
Wetland-to-Meadow Transition Alters Soil Microbial Networks and Stability in the Sanjiangyuan Region



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
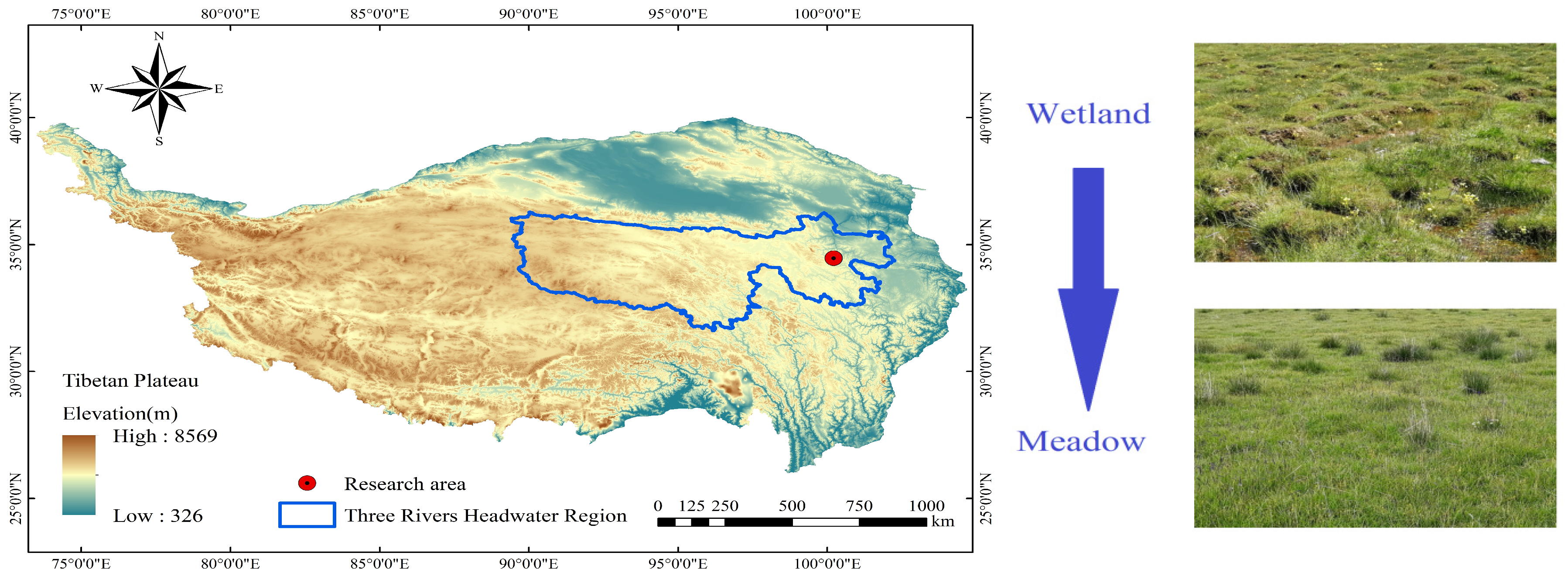
2.1. Study Area
2.2. Sample Collection
2.3. Data Analysis
- (1)
- Bacterial and Fungal Community Composition and Niche Breadth Indices
- (2)
- Soil Physicochemical Properties
- (3)
- Environmental Gradient Analysis
3. Results
3.1. Vegetation and Environmental Variables
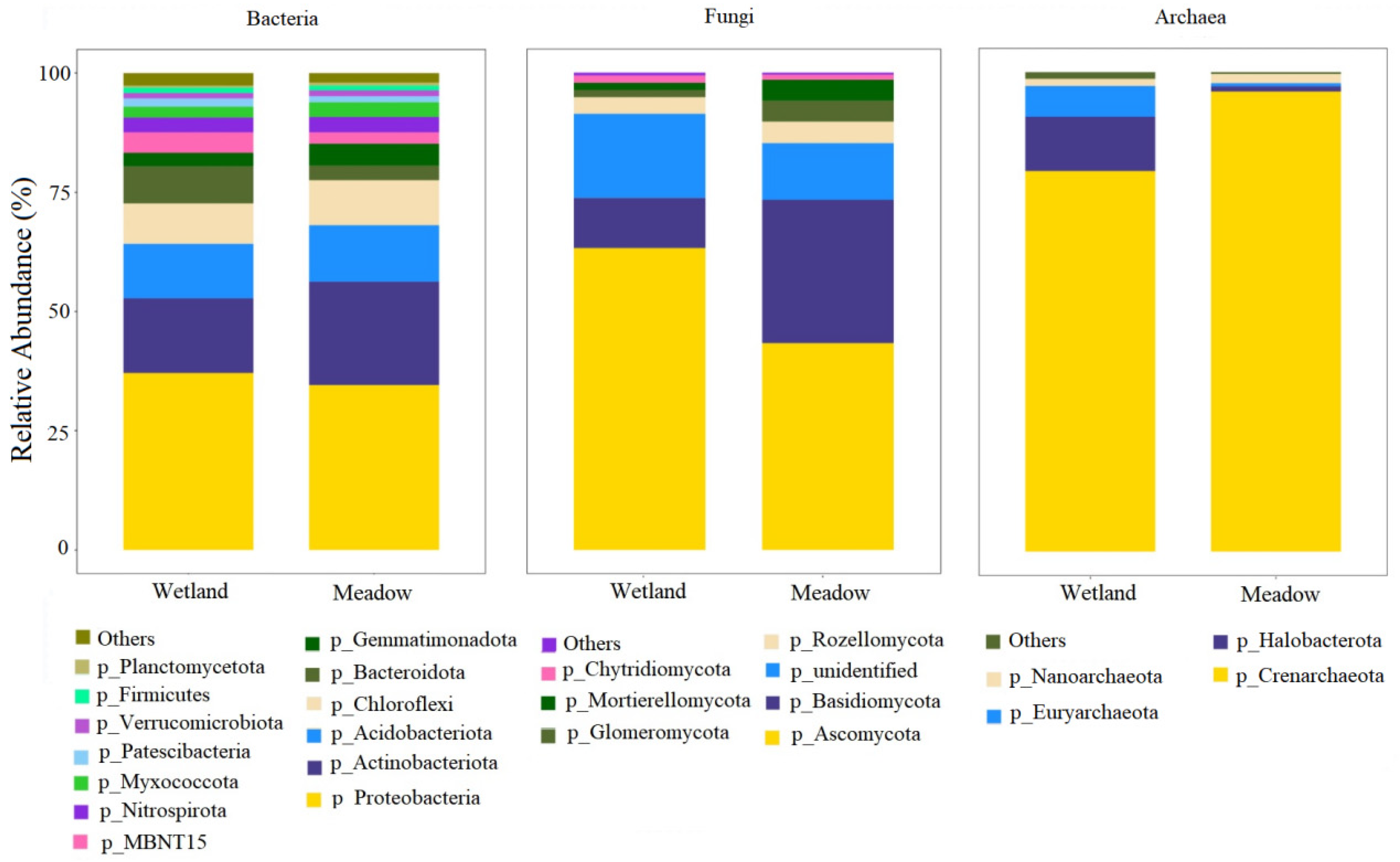
3.2. Characteristics of Soil Microbial Community Composition in Wetlands and Meadows
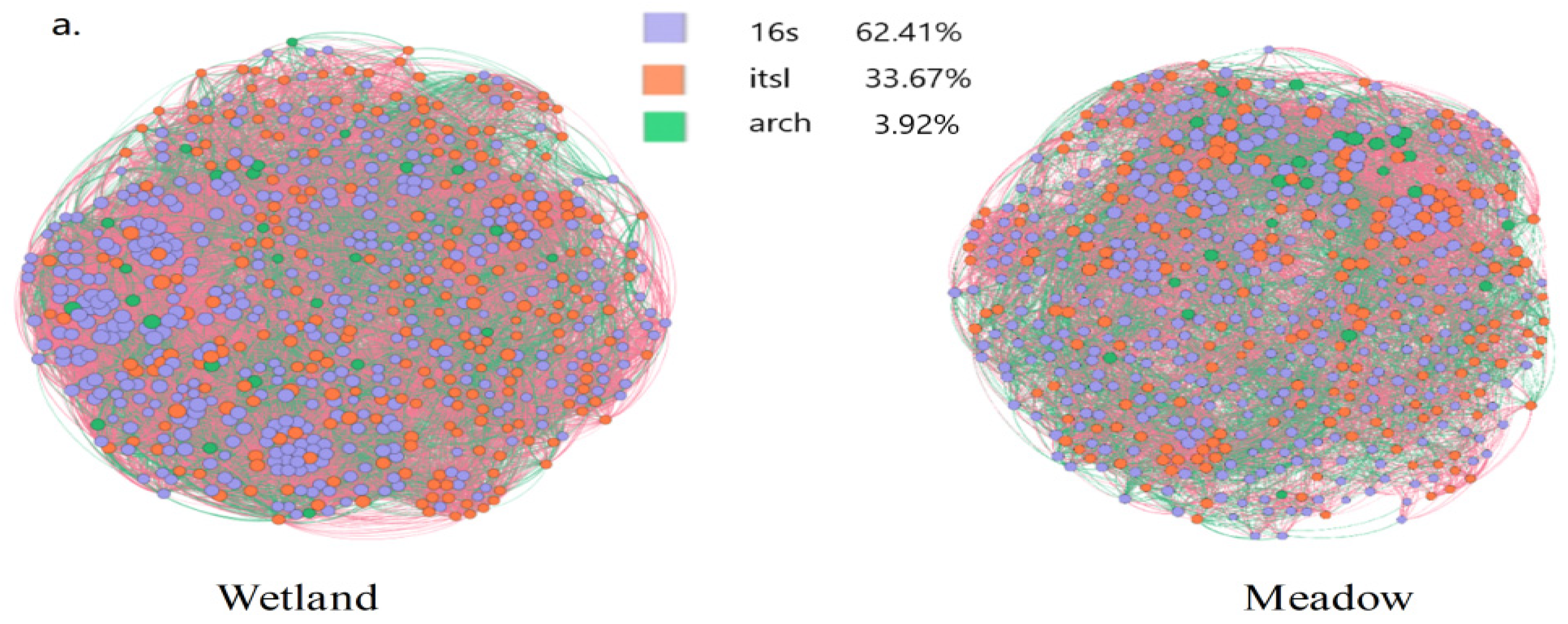
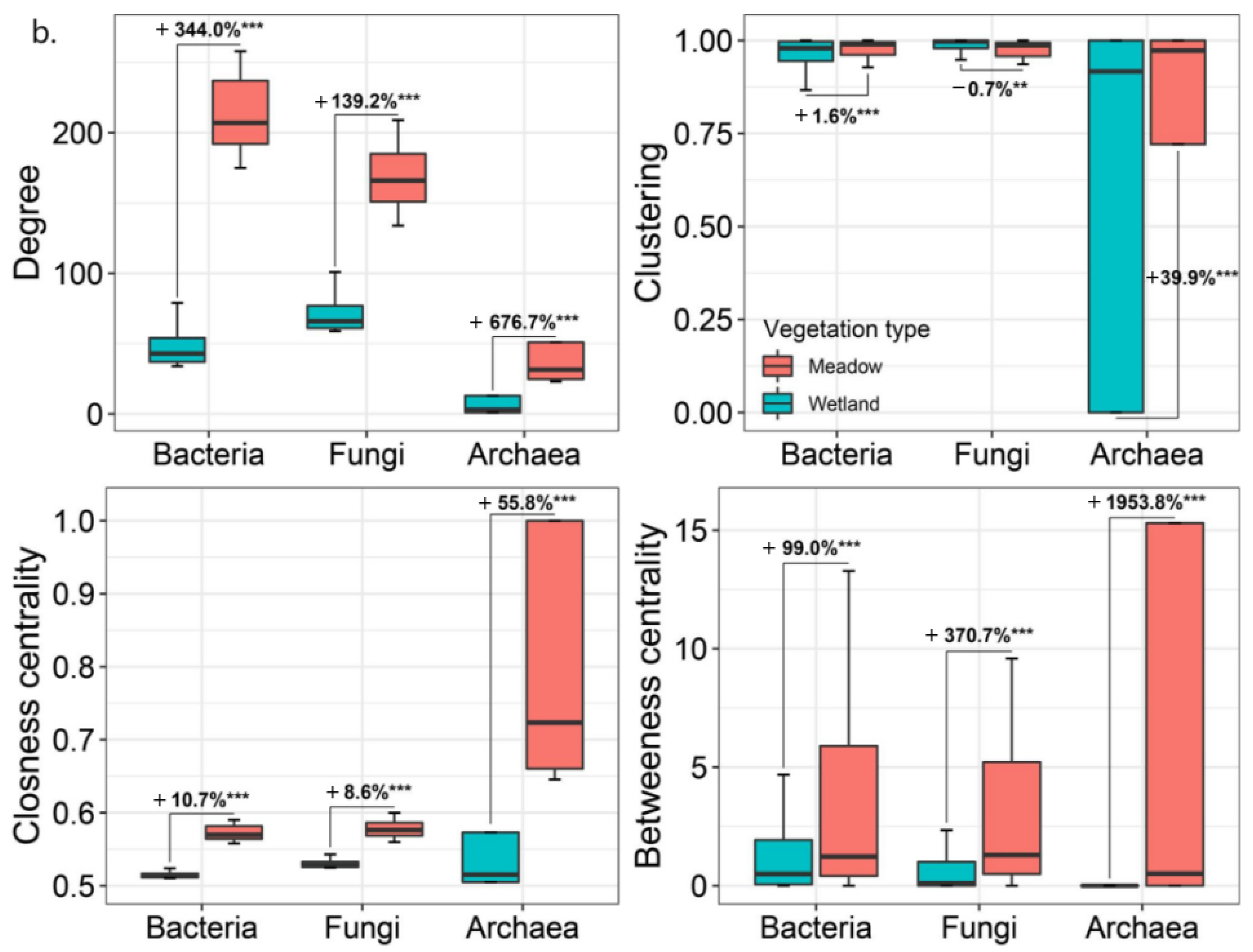
3.3. Microbial Network Structures and Niches in Wetlands and Meadows
3.4. Microbial Community Response to Environmental Gradients
3.5. Driving Effects of Environmental Factors on Microbial Communities
4. Discussion
4.1. Effects of the Transition from Wetland to Meadow on Microbial Communities and Their Ecological Adaptation Mechanisms
4.2. Contributions of Microorganisms to Soil Functions and Ecological Adaptation After Wetland Drying
4.3. Key Driving Factors of Wetland-to-Meadow Conversion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zeng, Y.; Zhong, L.; Wang, L. Changes in the Landscape Pattern in Sanjiangyuan during 1970–2020 and Implications for Ecotourism Development. J. Resour. Ecol. 2023, 14, 230–238. [Google Scholar] [CrossRef]
- Liu, T.; Peng, X.; Li, J. Evaluation of Ecological Sensitivity and Spatial Correlation Analysis of Landscape Patterns in Sanjiangyuan National Park. Sustainability 2024, 16, 5294. [Google Scholar] [CrossRef]
- Yang, J.; Li, X.; Zhu, G.; Zhang, W.; Chen, S.; Liu, H.; Li, Y.; He, J.; Zhang, Y.; Yu, G. Microbial community dynamics and nutrient cycling in alpine wetlands undergoing meadow degradation. J. Environ. Manag. 2022, 310, 114756. [Google Scholar] [CrossRef]
- Sun, Q.; Liu, W.; Gao, Y.; Li, J.; Yang, C. Spatiotemporal Variation and Climate Influence Factors of Vegetation Ecological Quality in the Sanjiangyuan National Park. Sustainability 2020, 12, 6634. [Google Scholar] [CrossRef]
- Gao, S.; Liu, L.; Tang, S.; Zhang, Y. Effects of wetland drainage on microbial-mediated carbon and nitrogen processes. Agric. Ecosyst. Environ. 2023, 354, 108392. [Google Scholar]
- Qiu, H.; Duan, Z.; Li, J.; Wang, Y. Changes in soil microbial community assembly processes with wetland loss and transition to meadows. Soil Ecol. Lett. 2023, 5, 32–44. [Google Scholar]
- Zhao, C.; Wang, S.; Liu, Y. Impacts of climate change on wetland drying and meadow expansion in the Sanjiangyuan region, Qinghai-Tibet Plateau. Sci. Total Environ. 2018, 634, 77–87. [Google Scholar]
- Zhao, H.; Wu, L.; Li, Z.; Zhang, Y. Vegetation and soil microbial diversity responses to wetland-to-meadow transitions in alpine ecosystems. Sci. Total Environ. 2022, 854, 158957. [Google Scholar] [CrossRef]
- Wu, J.; Wu, Y.; Fu, G.; Zhang, X. Long-term overgrazing impacts on vegetation, soil properties, and soil microbial communities in alpine wetlands. J. Environ. Manag. 2021, 279, 111747. [Google Scholar] [CrossRef]
- Yang, Y.; Zhang, Y.; Wang, H.; Chen, H. Effects of climate change on wetland ecosystems in the Sanjiangyuan region. J. Mt. Sci. 2018, 15, 560–573. [Google Scholar] [CrossRef]
- Chen, X.; Zhang, Y.; Liu, J.; Wang, L. Effects of wetland degradation on soil organic carbon storage and soil respiration intensity. J. Environ. Sci. 2021, 35, 102–112. [Google Scholar]
- Xu, Q.; Feng, X.; Zhao, Y. Responses of microbial communities to environmental gradients: Insights from wetland and meadow ecosystems. Appl. Soil Ecol. 2019, 138, 178–188. [Google Scholar]
- Wang, Y.; Wang, Z.; Liu, Q.; Li, M. Effects of wetland degradation on microbial community structure and soil carbon dynamics. Soil Biol. Biochem. 2023, 182, 108658. [Google Scholar]
- Zhou, Z.; Wang, C.; Luo, Y. Meta-analysis of the impacts of global change factors on soil microbial diversity and functionality. Nat. Commun. 2020, 11, 3072. [Google Scholar] [CrossRef] [PubMed]
- Waring, B.; Gee, A.; Liang, G.; Gee, S. A quantitative analysis of microbial community structure–function relationships in plant litter decay. iScience 2022, 25, 104523. [Google Scholar] [CrossRef]
- He, Z.; Zhao, W.; Zhang, Y.; Wang, C.; Li, H. Phosphorus limitation drives microbial functional shifts during wetland-to-meadow conversion. Geoderma 2023, 432, 116275. [Google Scholar]
- Zhou, J.; Wang, Y.; Wang, F. Decoupling soil community structure, functional composition, and environmental drivers. Soil Biol. Biochem. 2024, 190, 109148. [Google Scholar]
- Zhou, J.; Gu, Z.; Wang, B.; Wu, L. Network analysis reveals microbial community resilience to wetland drying and rewetting. ISME J. 2020, 14, 1331–1343. [Google Scholar]
- Yang, M.; Liu, N.; Wang, B.; Zhang, X.; Chen, L. Stepwise degradation of organic matters driven by microbial interac-tions in China’s coastal wetlands: Evidence from carbon isotope analysis. Water Res. 2023, 250, 121062. [Google Scholar] [CrossRef]
- Wu, G.; Li, X.; Gao, J. The evolution of hummock–depression micro-topography in an alpine marshy wetland in Sanjiangyuan as inferred from vegetation and soil characteristics. Ecol. Evol. 2021, 11, 3901–3916. [Google Scholar] [CrossRef]
- Zhang, T.; Chen, H.; Ruan, H.; Wang, L.; Li, X. Microbial responses to wetland drying: Implications for carbon cycling. Ecol. Lett. 2021, 24, 437–452. [Google Scholar] [CrossRef]
- Wu, D.; Bai, H.; Zhao, C.; Liu, Y.; Zhang, Q.; Liu, L. The characteristics of soil microbial co-occurrence networks across a high-latitude forested wetland ecotone in China. Front. Microbiol. 2023, 14, 1160683. [Google Scholar] [CrossRef] [PubMed]
- QIAGEN DNeasy PowerSoil Kit Handbook. 2021. Available online: https://www.qiagen.com (accessed on 15 March 2025).
- Emerson, J.B.; Adams, R.I.; Roman, C.M.B.; Brooks, B.; Coil, D.A.; Dahlhausen, K.; Ganz, H.H.; Hartmann, E.M.; Hsu, T.; Justice, N.B.; et al. Identifying and Removing Contaminant Sequences in Microbiome Data. Curr. Protoc. Microbiol. 2017, 46, 1–18. [Google Scholar]
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berg-Lyons, D.; Lozupone, C.A.; Huntley, J.; Fierer, N.; Owens, S.M.; Betley, J.; Fraser, L.; et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 2012, 6, 1621–1624. [Google Scholar] [CrossRef] [PubMed]
- White, T.J.; Bruns, T.; Lee, S.; Taylor, J. Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In PCR Protocols: A Guide to Methods and Applications; Innis, M.A., Gelfand, D.H., Sninsky, J.J., White, T.J., Eds.; Academic Press: Cambridge, MA, USA, 1990; pp. 315–322. [Google Scholar]
- Kozich, J.J.; Westcott, S.L.; Baxter, N.T.; Highlander, S.K.; Schloss, P.D. Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Appl. Environ. Microbiol. 2013, 79, 5112–5120. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, A.N.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef] [PubMed]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef]
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.R.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Huttenhower, C.; Langille, M.G.I. PICRUSt2: An improved and extensible approach for metagenome inference. Nat. Biotechnol. 2020, 38, 685–688. [Google Scholar] [CrossRef]
- Yan, Z.; Kang, E.; Zhang, K.; Li, M.; Wang, J.; Zhang, Y. Asynchronous responses of microbial CAZymes genes and the net CO2 exchange in alpine peatland following 5 years of continuous extreme drought events. ISME Commun. 2022, 2, 115. [Google Scholar] [CrossRef]
- Liu, X.; Feng, Q.; Wang, R.; Zhang, L.; Huang, L.; Yang, Y.; Chen, J. Long-term changes in microbial community structure during wetland-to-meadow transition under climate warming. Ecol. Indic. 2023, 154, 110456. [Google Scholar] [CrossRef]
- Oksanen, J.; Simpson, G.L.; Blanchet, F.G.; Kindt, R.; Legendre, P.; Minchin, P.R.; O’Hara, R.B.; Solymos, P.; Stevens, M.H.H.; Szoecs, E.; et al. vegan: Community Ecology Package (R Package Version 2.6-6). 2023. Available online: https://CRAN.R-project.org/package=vegan (accessed on 15 March 2025).
- Ramette, A. Multivariate analyses in microbial ecology. FEMS Microbiol. Ecol. 2007, 62, 142–160. [Google Scholar] [CrossRef] [PubMed]
- Anderson, M.J.; Willis, T.J. Canonical analysis of principal coordinates: A useful method of constrained ordination for ecology. Ecology 2003, 84, 511–525. [Google Scholar] [CrossRef]
- Zhou, J.; Wang, Y.; Wang, F. Soil microbial community structure and function mainly respond to indirect effects of future climate change scenarios. Soil Biol. Biochem. 2020, 144, 107780. [Google Scholar] [CrossRef]
- Wu, M.H.; Xue, K.; Wei, P.J.; Li, W.; Li, W.Z.; Zhou, J.Z. Soil microbial distribution and assembly are related to vegetation biomassin the alpine permafrost regions of the Qinghai-Tibet Plateau. Sci. Total Environ. 2022, 834, 155259. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Wang, Y.; Wang, S.; Zhang, Y.; Liu, X.; Wang, H. Effects of wetland degradation on the diversity and composition of microbial communities involved in carbon and nitrogen cycles. Sci. Total Environ. 2020, 727, 138153. [Google Scholar] [CrossRef]
- Ma, J.; Xu, X.F.; He, N.P.; Wang, Y.F.; Yu, G.R. Effects of wetland drying on microbial communities and soil carbon storage in subtropical wetlands. Glob. Change Biol. 2019, 25, 3348–3360. [Google Scholar] [CrossRef]
- Zhou, X.Q.; Guo, Z.Y.; Chen, C.R.; Jia, Z.J. Soil microbial community structure and diversity are largely influenced by soil pH and nutrient quality in78-year-old tree plantations. Biogeosciences 2017, 14, 2101–2111. [Google Scholar] [CrossRef]
- Banerjee, S.; Schlaeppi, K.; van der Heijden, M.G.A. Keystone taxa as drivers of microbiome structure and functioning. Nat. Rev. Microbiol. 2019, 16, 567–576. [Google Scholar] [CrossRef]
- Zhang, X.; Liu, S.; Wang, J.; Huang, Y.; Freedman, Z.; Fu, S.; Liu, K.; Wang, H.; Li, X.; Yao, M.; et al. Local community assembly mechanisms shape soil bacterial β diversity patterns along a latitudinal gradient. Nat. Commun. 2020, 11, 5428. [Google Scholar] [CrossRef]
- Li, M.; Hao, Y.; Yan, Z.; Kang, E. Long-term degradation from marshes into meadows shifts microbial functional diversity of soil phosphorus cycling in an alpine wetland of the Tibetan Plateau. Land Degrad. Dev. 2022, 33, 628–637. [Google Scholar] [CrossRef]
- Tian, H.; Liu, X.; Zhao, Y.; Zhang, Y.; Wang, Y.; Fang, W.; Li, M.; Chen, S.; Zhang, W.; Yu, G. Carbon cycling implications of microbial community shifts in wetlands transitioning to grasslands. Glob. Change Biol. 2022, 28, 3674–3689. [Google Scholar] [CrossRef]
- Zhou, J.; Xue, K.; Xie, J.; Deng, Y.; Wu, L.; Cheng, X.; Fei, S.; Deng, S.; He, Z.; Van Nostrand, J.D.; et al. Microbial mediation of carbon-cycle feedbacks to climate warming. Nat. Clim. Change 2012, 2, 106–110. [Google Scholar] [CrossRef]
- Ma, Z.; Liu, H.; Mi, Z.; Zhang, Z.; Wang, Y.; Xu, W.; Jiang, L.; He, J.S. Climate warming reduces the temporal stability of plant community biomass production. Nat. Commun. 2017, 8, 15378. [Google Scholar] [CrossRef]
- Liu, S.; Garcia-Palacios, P.; Tedersoo, L.; Guirado, E.; van der Heijden, M.G.A.; Wagg, C.; Chen, D.; Wang, Q.; Wang, J.; Singh, B.K.; et al. Phylotype diversity within soil fungal functional groups drives ecosystem stability. Nat. Ecol. Evol. 2022, 7, 6. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Li, H.; Zhou, J.; Huang, X.; Zhang, L.; Liu, X. Diversity and niche width of microbial communities in wetland ecosystems: Implications for nutrient decomposition and carbon storage. Soil Biol. Biochem. 2020, 141, 107688. [Google Scholar]
- Zhang, Y.; Li, B.; Liu, L. Changes in Wetland Ecosystems and Their Environmental Drivers in the Sanjiangyuan Region. Wetl. Ecol. Manag. 2021, 29, 707–722. [Google Scholar]
- Yu, Z.; Li, Y.; Wu, S.; Wang, F.; Xu, X.; Chen, L. Response of microbial communities to wetland degradation in the Tibetan Plateau. Soil Biol. Biochem. 2018, 119, 15–26. [Google Scholar] [CrossRef]
- Clemmensen, K.E.; Bahr, A.; Ovaskainen, O.; Dahlberg, A.; Ekblad, A.; Wallander, H.; Stenlid, J.; Finlay, R.D.; Wardle, D.A.; Lindahl, B.D. Roots and associated fungi drive long-term carbon sequestration in boreal forest. Science 2013, 339, 1615–1618. [Google Scholar] [CrossRef] [PubMed]
- Yuan, M.M.; Guo, X.; Wu, L.; Zhang, Y.; Xiao, N.; Ning, D.; Shi, Z.J.; Zhou, X.; Wu, L.; Yang, Y.; et al. Climate warming enhances microbial network complexity and stability. Nat. Clim. Change 2021, 11, 343–348. [Google Scholar] [CrossRef]
- Nguyen, N.H.; Song, Z.; Bates, S.T.; Branco, S.; Tedersoo, L.; Menke, J.; Schilling, J.S.; Kennedy, P.G. FUNGuild: An open annotation tool for parsing fungal community datasets by ecological guild. Fungal Ecol. 2016, 20, 241–248. [Google Scholar] [CrossRef]
- Yang, W.; Guo, Y.; Wang, X.; Chen, C.; Hu, Y.; Cheng, L.; Gu, S.; Xu, X. Temporal variations of soil microbial community under compost addition in black soil of Northeast China. Appl. Soil Ecol. 2017, 121, 214–222. [Google Scholar] [CrossRef]
- Ma, B.; Wang, H.; Dsouza, M.; Lou, J.; He, Y.; Dai, Z.; Brookes, P.C.; Xu, J.; Gilbert, J.A. Geographic patterns of co-occurrence network topological features for soil microbiota at continental scale in eastern China. ISME J. 2015, 10, 1891. [Google Scholar] [CrossRef] [PubMed]
- Six, J.; Frey, S.D.; Thiet, R.K.; Batten, K.M. Stabilization mechanisms of soil organic matter: Implications for C-saturation of soils. Plant Soil 2004, 241, 155–176. [Google Scholar] [CrossRef]
- Xue, K.; Yuan, M.M.; Shi, Z.; Qin, Y.; Deng, Y.; Cheng, L.; Wu, L.; He, Z.; Van Nostrand, J.D.; Bracho, R.; et al. Tundra soil carbon is vulnerable to rapid microbial decomposition under climate warming. Nat. Clim. Change 2016, 6, 595–600. [Google Scholar] [CrossRef]
- Reddy, K.R.; DeLaune, R.D. Biogeochemistry of Wetlands: Science and Applications; CRC Press: Boca Raton, FL, USA, 2008. [Google Scholar] [CrossRef]
- Chen, L.; Zheng, Y.; Zhang, J.; He, J. Transition of wetlands to meadows changes soil microbial communities and their functional potential: A meta-analysis. Appl. Soil Ecol. 2023, 186, 104774. [Google Scholar]
- Tong, L.; Xu, X.; Fu, Y.; Li, S. Wetland changes and their responses to climate change in the “Three-River Headwaters” region of China since the 1990s. Energies 2014, 7, 2515–2534. [Google Scholar] [CrossRef]
- Li, X.; Brierley, G.; Shi, D.; Xie, Y.; Sun, H. Ecological Protection and Restoration in Sanjiangyuan National Nature Reserve, Qinghai Province, China. In Perspectives on Environmental Management and Technology in Asian River Basins; Springer: Berlin/Heidelberg, Germany, 2011. [Google Scholar] [CrossRef]
- Mori, A.S.; Forest, I.; Rupert, S. β-Diversity, Community Assembly, and Ecosystem Functioning. Trends Ecol. Evol. 2018, 33, 549–564. [Google Scholar] [CrossRef]
- Yuan, W.; Li, F.; Yu, J.; Wang, Y.; Liu, S. Relationship Between Vegetation Restoration and Soil Microbial Characteristics in Degraded Karst Regions: A Case Study. Pedosphere 2011, 21, 132–138. [Google Scholar]
- Wu, H.; Yang, H.; Zhang, Z.; Li, B.; Liu, W. Effects of vegetation degradation on the functional diversity and stability of soil microbial communities in alpine wetlands. Funct. Ecol. 2021, 35, 343–356. [Google Scholar] [CrossRef]
- Zhou, J.; Deng, Y.; Zhang, P.; Xue, K.; Liang, Y.; Van Nostrand, J.D.; Yang, Y.; He, Z.; Wu, L.; Stahl David, A.; et al. Stochastic assembly leads to alternative communities with distinct functions in a high-diversity ecosystem. mBio 2017, 8, e00482-17. [Google Scholar]
- Xu, X.; Zhang, W.; He, Y.; Li, M.; Liu, S.; Wang, F.; Chen, Y.; Zhao, Y.; Wang, Z.; Zhang, Q.; et al. Soil microbial responses to wetland reclamation and conversion to agriculture and grassland. Front. Microbiol. 2022, 13, 923158. [Google Scholar]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, G.; Gao, J.; Wang, Z.; Du, Y. Wetland-to-Meadow Transition Alters Soil Microbial Networks and Stability in the Sanjiangyuan Region. Microorganisms 2025, 13, 1263. https://doi.org/10.3390/microorganisms13061263
Wu G, Gao J, Wang Z, Du Y. Wetland-to-Meadow Transition Alters Soil Microbial Networks and Stability in the Sanjiangyuan Region. Microorganisms. 2025; 13(6):1263. https://doi.org/10.3390/microorganisms13061263
Chicago/Turabian StyleWu, Guiling, Jay Gao, Zhaoqi Wang, and Yangong Du. 2025. "Wetland-to-Meadow Transition Alters Soil Microbial Networks and Stability in the Sanjiangyuan Region" Microorganisms 13, no. 6: 1263. https://doi.org/10.3390/microorganisms13061263
APA StyleWu, G., Gao, J., Wang, Z., & Du, Y. (2025). Wetland-to-Meadow Transition Alters Soil Microbial Networks and Stability in the Sanjiangyuan Region. Microorganisms, 13(6), 1263. https://doi.org/10.3390/microorganisms13061263