Appl. Environ. Microbiol. 2012, 78, 2179–2189. [Google Scholar] [CrossRef]
- Bai, X.; Zhang, J.; Ambikan, A.; Jernberg, C.; Ehricht, R.; Scheutz, F.; Xiong, Y.; Matussek, A. Molecular characterization and comparative genomics of clinical hybrid Shiga toxin-producing and enterotoxigenic Escherichia coli (STEC/ETEC) strains in Sweden. Sci. Rep. 2019, 9, 5619. [Google Scholar] [CrossRef] [PubMed]
- Ateba, C.N.; Mbewe, M. Detection of Escherichia coli O157:H7 virulence genes in isolates from beef, pork, water, human and animal species in the Northwest Province, South Africa: Public health implications. Res. Microbiol. 2011, 162, 240–248. [Google Scholar] [CrossRef] [PubMed]
- Kumawat, M.; Nabi, B.; Daswani, M.; Viquar, I.; Pal, N.; Sharma, P.; Tiwari, S.; Sarma, D.K.; Shubham, S.; Kumar, M. Role of bacterial efflux pump proteins in antibiotic resistance across microbial species. Microb. Pathog. 2023, 181, 106182. [Google Scholar] [CrossRef] [PubMed]
- Pulingam, T.; Parumasivam, T.; Gazzali, A.M.; Sulaiman, A.M.; Chee, J.Y.; Lakshmanan, M.; Chin, C.F.; Sudesh, K. Antimicrobial resistance: Prevalence, economic burden, mechanisms of resistance and strategies to overcome. Eur. J. Pharm. Sci. 2022, 170, 106103. [Google Scholar] [CrossRef]
- Yang, J.; Wang, H.; Roberts, D.J.; Du, H.N.; Yu, X.F.; Zhu, N.Z.; Meng, X.Z. Persistence of antibiotic resistance genes from river water to tap water in the Yangtze River Delta. Sci. Total Environ. 2020, 742, 140592. [Google Scholar] [CrossRef]
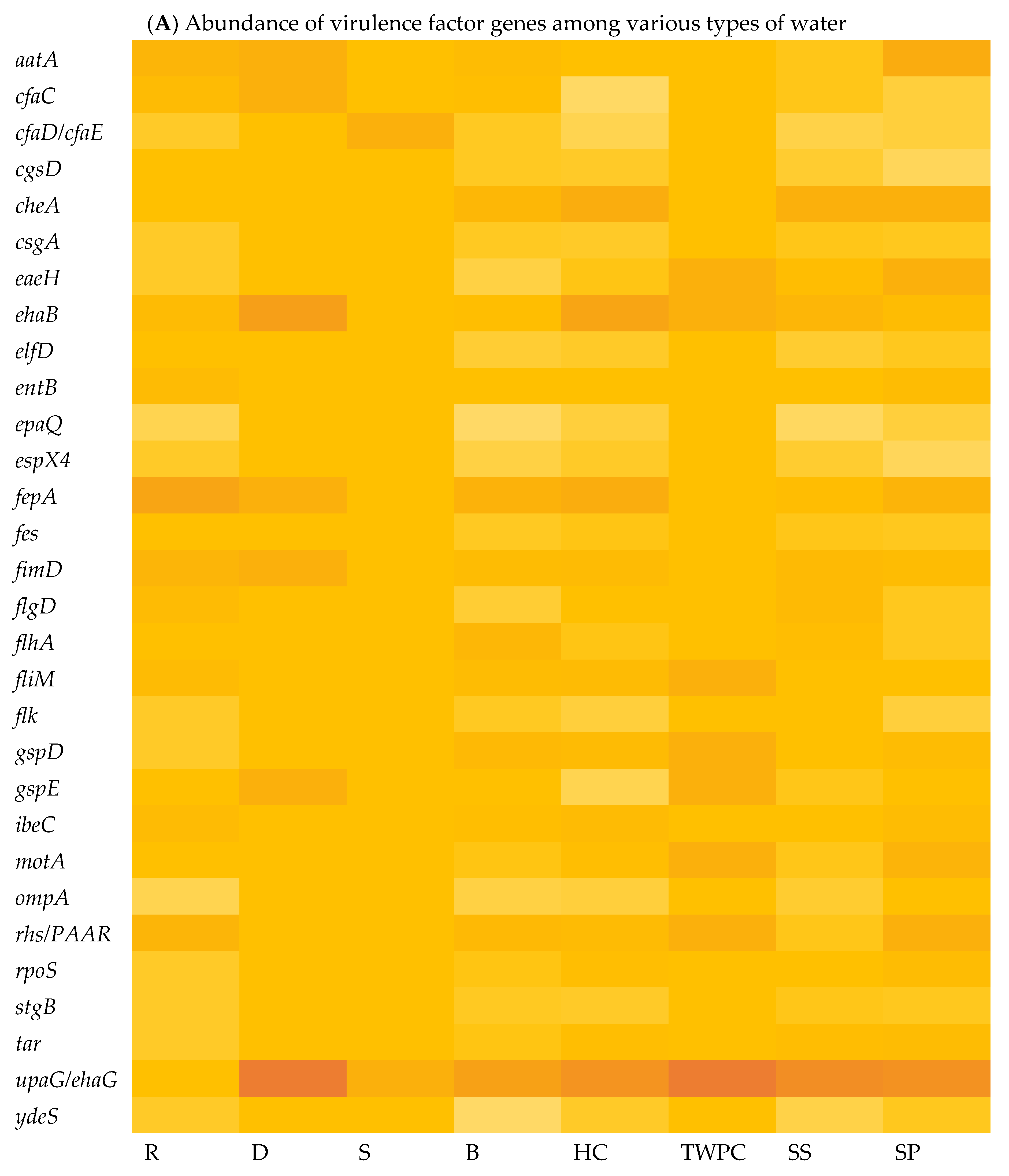
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).