
Optimization of Mapping Tools and Investigation of Ribosomal RNA Influence for Data-Driven Gene Expression Analysis in Complex Microbiomes


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
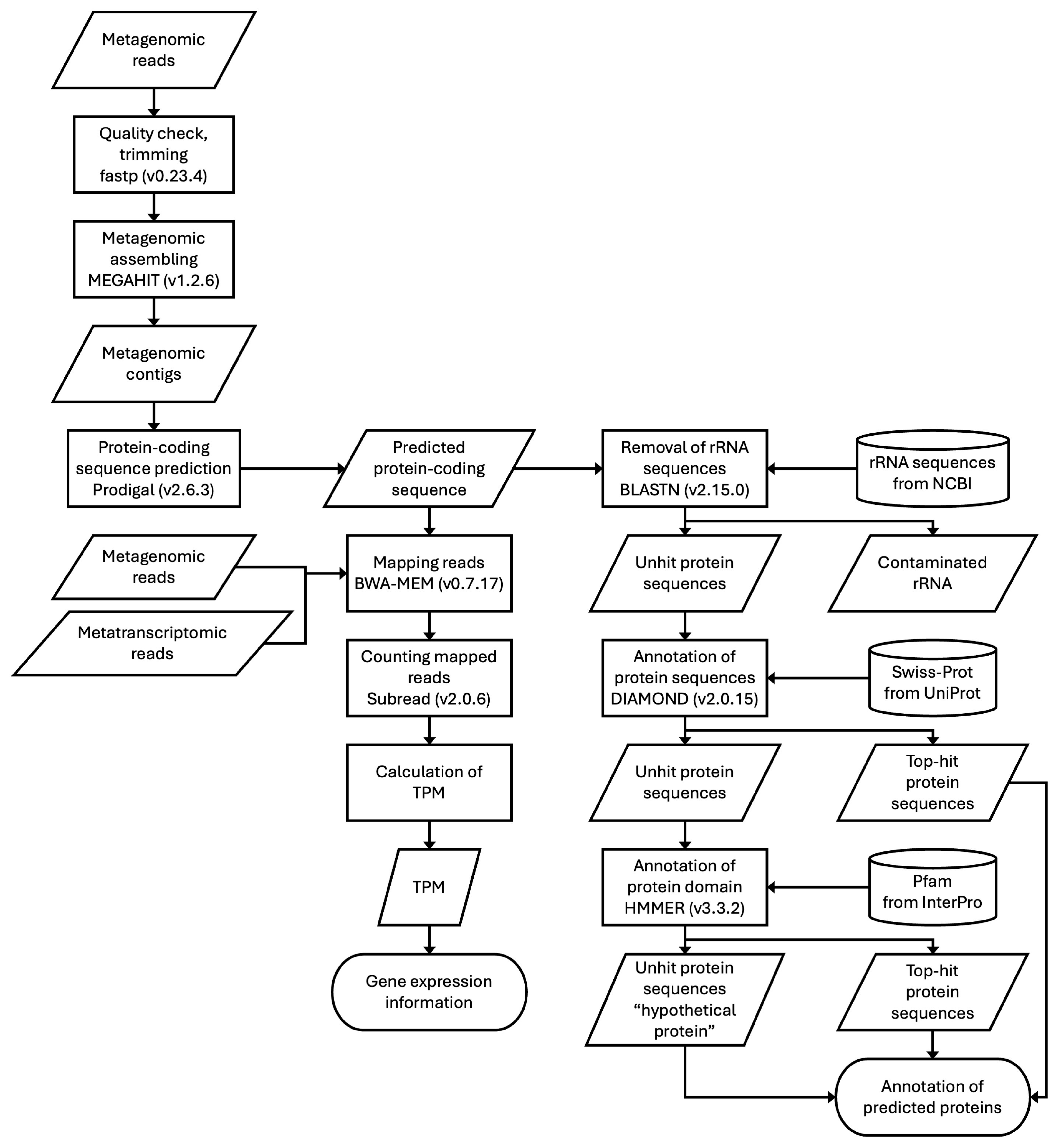
2. Materials and Methods
2.1. Computational Resources
2.2. Optimization of Mapping Tools
2.3. Gene Expression Analysis
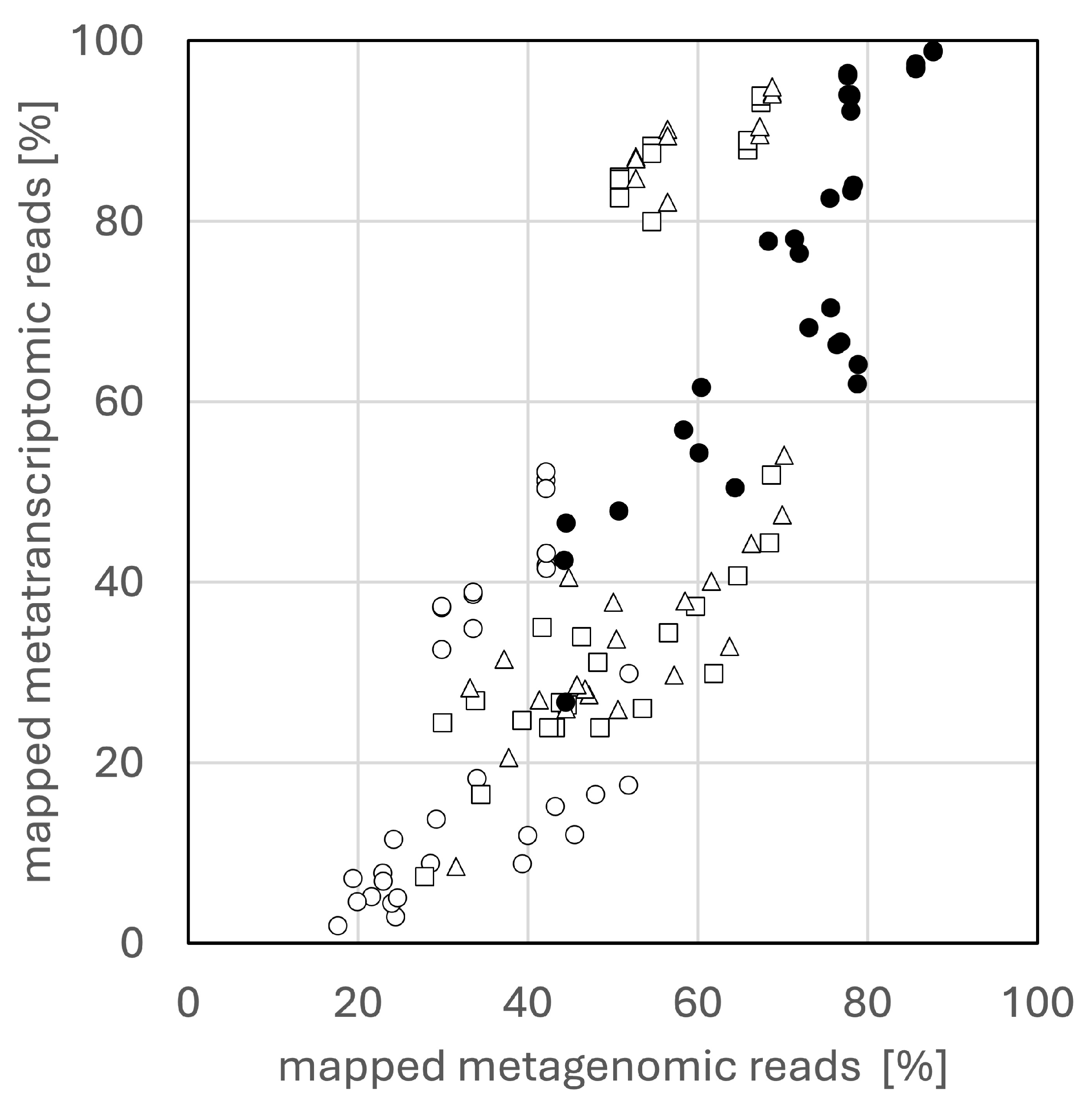
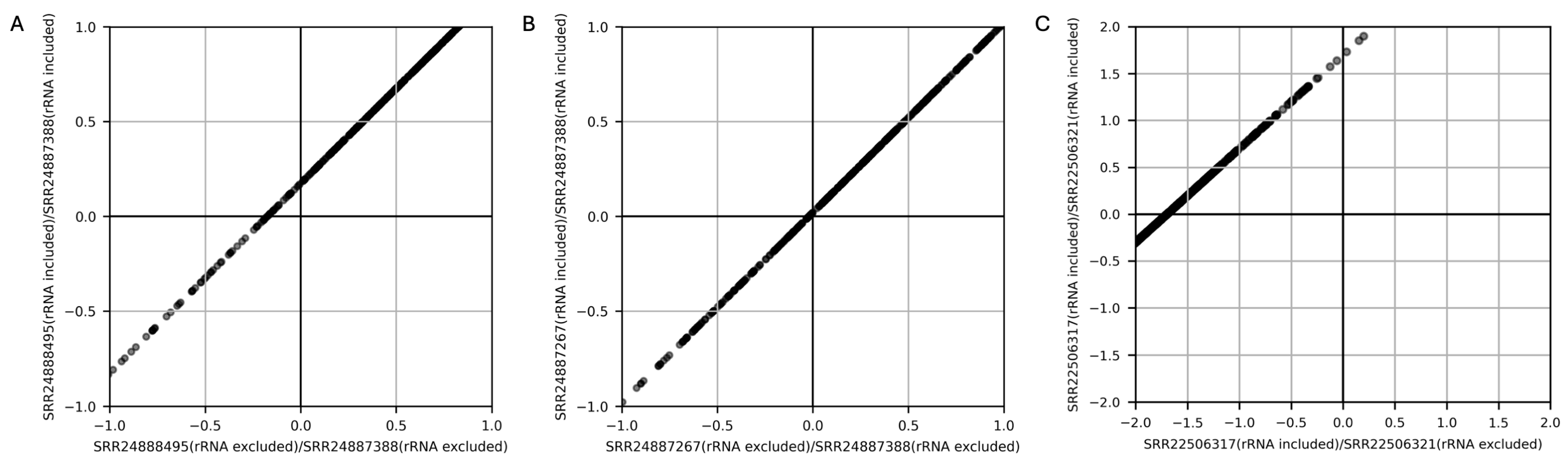
3. Results and Discussion
3.1. Mapping Rates to Metagenomic Contigs
3.2. Influence of rRNA on TPM Calculation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| NGS | Next-Generation Sequencing |
| MAGs | Metagenome-Assembled Genomes |
| SAGs | Single-Amplified Genomes |
| TPM | Transcripts Per Million |
| rRNA | Ribosomal RNA |
| BWA-MEM | Burrows–Wheeler Aligner–Maximal Exact Matches |
| NCBI | National Center for Biotechnology Information |
| SRA | Sequence Read Archive |
| SAM | Sequence Alignment/Map |
| BAM | Binary Alignment/Map |
References
- Sato, Y.; Hori, T.; Koike, H.; Navarro, R.R.; Ogata, A.; Habe, H. Transcriptome Analysis of Activated Sludge Microbiomes Reveals an Unexpected Role of Minority Nitrifiers in Carbon Metabolism. Commun. Biol. 2019, 2, 179. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Zhang, W.; Yang, B.; Feng, X.; Li, Y.; Li, X.; Liu, Q. Metagenomic Evidence for Antibiotic-Associated Actinomycetes in the Karamay Gobi Region. Front. Microbiol. 2024, 15, 1330880. [Google Scholar] [CrossRef]
- Nelson, M.B.; Martiny, A.C.; Martiny, J.B.H. Global Biogeography of Microbial Nitrogen-Cycling Traits in Soil. Proc. Natl. Acad. Sci. USA 2016, 113, 8033–8040. [Google Scholar] [CrossRef] [PubMed]
- Hartman, W.H.; Ye, R.; Horwath, W.R.; Tringe, S.G. A Genomic Perspective on Stoichiometric Regulation of Soil Carbon Cycling. ISME J. 2017, 11, 2652–2665. [Google Scholar] [CrossRef] [PubMed]
- Albertsen, M.; Hugenholtz, P.; Skarshewski, A.; Nielsen, K.L.; Tyson, G.W.; Nielsen, P.H. Genome Sequences of Rare, Uncultured Bacteria Obtained by Differential Coverage Binning of Multiple Metagenomes. Nat. Biotechnol. 2013, 31, 533–538. [Google Scholar] [CrossRef] [PubMed]
- Lasken, R.S.; McLean, J.S. Recent Advances in Genomic DNA Sequencing of Microbial Species from Single Cells. Nat. Rev. Genet. 2014, 15, 577–584. [Google Scholar] [CrossRef]
- The University of Queenland. The Genome Taxonomy Database. Available online: https://gtdb.ecogenomic.org/ (accessed on 29 December 2024).
- Zhang, Z.; Wang, J.; Wang, J.; Wang, J.; Li, Y. Estimate of the Sequenced Proportion of the Global Prokaryotic Genome. Microbiome 2020, 8, 134. [Google Scholar] [CrossRef]
- Honeker, L.K.; Pugliese, G.; Ingrisch, J.; Fudyma, J.; Gil-Loaiza, J.; Carpenter, E.; Singer, E.; Hildebrand, G.; Shi, L.; Hoyt, D.W.; et al. Drought Re-Routes Soil Microbial Carbon Metabolism towards Emission of Volatile Metabolites in an Artificial Tropical Rainforest. Nat. Microbiol. 2023, 8, 1480–1494. [Google Scholar] [CrossRef]
- Mendes, L.W.; Raaijmakers, J.M.; De Hollander, M.; Sepo, E.; Gómez Expósito, R.; Chiorato, A.F.; Mendes, R.; Tsai, S.M.; Carrión, V.J. Impact of the Fungal Pathogen Fusarium Oxysporum on the Taxonomic and Functional Diversity of the Common Bean Root Microbiome. Environ. Microbiome 2023, 18, 68. [Google Scholar] [CrossRef]
- Yu, K.; Zhang, T. Metagenomic and Metatranscriptomic Analysis of Microbial Community Structure and Gene Expression of Activated Sludge. PLoS ONE 2012, 7, e38183. [Google Scholar] [CrossRef]
- Zhang, Y.; Thompson, K.N.; Huttenhower, C.; Franzosa, E.A. Statistical Approaches for Differential Expression Analysis in Metatranscriptomics. Bioinformatics 2021, 37, i34–i41. [Google Scholar] [CrossRef]
- Freeman, E.C.; Emilson, E.J.S.; Dittmar, T.; Braga, L.P.P.; Emilson, C.E.; Goldhammer, T.; Martineau, C.; Singer, G.; Tanentzap, A.J. Universal Microbial Reworking of Dissolved Organic Matter along Environmental Gradients. Nat. Commun. 2024, 15, 187. [Google Scholar] [CrossRef] [PubMed]
- Jaillard, M.; Tournoud, M.; Meynier, F.; Veyrieras, J.-B. Optimization of Alignment-Based Methods for Taxonomic Binning of Metagenomics Reads. Bioinformatics 2016, 32, 1779–1787. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, G.; Martino, C.; Morris, J.; Khaleghi, B.; Kang, J.; DeReus, J.; Zhu, Q.; Roush, D.; McDonald, D.; Gonazlez, A.; et al. Swapping Metagenomics Preprocessing Pipeline Components Offers Speed and Sensitivity Increases. mSystems 2022, 7, e01378-21. [Google Scholar] [CrossRef]
- Wang, L.; Ding, R.; He, S.; Wang, Q.; Zhou, Y. A Pipeline for Constructing Reference Genomes for Large Cohort-Specific Metagenome Compression. Microorganisms 2023, 11, 2560. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Fernandez, M.; Simone, D.; Wu, X.; Soler, L.; Nilsson, E.; Holmfeldt, K.; Lantz, H.; Bertilsson, S.; Dopson, M. Metatranscriptomes Reveal That All Three Domains of Life Are Active but Are Dominated by Bacteria in the Fennoscandian Crystalline Granitic Continental Deep Biosphere. mBio 2018, 9, e01792-18. [Google Scholar] [CrossRef]
- Braga, L.P.P.; Pereira, R.V.; Martins, L.F.; Moura, L.M.S.; Sanchez, F.B.; Patané, J.S.L.; Da Silva, A.M.; Setubal, J.C. Genome-Resolved Metagenome and Metatranscriptome Analyses of Thermophilic Composting Reveal Key Bacterial Players and Their Metabolic Interactions. BMC Genom. 2021, 22, 652. [Google Scholar] [CrossRef]
- Li, X.; Bei, Q.; Rabiei Nematabad, M.; Peng, J.; Liesack, W. Time-Shifted Expression of Acetoclastic and Methylotrophic Methanogenesis by a Single Methanosarcina Genomospecies Predominates the Methanogen Dynamics in Philippine Rice Field Soil. Microbiome 2024, 12, 39. [Google Scholar] [CrossRef]
- SRA Toolkit Development Team. SRA Toolkit. Available online: https://github.com/ncbi/sra-tools (accessed on 29 December 2024).
- Chen, S. Ultrafast One-pass FASTQ Data Preprocessing, Quality Control, and Deduplication Using Fastp. iMeta 2023, 2, e107. [Google Scholar] [CrossRef]
- Li, D.; Liu, C.-M.; Luo, R.; Sadakane, K.; Lam, T.-W. MEGAHIT: An Ultra-Fast Single-Node Solution for Large and Complex Metagenomics Assembly via Succinct de. Bruijn Graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef]
- Li, D.; Luo, R.; Liu, C.-M.; Leung, C.-M.; Ting, H.-F.; Sadakane, K.; Yamashita, H.; Lam, T.-W. MEGAHIT v1.0: A Fast and Scalable Metagenome Assembler Driven by Advanced Methodologies and Community Practices. Methods 2016, 102, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Hyatt, D.; Chen, G.-L.; LoCascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic Gene Recognition and Translation Initiation Site Identification. BMC Bioinform. 2010, 11, 119. [Google Scholar] [CrossRef]
- Li, H. Aligning Sequence Reads, Clone Sequences and Assembly Contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and Memory-Efficient Alignment of Short DNA Sequences to the Human Genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast Gapped-Read Alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Langmead, B.; Wilks, C.; Antonescu, V.; Charles, R. Scaling Read Aligners to Hundreds of Threads on General-Purpose Processors. Bioinformatics 2019, 35, 421–432. [Google Scholar] [CrossRef] [PubMed]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve Years of SAMtools and BCFtools. GigaScience 2021, 10, giab008. [Google Scholar] [CrossRef] [PubMed]
- Okonechnikov, K.; Conesa, A.; García-Alcalde, F. Qualimap 2: Advanced Multi-Sample Quality Control for High-Throughput Sequencing Data. Bioinformatics 2016, 32, 292–294. [Google Scholar] [CrossRef]
- NCBI rRNA Sequences. Available online: https://ftp.ncbi.nlm.nih.gov/blast/db/ (accessed on 29 December 2024).
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and Applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef]
- UniProt-EMBL-EBI Swiss-Prot Database. Available online: https://ftp.uniprot.org/pub/databases/uniprot/current_release/knowledgebase/complete/uniprot_sprot.fasta.gz (accessed on 29 December 2024).
- Buchfink, B.; Reuter, K.; Drost, H.-G. Sensitive Protein Alignments at Tree-of-Life Scale Using DIAMOND. Nat. Methods 2021, 18, 366–368. [Google Scholar] [CrossRef]
- InterPro-EMBL-EBI Pfam Database. Available online: https://ftp.ebi.ac.uk/pub/databases/Pfam/current_release/Pfam-A.hmm.gz (accessed on 29 December 2024).
- HMMER. Available online: http://hmmer.org/ (accessed on 29 December 2024).
- Tange, O. GNU Parallel 20230922 (‘Derna’). Available online: https://zenodo.org/records/8374296 (accessed on 22 April 2025).
- Liao, Y.; Smyth, G.K.; Shi, W. featureCounts: An Efficient General Purpose Program for Assigning Sequence Reads to Genomic Features. Bioinformatics 2014, 30, 923–930. [Google Scholar] [CrossRef]
- Ferragina, P.; Manzini, G. Opportunistic Data Structures with Applications. In Proceedings of the 41st Annual Symposium on Foundations of Computer Science, Redondo Beach, CA, USA, 12–14 November 2000; IEEE Computer Society: Washington, DC, USA, 2000; pp. 390–398. [Google Scholar]
- Burrows, M.; Wheeler, D.J. A Block-Sorting Lossless Data Compression Algorithm 1994. Available online: https://www.cl.cam.ac.uk/teaching/2003/DSAlgs/SRC-124.pdf (accessed on 22 April 2025).
- Hatem, A.; Bozdag, D.; Toland, A.E.; Çatalyürek, Ü.V. Benchmarking Short Sequence Mapping Tools. BMC Bioinform. 2013, 14, 184. [Google Scholar] [CrossRef] [PubMed]
- Stewart, F.J.; Ottesen, E.A.; DeLong, E.F. Development and Quantitative Analyses of a Universal rRNA-Subtraction Protocol for Microbial Metatranscriptomics. ISME J. 2010, 4, 896–907. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Wurtzel, O.; Singh, K.; Froula, J.L.; Yilmaz, S.; Tringe, S.G.; Wang, Z.; Chen, F.; Lindquist, E.A.; Sorek, R.; et al. Validation of Two Ribosomal RNA Removal Methods for Microbial Metatranscriptomics. Nat. Methods 2010, 7, 807–812. [Google Scholar] [CrossRef]
- Zhao, Y.; Li, M.-C.; Konaté, M.M.; Chen, L.; Das, B.; Karlovich, C.; Williams, P.M.; Evrard, Y.A.; Doroshow, J.H.; McShane, L.M.; et al. A Comparative Study of Quantification Measures for the Analysis of RNA-Seq Data from the NCI Patient-Derived Models Repository. J. Transl. Med. 2021, 19, 269. [Google Scholar] [CrossRef] [PubMed]
- Kopylova, E.; Noé, L.; Touzet, H. SortMeRNA: Fast and Accurate Filtering of Ribosomal RNAs in Metatranscriptomic Data. Bioinformatics 2012, 28, 3211–3217. [Google Scholar] [CrossRef]
- Klingenberg, H.; Meinicke, P. How to Normalize Metatranscriptomic Count Data for Differential Expression Analysis. PeerJ 2017, 5, e3859. [Google Scholar] [CrossRef]
- Hardwick, S.A.; Chen, W.Y.; Wong, T.; Kanakamedala, B.S.; Deveson, I.W.; Ongley, S.E.; Santini, N.S.; Marcellin, E.; Smith, M.A.; Nielsen, L.K.; et al. Synthetic Microbe Communities Provide Internal Reference Standards for Metagenome Sequencing and Analysis. Nat. Commun. 2018, 9, 3096. [Google Scholar] [CrossRef]
- Mori, H.; Kato, T.; Ozawa, H.; Sakamoto, M.; Murakami, T.; Taylor, T.D.; Toyoda, A.; Ohkuma, M.; Kurokawa, K.; Ohno, H. Assessment of Metagenomic Workflows Using a Newly Constructed Human Gut Microbiome Mock Community. DNA Res. 2023, 30, dsad010. [Google Scholar] [CrossRef]
- Thorn, C.E.; Bergesch, C.; Joyce, A.; Sambrano, G.; McDonnell, K.; Brennan, F.; Heyer, R.; Benndorf, D.; Abram, F. A Robust, Cost-effective Method for DNA, RNA and Protein Co-extraction from Soil, Other Complex Microbiomes and Pure Cultures. Mol. Ecol. Resour. 2019, 19, 439–455. [Google Scholar] [CrossRef]
- Shaffer, J.P.; Marotz, C.; Belda-Ferre, P.; Martino, C.; Wandro, S.; Estaki, M.; Salido, R.A.; Carpenter, C.S.; Zaramela, L.S.; Minich, J.J.; et al. A Comparison of DNA/RNA Extraction Protocols for High-Throughput Sequencing of Microbial Communities. BioTechniques 2021, 70, 149–159. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mameda, R.; Bono, H. Optimization of Mapping Tools and Investigation of Ribosomal RNA Influence for Data-Driven Gene Expression Analysis in Complex Microbiomes. Microorganisms 2025, 13, 995. https://doi.org/10.3390/microorganisms13050995
Mameda R, Bono H. Optimization of Mapping Tools and Investigation of Ribosomal RNA Influence for Data-Driven Gene Expression Analysis in Complex Microbiomes. Microorganisms. 2025; 13(5):995. https://doi.org/10.3390/microorganisms13050995
Chicago/Turabian StyleMameda, Ryo, and Hidemasa Bono. 2025. "Optimization of Mapping Tools and Investigation of Ribosomal RNA Influence for Data-Driven Gene Expression Analysis in Complex Microbiomes" Microorganisms 13, no. 5: 995. https://doi.org/10.3390/microorganisms13050995
APA StyleMameda, R., & Bono, H. (2025). Optimization of Mapping Tools and Investigation of Ribosomal RNA Influence for Data-Driven Gene Expression Analysis in Complex Microbiomes. Microorganisms, 13(5), 995. https://doi.org/10.3390/microorganisms13050995