
Marine-Derived Peptides from Phaeodactylum tricornutum as Potential SARS-CoV-2 Mpro Inhibitors: An In Silico Approach

 , ,
, ,  , , ,
, , ,  ,
,  ,
,  , ,
, ,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Retrieving and Aligning Peptide Sequences
2.2. Peptidic Modeling
2.3. Molecular Docking
2.4. Residue Interaction Analysis
2.5. Immunogenicity
2.6. Molecular Dynamics
2.7. Use of Antiviral Peptide Prediction Server (AVPpred)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide Reference | UniprotID | Score | Name | Peptide Alignment |
|---|---|---|---|---|
| QTFSVLACY [23] | A0A8J9SAR9 | 9 | Unknown | NESEQGRIQGALFSLQALASATGPMLLRFIYHLTKDG AFLGPGSMFVVASGIYLIAVYCAYSLP |
| QTFSVLACY [23] | A0A8J9TDK2 | 9 | Unknown | QIGTVVKANYCLWPFFQYINFTFVPSSLRV LATNLMSVLWNCYFCSCIA |
| QTFSVLACY [23] | A0A172E6W5 | 8 | H(+) export diphosphatase | VRGAPDSELQGKGSDIHKAAVVGDTVGDPFKD TSGPALNIVMKLMAVLSLVFADTFAVNNGQGLLNLA |
| TVNVLAWLY [23] | A0A8J9SDW0 | 9 | Unknown | MTVSNEESPDVIELDASTTVETIKIAPTEWIKRLQSTW GEPLVVPEWEDDTEGYRAKNGWQA |
| TVNVLAWLY [23] | A0A8J9SA87 | 8 | G-domain protein | MKLQTAIVGLPNVGKSTLFNALTETQGAEAANYPFCTIEPN |
| TVNVLAWLY [23] | A0A8J9X3P8 | 8 | Protein with protein kinase domain | NPKRTTKVNLGRVLKTLVHVHGLQLMQDGVFNAD PHPGNVLVLPDGRLGLLDYGMV |
| YLQYAVLRHKRREC [24] | A0A8J9SA66 | 11 | Protein with protein kinase domain | VYL-AADVMLPLLQRMHEAGVVHRDVKPSNCV RSTGERDFCIVDFGLSK |
| YLRYKCLCTWQITVC [24] | A0A8J9X2W5 | 11 | Unknown | MQSGAMYEFLFSYKSTQTLPGIPTSGVPRNWRGPLGAQE WAVRRTDRNQWEDGLVFLCTS |
| YLRYKCLCTWQITVC [24] | A0A8J9X430 | 11 | Impact N-terminal domain-containing protein | FAYRLTETISDGTRVSKHDNDDDGEYGAGSKLAHLLQ VRDEKDVVVLVARWFGGVHL |
| YSVAQKRKYWLFVLC [24] | A0A8J9SNS2 | 11 | Bromine domain protein | FLNPVTDEIAPGYSKVIKHPICIA-AMEDK-VESHKYNSPSDW —EGDVNLMYKNCIDYNRGN |
| YSWTYLGRDYYWSC [24] | A0A8J9TRC1 | 10 | Protein with Arf-GAP domain | MAIYEKELNTAS-NTVYEMKTADYAELLSMPG-NSVCAD —CGAVNPNWGSPKLGILFCTDCSGKH |
| YSWTYLGRDYYWSC [24] | A0A8J9T715 | 10 | N-terminal SNF2 protein | AKEAWLEFRDKLYDPNEPHS—Y-KNGNRLRD -YQVEGVNWLASTWYKKQGCILADEMGLGK |
| YTKHVYYHITYILYVC [24] | A0A8J9T326 | 11 | USP domain protein | DGHY-KCHV-QHQATRQWYEIQDL- HVQEIMPQQIGLSECYLLIFRKSGL |
| YVHPKLHKCCIYIVWC [24] | A0A8J9TWV5 | 10 | Protein with protein kinase domain | VYLAADV-MLP-LLQRMHEAGVVHRDVKPSNCVRSTGERDFC —IV—DFGLSK |
3. Results
3.1. Alignment
3.2. Peptidic Modeling
3.3. Molecular Docking
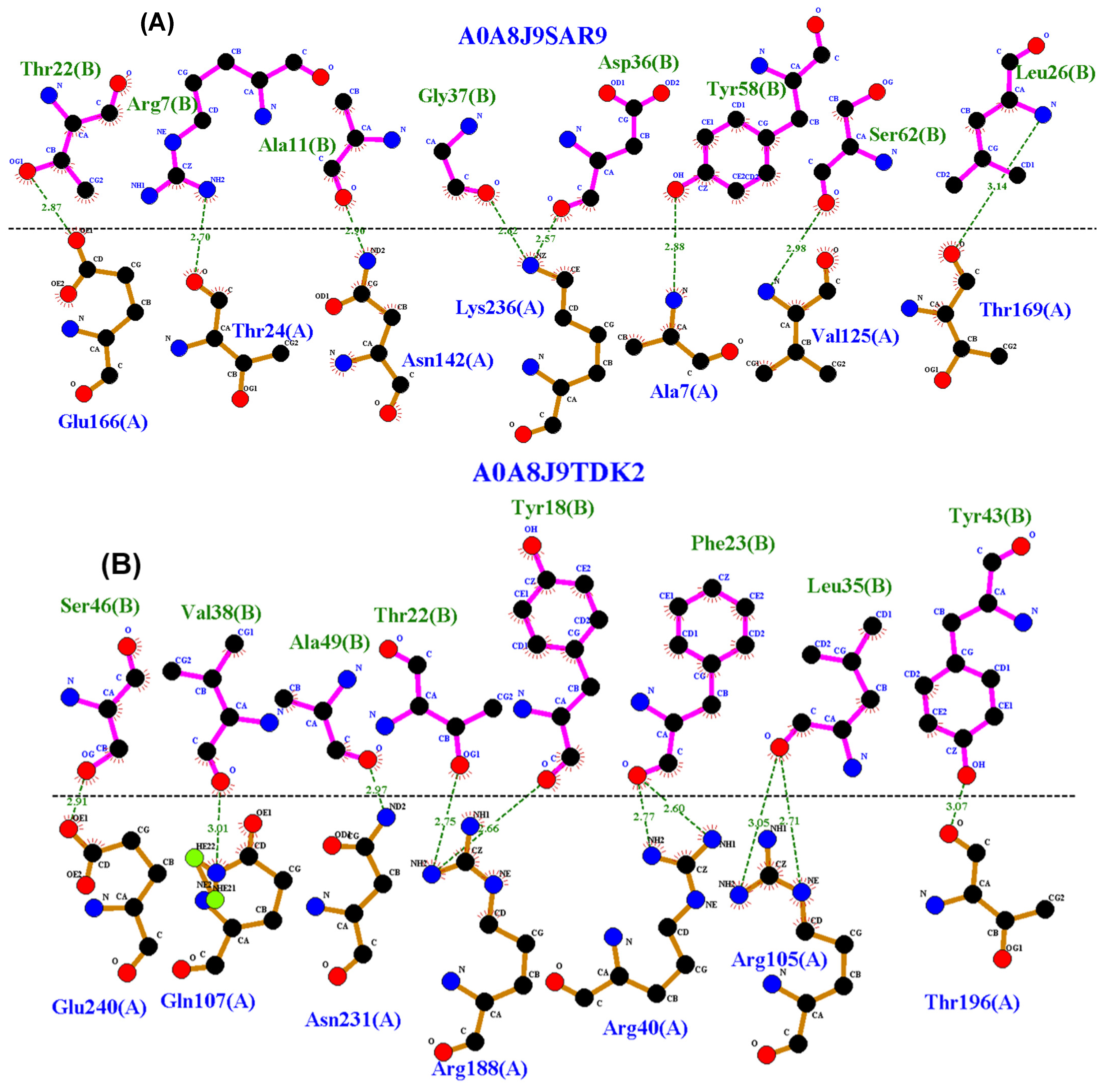
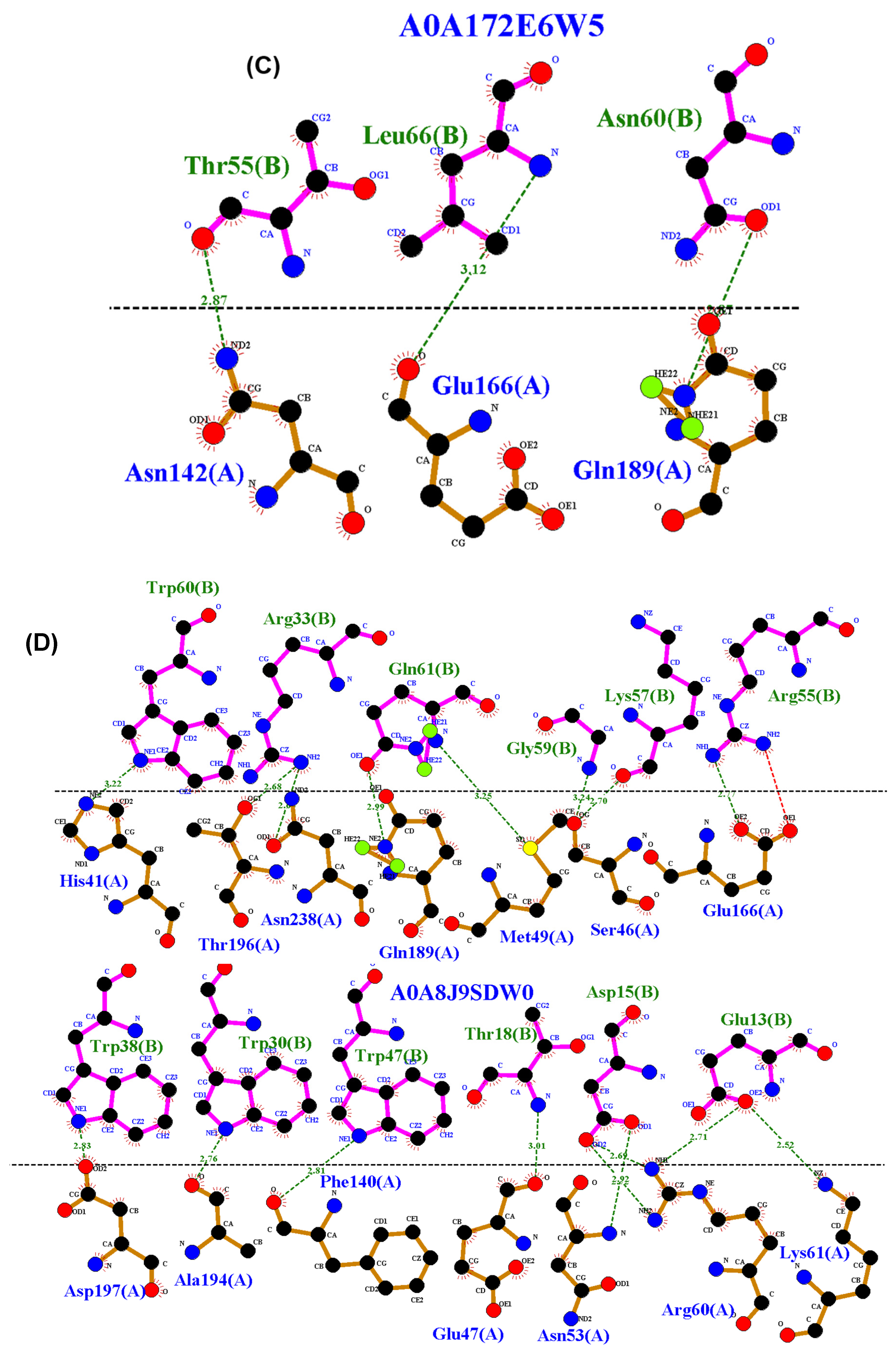
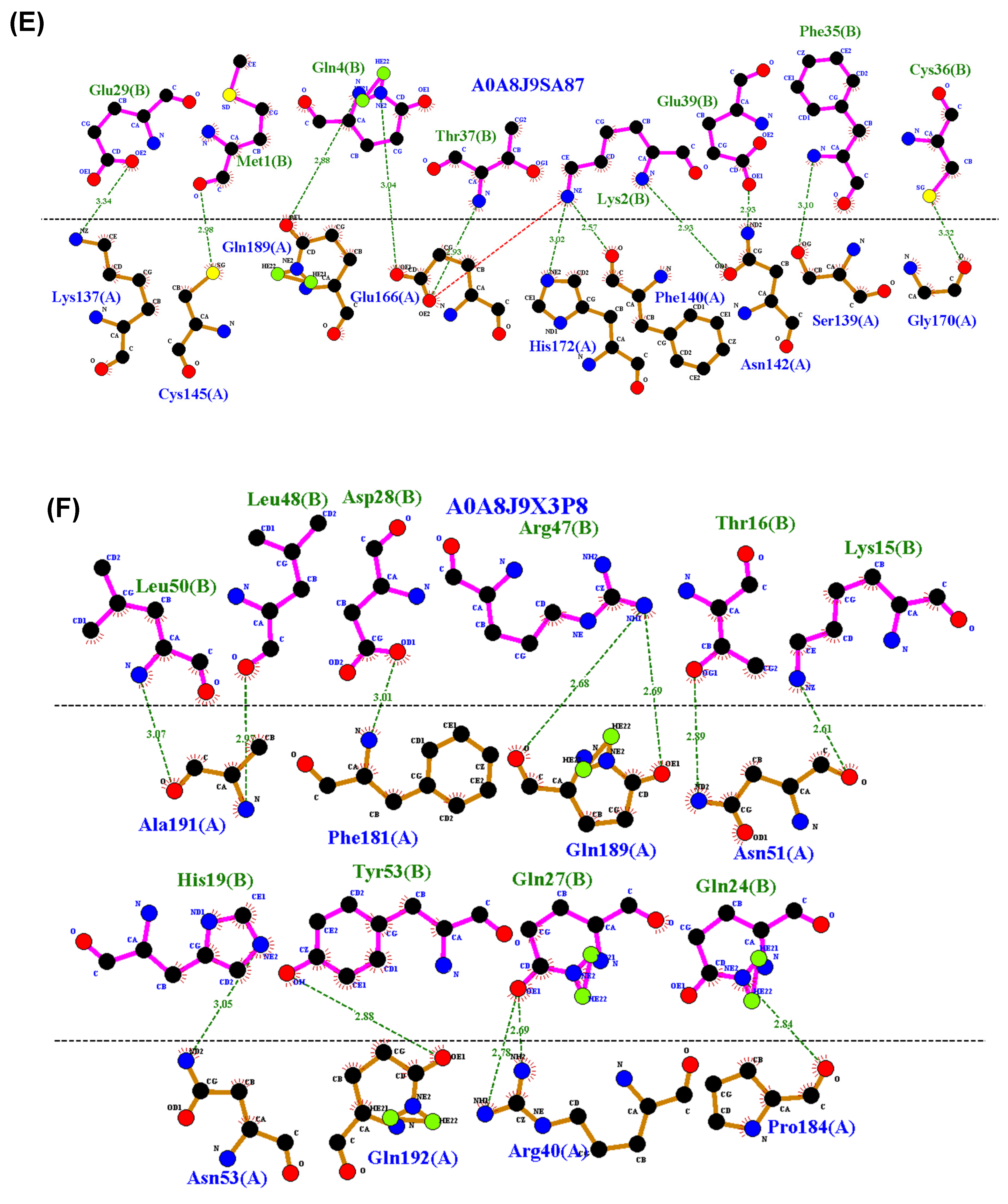
3.4. Residue Interactions
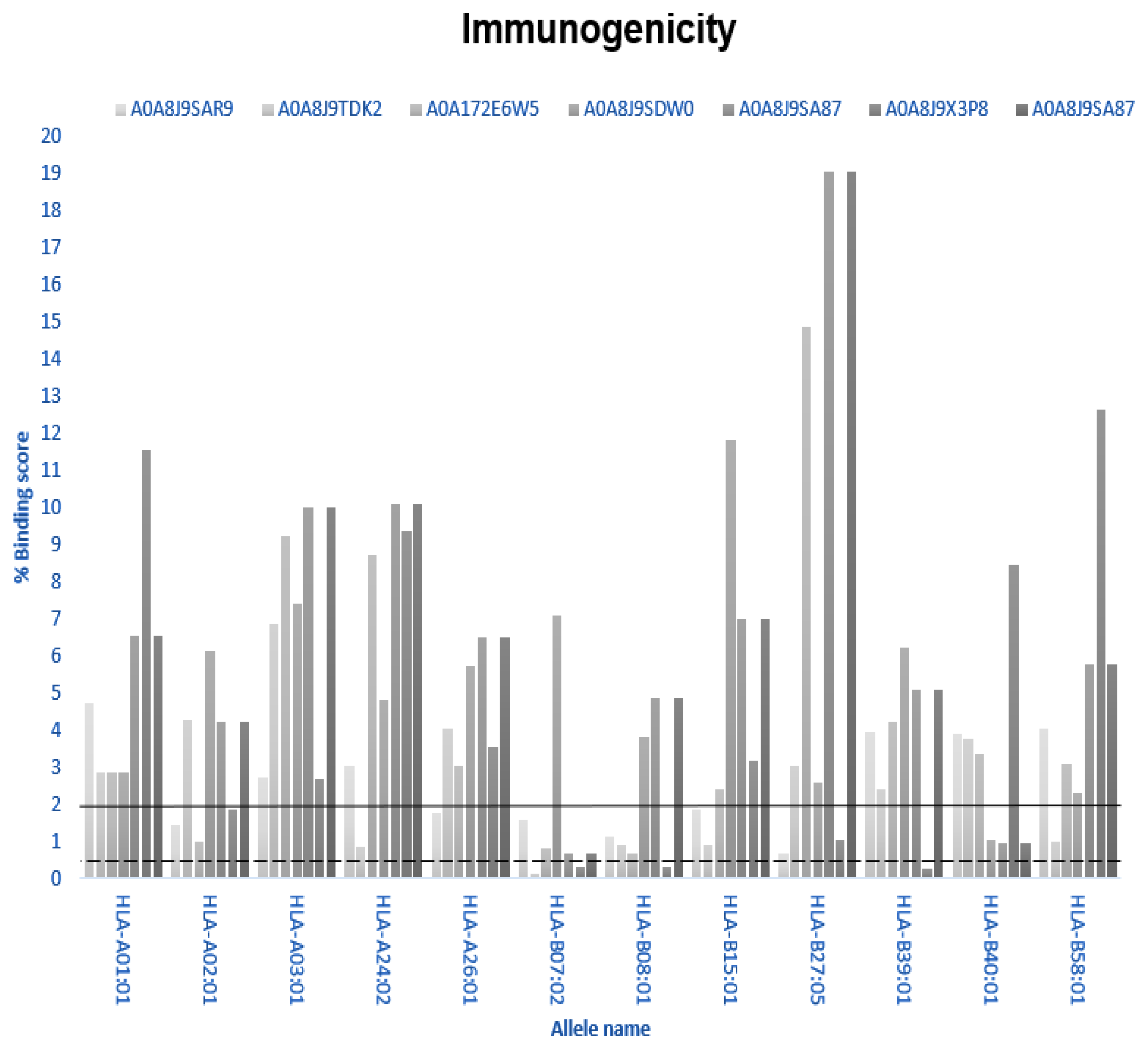
3.5. Immunogenicity
3.6. Use of Antiviral Peptide Predicton Server (AVPpred)
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pan, J.; Villalan, A.K.; Ni, G.; Wu, R.; Sui, S.; Wu, X.; Wang, X. Assessing Eco-Geographic Influences on COVID-19 Transmission: A Global Analysis. Sci. Rep. 2024, 14, 62360. [Google Scholar] [CrossRef] [PubMed]
- De Wit, E.; Van Doremalen, N.; Falzarano, D.; Munster, V.J. SARS and MERS: Recent Insights into Emerging Coronaviruses. Nat. Rev. Microbiol. 2016, 14, 523–534. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization (WHO). Number of COVID-19 Deaths Reported to WHO. 2024. Available online: https://data.who.int/dashboards/covid19/deaths?n=c (accessed on 15 May 2024).
- Su, S.; Wong, G.; Shi, W.; Liu, J.; Lai, A.C.K.; Zhou, J.; Liu, W.; Bi, Y.; Gao, G.F. Epidemiology, Genetic Recombination, and Pathogenesis of Coronaviruses. Trends Microbiol. 2016, 24, 490–502. [Google Scholar] [CrossRef] [PubMed]
- Cortellini, A.; Tabernero, J.; Mukherjee, U.; Salazar, R.; Sureda, A.; Maluquer, C. SARS-CoV-2 Omicron (B.1.1.529)-Related COVID-19 Sequelae in Vaccinated and Unvaccinated Patients with Cancer: Results from the OnCovid Registry. Lancet Oncol. 2023, 24, 335–346. [Google Scholar] [CrossRef]
- Petrosillo, N.; Viceconte, G.; Ergönül, Ö.; Ippolito, G.; Petersen, E. COVID-19, SARS and MERS: Are They Closely Related? Clin. Microbiol. Infect. 2020, 26, 729–734. [Google Scholar] [CrossRef]
- Tiwari, A.; Melchor-Martínez, E.M.; Saxena, A.; Kapoor, N.; Singh, K.J.; Saldarriaga-Hernández, S.; Parra-Saldívar, R.; Iqbal, H.M.N. Therapeutic Attributes and Applied Aspects of Biological Macromolecules from Diatoms—A Review. Int. J. Biol. Macromol. 2021, 171, 398–413. [Google Scholar] [CrossRef]
- Bhattacharjya, R.; Singh, P.K.; Tiwari, A. Aquaculture Water as a Source of Sustainable Growth Medium for Diatom Cultivation and Its Nutritive Suitability as a Potential Aqua Feed. Environ. Technol. Innov. 2021, 24, 101987. [Google Scholar] [CrossRef]
- Ribeiro, M.C.M.; Salles, T.S.; Moreira, M.F.; Barbarino, E.; Valle, A.F.D.; Couto, M.A.P.G. Antiviral Activity of Microalgae Extracts against Mayaro Virus. Algal Res. 2022, 61, 102577. [Google Scholar] [CrossRef]
- Pradhan, B.; Nayak, R.; Patra, S.; Bhuyan, P.P.; Dash, S.R.; Ki, J.; Adhikary, S.P.; Ragusa, A.; Jena, M. Cyanobacteria and Algae-Derived Bioactive Metabolites as Antiviral Agents: Evidence, Mode of Action, and Scope for Further Expansion; A Comprehensive Review in Light of the SARS-CoV-2 Outbreak. Antioxidants 2022, 11, 354. [Google Scholar] [CrossRef]
- Bortolini, D.G.; Maciel, G.M.; Fernandes, I.D.A.A.; Pedro, A.C.; Rúbio, F.; Branco, I.G.; Haminiuk, C.W.I. Functional Properties of Bioactive Compounds from Spirulina spp.: Current Status and Future Trends. Food Chem. Mol. Sci. 2022, 5, 100134. [Google Scholar] [CrossRef]
- Rivera-Serrano, B.V.; Cabanillas-Salcido, S.L.; Cordero-Rivera, C.D.; Jiménez-Camacho, R.; Norzagaray-Valenzuela, C.D.; Calderón-Zamora, L.; De Jesús-González, L.A.; Reyes-Ruiz, J.M.; Farfan-Morales, C.N.; Romero-Utrilla, A.; et al. Antiviral Effect of Microalgae Phaeodactylum tricornutum Protein Hydrolysates against Dengue Virus Serotype 2. Mar. Drugs 2024, 22, 369. [Google Scholar] [CrossRef]
- Mariani, V.; Biasini, M.; Barbato, A.; Schwede, T. LDDT: A Local Superposition-Free Score for Comparing Protein Structures and Models Using Distance Difference Tests. Bioinformatics 2013, 29, 2722–2728. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Skolnick, J. Scoring Function for Automated Assessment of Protein Structure Template Quality. Proteins 2004, 57, 702–710. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Zhang, Y. How Significant Is a Protein Structure Similarity with TM-Score = 0.5? Bioinformatics 2010, 26, 889–895. [Google Scholar] [CrossRef]
- Scardino, V.; Di Filippo, J.I.; Cavasotto, C.N. How Good Are AlphaFold Models for Docking-Based Virtual Screening? iScience 2023, 26, 105920. [Google Scholar] [CrossRef] [PubMed]
- Wallace, A.C.; Laskowski, R.A.; Thornton, J.M. LIGPLOT: A Program to Generate Schematic Diagrams of Protein-Ligand Interactions. Protein Eng. Des. Sel. 1995, 8, 127–134. [Google Scholar] [CrossRef]
- Jurtz, V.I.; Paul, S.; Andreatta, M.; Marcatili, P.; Peters, B.; Nielsen, M. NetMHCpan-4.0: Improved Peptide–MHC Class Interaction Predictions Integrating Eluted Ligand and Peptide Binding Affinity Data. J. Immunol. 2017, 199, 3360–3368. [Google Scholar] [CrossRef]
- Arantes, P.R.; Polêto, M.D.; Pedebos, C.; Ligabue-Braun, R. Making It Rain: Cloud-Based Molecular Simulations for Everyone. J. Chem. Inf. Model. 2021, 61, 4852–4856. [Google Scholar] [CrossRef]
- Tian, C.; Kasavajhala, K.; Belfon, K.; Raguette, L.; Huang, H.; Migues, A.N.; Bickel, J.; Wang, Y.; Pincay, J.; Wu, Q.; et al. ff19SB: Amino-Acid-Specific Protein Backbone Parameters Trained against Quantum Mechanics Energy Surfaces in Solution. J. Chem. Theory Comput. 2020, 16, 528–552. [Google Scholar] [CrossRef]
- Robustelli, P.; Piana, S.; Shaw, D.E. Developing a Molecular Dynamics Force Field for Both Folded and Disordered Protein States. Proc. Natl. Acad. Sci. USA 2018, 115, E4758–E4766. [Google Scholar] [CrossRef]
- Thakur, N.; Qureshi, A.; Kumar, M. AVPpred: Collection and Prediction of Highly Effective Antiviral Peptides. Nucleic Acids Res. 2012, 40, W199–W204. [Google Scholar] [CrossRef] [PubMed]
- Marriam, S.; Afghan, M.S.; Nadeem, M.; Sajid, M.; Ahsan, M.; Basit, A.; Wajid, M.; Sabri, S.; Sajid, M.; Zafar, I.; et al. Elucidation of Novel Compounds and Epitope-Based Peptide Vaccine Design against C30 Endopeptidase Regions of SARS-CoV-2 Using Immunoinformatics Approaches. Front. Cell. Infect. Microbiol. 2023, 13, 1134802. [Google Scholar] [CrossRef]
- Johansen-Leete, J.; Ullrich, S.; Fry, S.E.; Frkic, R.L.; Bedding, M.J.; Aggarwal, A.; Ashhurst, A.S.; Ekanayake, K.B.; Mahawaththa, M.C.; Sasi, V.M.; et al. Antiviral Cyclic Peptides Targeting the Main Protease of SARS-CoV-2. Chem. Sci. 2022, 13, 3826–3836. [Google Scholar] [CrossRef]
- Abramson, J.; Adler, J.; Dunger, J.; Evans, R.; Green, T.; Pritzel, A.; Ronneberger, O.; Willmore, L.; Ballard, A.J.; Bambrick, J.; et al. Accurate Structure Prediction of Biomolecular Interactions with AlphaFold 3. Nature 2024, 630, 493–500. [Google Scholar] [CrossRef]
- Yueh, C.; Beglov, D.; Bohnuud, T.; Mottarella, S.E.; Xia, B.; Kozakov, D. New Additions to the ClusPro Server Motivated by CAPRI. Proteins 2017, 85, 435–444. [Google Scholar] [CrossRef]
- Jones, G.; Jindal, A.; Ghani, U.; Kotelnikov, S.; Egbert, M.; Hashemi, N.; Vajda, S.; Padhorny, D.; Kozakov, D. Elucidation of Protein Function Using Computational Docking and Hotspot Analysis by ClusPro and FTMap. Acta Crystallogr. D Struct. Biol. 2022, 78, 690–697. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of Mpro from SARS-CoV-2 and Discovery of Its Inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef]
- Świderek, K.; Moliner, V. Revealing the Molecular Mechanisms of Proteolysis of SARS-CoV-2 Mpro by QM/MM Computational Methods. Chem. Sci. 2020, 11, 10626–10630. [Google Scholar] [CrossRef]
- Guedes, I.A.; Silva, M.M.P.D.; Galheigo, M.; Krempser, E.; Magalhães, C.S.D.; Barbosa, H.J.C.; Dardenne, L.E. DockThor-VS: A Free Platform for Receptor-Ligand Virtual Screening. J. Mol. Biol. 2024, 436, 168548. [Google Scholar] [CrossRef]
- Zhao, H.; Song, G. Antiviral Peptide-Generative Pre-Trained Transformer (AVP-GPT): A Deep Learning-Powered Model for Antiviral Peptide Design with High-Throughput Discovery and Exceptional Potency. Viruses 2024, 16, 1673. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, Z.; Zheng, Z. Rational Design of an Epidermal Growth Factor Receptor Vaccine: Immunogenicity and Antitumor Research. Biomolecules 2024, 14, 1620. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Wang, W.; Zhang, J.; Zhou, B.; Zhao, W.; Su, Z.; Gu, X.; Wu, J.; Zhou, Z.; Chen, S. DeepHLAPan: A Deep Learning Approach for Neoantigen Prediction Considering Both HLA-Peptide Binding and Immunogenicity. Front. Immunol. 2019, 10, 2559. [Google Scholar] [CrossRef] [PubMed]
- Muteeb, G.; Alshoaibi, A.; Aatif, M.; Rehman, M.T. Screening Marine Algae Metabolites as High-Affinity Inhibitors of SARS-CoV-2 Main Protease (3CLpro): An In Silico Analysis to Identify Novel Drug Candidates to Combat COVID-19. J. Mol. Model. 2020, 26, 217. [Google Scholar] [CrossRef]
- Singh, S.; Chauhan, P.; Sharma, V.; Rao, A.; Kumbhar, B.V.; Prajapati, V.K. Identification of Multi-Targeting Natural Antiviral Peptides to Impede SARS-CoV-2 Infection. Struct. Chem. 2023, 34, 1743–1758. [Google Scholar] [CrossRef]
- Sansone, C.; Pistelli, L.; Del Mondo, A.; Calabrone, L. The Microalgal Diatoxanthin Inflects the Cytokine Storm in SARS-CoV-2 Stimulated ACE2 Overexpressing Lung Cells. Antioxidants 2022, 11, 1515. [Google Scholar] [CrossRef] [PubMed]
- Karagöl, T.; Karagöl, A. Benchmarking GROMACS on Optimized Colab Processors and the Flexibility of Cloud Computing for Molecular Dynamics. bioRxiv 2024. [CrossRef]
- MubarakAli, D.; MohamedSaalis, J.; Sathya, R.; Irfan, N. An Evidence of Microalgal Peptides to Target Spike Protein of COVID-19: In Silico Approach. Mol. Simul. 2021, 48, 178–189. [Google Scholar] [CrossRef]
- Arunkumar, M.; Gunaseelan, S.; Kubendran, A.M.; Mohankumar, V.; Anupam, P.; Harikrishnan, M.; Siva, A.; Ashokkumar, B.; Varalakshmi, P. Marine Algal Antagonists Targeting 3CL Protease and Spike Glycoprotein of SARS-CoV-2: A Computational Approach for Anti-COVID-19 Drug Discovery. J. Biomol. Struct. Dyn. 2023, 40, 10109–10127. [Google Scholar] [CrossRef]
- Chuntakaruk, H.; Hengphasatporn, K.; Shigeta, Y.; Aonbangkhen, C.; Lee, V.S.; Khotavivattana, T.; Rungrotmongkol, T.; Hannongbua, S. FMO-Guided Design of Darunavir Analogs as HIV-1 Protease Inhibitors. Sci. Rep. 2024, 14, 53940. [Google Scholar] [CrossRef]
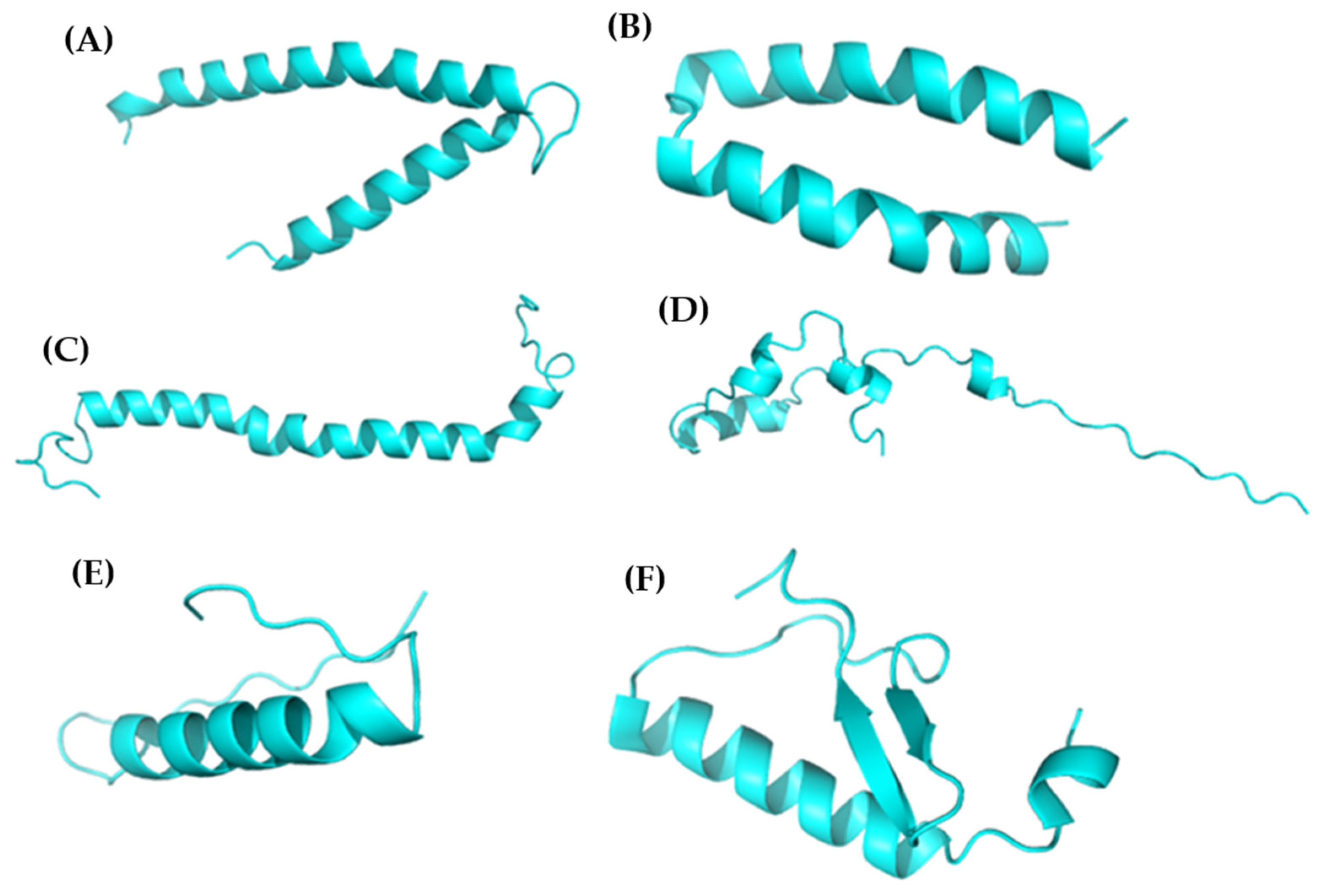
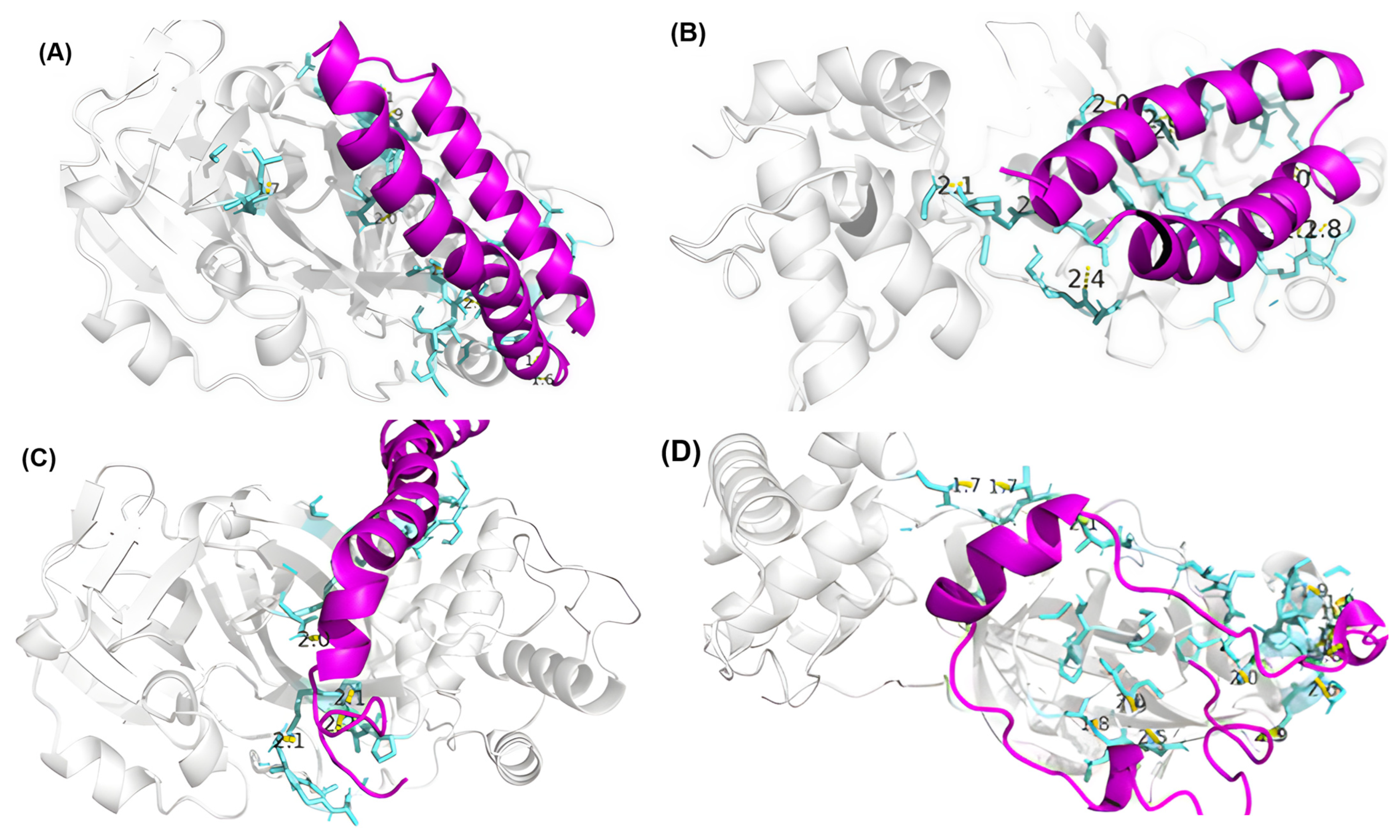
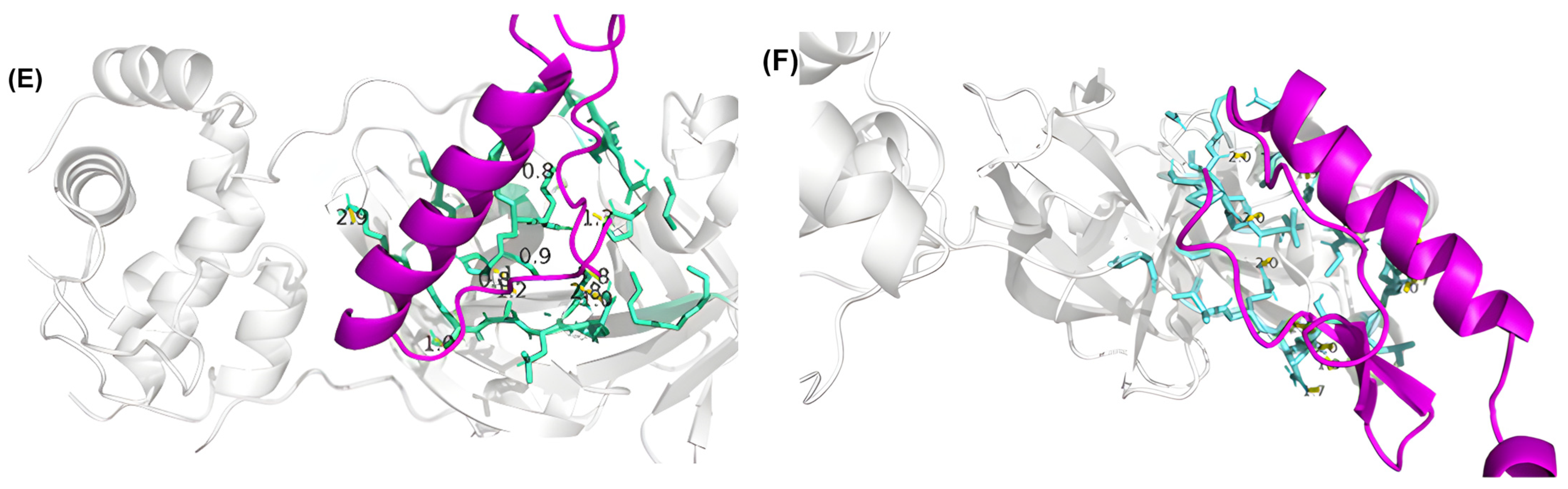
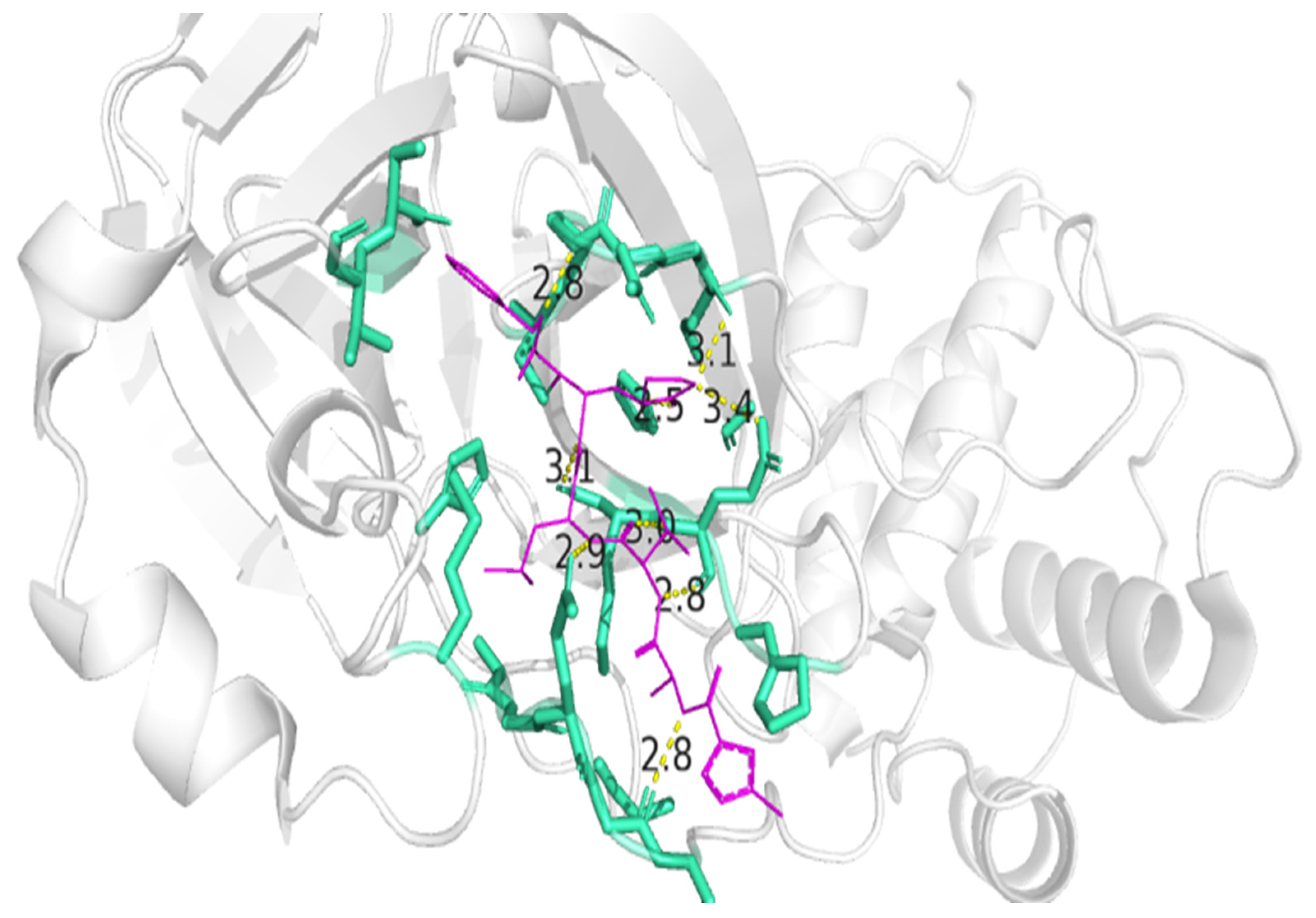
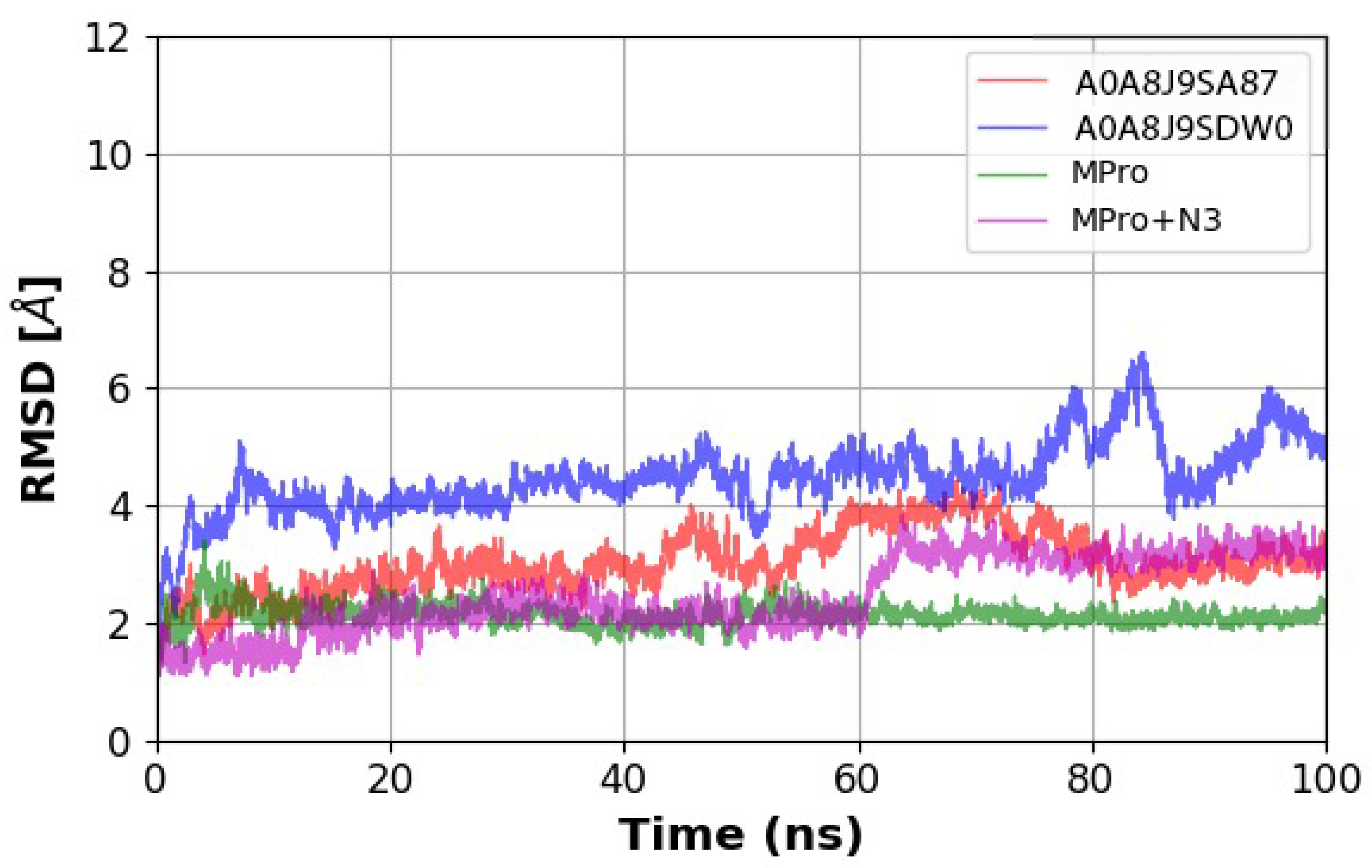
| Peptide ID | Seed | Interface Predicted Template Modeling (ipTM) |
|---|---|---|
| A0A8J9SAR9 | 1743890153 | 0.52 |
| A0A8J9TDK2 | 1142432823 | 0.51 |
| A0A172E6W5 | 143176737 | 0.56 |
| A0A8J9SDW0 | 854280539 | 0.25 |
| A0A8J9SA87 | 1972657144 | 0.5 |
| A0A8J9X3P8 | 2021447260 | 0.71 |
| Peptide ID | GMQE | QMEANDiscCo | % Identity |
|---|---|---|---|
| A0A8J9SAR9 | 0.88 | 0.68 ± 0.11 | 100 |
| A0A8J9TDK2 | 0.80 | 0.45 ± 0.11 | 100 |
| A0A172E6W5 | 0.92 | 0.60 ± 0.11 | 98.53 |
| A0A8J9SDW0 | 0.61 | 0.29 ± 0.11 | 100 |
| A0A8J9SA87 | 0.80 | 0.54 ± 0.12 | 82.93 |
| A0A8J9X3P8 | 0.83 | 0.57 ± 0.11 | 100 |
| Peptide ID | Members | Representative | Weighted Score (kcal/mol) |
|---|---|---|---|
| A0A8J9SAR9 | 122 | Center | −831.2 |
| Lowest energy | −1214.2 | ||
| A0A8J9TDK2 | 132 | Center | −867.3 |
| Lowest energy | −1042.3 | ||
| A0A172E6W5 | 90 | Center | −799.8 |
| Lowest energy | −964.1 | ||
| A0A8J9SDW0 | 94 | Center | −1262.5 |
| Lowest energy | −1371.5 | ||
| A0A8J9SA87 | 103 | Center | −805.2 |
| Lowest energy | −944.3 | ||
| A0A8J9X3P8 | 71 | Center | −672.6 |
| Lowest energy | −719.3 |
| Weak Interactions | |
|---|---|
| Peptide ID | Allele Name |
| A0A8J9SAR9 | HLA-A02:01, HLA-A26:01, HLA-B07:02, HLA-B08-01, HLA-B15:01, HLA-B27:05 |
| A0A8J9TDK2 | HLA-24:02, HLA-B08:01, HLA-B15:01, HLA-B58:01 |
| A0A172E6W5 | HLA-A02:01, HLA-B07:02, HLA-B08:0 |
| A0A8J9SDW0 | HLA-B40:01 |
| A0A8J9SA87 | HLA-B07:02, HLA-B40:01 |
| A0A8J9X3P8 | HLA-A02;01, HLA-B27:05 |
| Name | Lenght | Align | Comp | Physico | AVP | Polarity |
|---|---|---|---|---|---|---|
| Reference | 9 | Non-AVP | 49.38% | 31.83% | By no method | Hydrophobic = 77% Hydrophilic = 23% |
| A0A8J9SDW0 | 62 | Non-AVP | 18.79% | 64.08% | By 1 | Hydrophobic = 45% Hydrophilic = 55% |
| A0A8J9SA87 | 41 | Non-AVP | 21.88% | 63.38% | By 1 | Hydrophobic = 53% Hydrophilic = 47% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cañedo-Figueroa, D.M.; Valdez-Flores, M.A.; Norzagaray-Valenzuela, C.D.; Calderón-Zamora, L.; Rábago-Monzón, Á.R.; Camberos-Barraza, J.; Guadrón-Llanos, A.M.; Herrán-Arita, A.K.D.l.; Picos-Cárdenas, V.J.; Camacho-Zamora, A.; et al. Marine-Derived Peptides from Phaeodactylum tricornutum as Potential SARS-CoV-2 Mpro Inhibitors: An In Silico Approach. Microorganisms 2025, 13, 1271. https://doi.org/10.3390/microorganisms13061271
Cañedo-Figueroa DM, Valdez-Flores MA, Norzagaray-Valenzuela CD, Calderón-Zamora L, Rábago-Monzón ÁR, Camberos-Barraza J, Guadrón-Llanos AM, Herrán-Arita AKDl, Picos-Cárdenas VJ, Camacho-Zamora A, et al. Marine-Derived Peptides from Phaeodactylum tricornutum as Potential SARS-CoV-2 Mpro Inhibitors: An In Silico Approach. Microorganisms. 2025; 13(6):1271. https://doi.org/10.3390/microorganisms13061271
Chicago/Turabian StyleCañedo-Figueroa, David Mauricio, Marco Antonio Valdez-Flores, Claudia Desireé Norzagaray-Valenzuela, Loranda Calderón-Zamora, Ángel Radamés Rábago-Monzón, Josué Camberos-Barraza, Alma Marlene Guadrón-Llanos, Alberto Kousuke De la Herrán-Arita, Verónica Judith Picos-Cárdenas, Alejandro Camacho-Zamora, and et al. 2025. "Marine-Derived Peptides from Phaeodactylum tricornutum as Potential SARS-CoV-2 Mpro Inhibitors: An In Silico Approach" Microorganisms 13, no. 6: 1271. https://doi.org/10.3390/microorganisms13061271
APA StyleCañedo-Figueroa, D. M., Valdez-Flores, M. A., Norzagaray-Valenzuela, C. D., Calderón-Zamora, L., Rábago-Monzón, Á. R., Camberos-Barraza, J., Guadrón-Llanos, A. M., Herrán-Arita, A. K. D. l., Picos-Cárdenas, V. J., Camacho-Zamora, A., Romero-Utrilla, A., Cordero-Rivera, C. D., Ángel, R. M. d., León-Juárez, M., Reyes-Ruiz, J. M., Farfan-Morales, C. N., De Jesús-González, L. A., & Osuna-Ramos, J. F. (2025). Marine-Derived Peptides from Phaeodactylum tricornutum as Potential SARS-CoV-2 Mpro Inhibitors: An In Silico Approach. Microorganisms, 13(6), 1271. https://doi.org/10.3390/microorganisms13061271