
Microbial Contamination in Urban Marine Sediments: Source Identification Using Microbial Community Analysis and Fecal Indicator Bacteria
 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Material and Methods
2.1. Study Site and Sampling
2.2. Sample Preparations and Viable E. coli Quantification
2.3. DNA Extraction and 16S rRNA Sequencing
2.4. ASV Analysis
2.5. Core Community
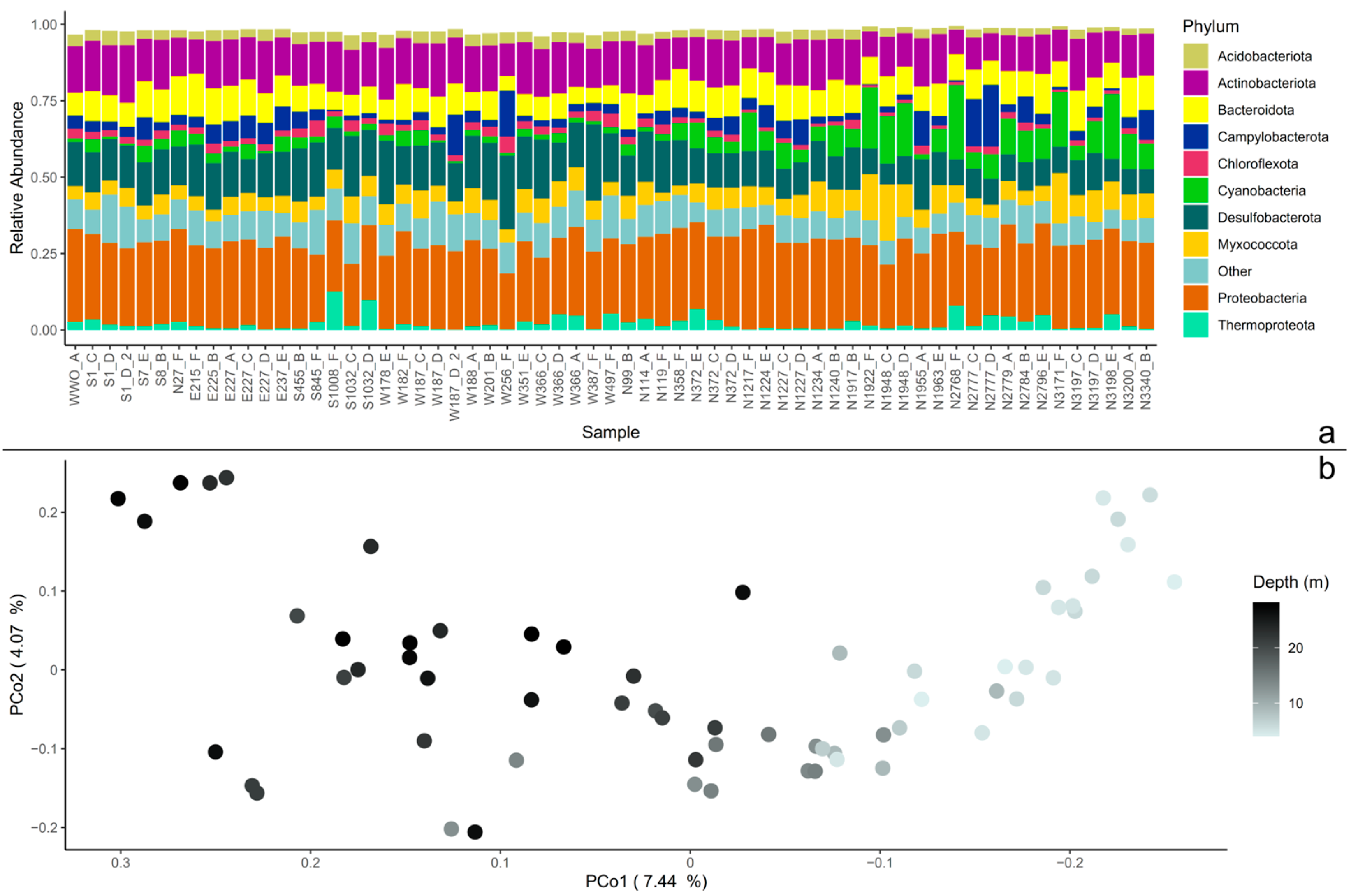
2.6. Beta-Diversity
2.7. Impact of Sewage Taxa
3. Results
3.1. Core and Non-Core Microbial Community in the Coastal Sediments
3.2. Viable E. coli
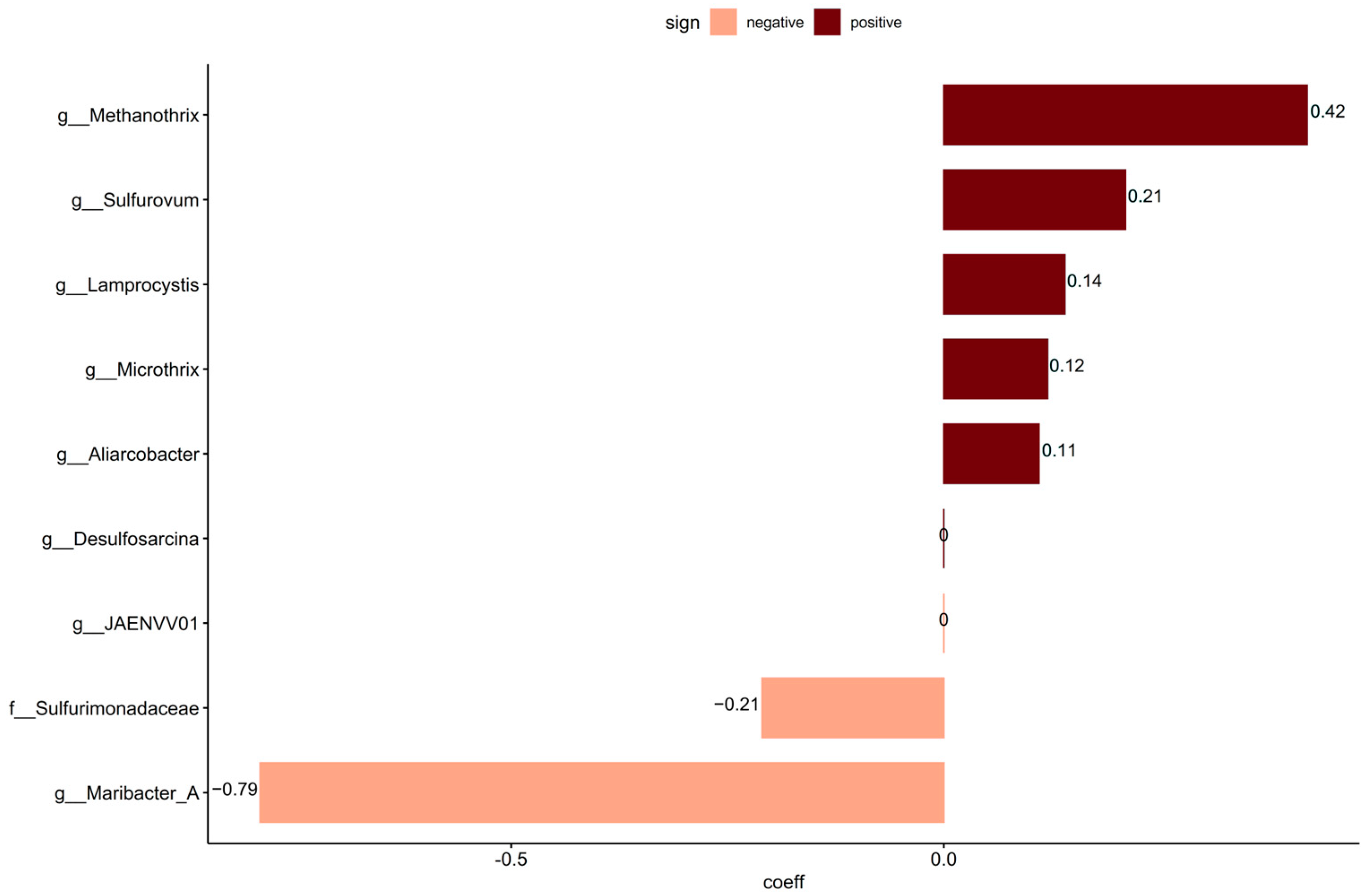
3.3. Impact of Sewage Taxa
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cabral, A.C.; Stark, J.S.; Kolm, H.E.; Martins, C.C. An integrated evaluation of some faecal indicator bacteria (FIB) and chemical markers as potential tools for monitoring sewage contamination in subtropical estuaries. Environ. Pollut. 2018, 235, 739–749. [Google Scholar] [CrossRef] [PubMed]
- Dudley, B.D.; Burge, O.R.; Plew, D.; Zeldis, J. Effects of agricultural and urban land cover on New Zealand’s estuarine water quality. N. Z. J. Mar. Freshw. Res. 2020, 54, 372–392. [Google Scholar] [CrossRef]
- Saingam, P.; Li, B.; Yan, T. Fecal indicator bacteria, direct pathogen detection, and microbial community analysis provide different microbiological water quality assessment of a tropical urban marine estuary. Water Res. 2020, 185, 116280. [Google Scholar] [CrossRef] [PubMed]
- Uniyal, A. Chapter 1: Deteriorating impacts of emerging water pollutants on biological diversity. In Advances in Environmental Pollution Management. Wastewater Impacts and Treatment Technologies; Agro Environ Media, Publication Cell of AESA, Agriculture and Environmental Science Academy: Haridwar, India, 2020; Volume 1, pp. 1–9. [Google Scholar]
- Malik, D.S.; Sharma, A.K.; Sharma, A.K.; Thakur, R.; Sharma, M. Chapter 2: A review on impact of water pollution on freshwater fish species and their aquatic environment. In Advances in Environmental Pollution Management. Wastewater Impacts and Treatment Technologies; Agro Environ Media, Publication Cell of AESA, Agriculture and Environmental Science Academy: Haridwar, India, 2020; Volume 1, pp. 10–28. [Google Scholar]
- Leonard, A.F.C.; Singer, A.; Ukoumunne, O.C.; Gaze, W.H.; Garside, R. Is it safe to go back into the water? A systematic review and meta-analysis of the risk of acquiring infections from recreational exposure to seawater. Int. J. Epidemiol. 2018, 47, 572–586. [Google Scholar] [CrossRef]
- Prüss, A. Review of epidemiological studies on health effects from exposure to recreational water. Int. J. Epidemiol. 1998, 27, 1–9. [Google Scholar] [CrossRef]
- Abia, A.L.; Ubomba-Jaswa, E.; Momba, M.N. Impact of seasonal variation on Escherichia coli concentrations in the riverbed sediments in the Apies River, South Africa. Sci. Total Environ. 2015, 537, 462–469. [Google Scholar] [CrossRef] [PubMed]
- Frank, E.M.; Ahlinder, J.; Jephson, T.; Persson, K.M.; Lindberg, E.; Paul, C.J. Marine sediments are identified as an environmental reservoir for Escherichia coli: Comparing signature-based and novel amplicon sequencing approaches for microbial source tracking. Sci. Total Environ. 2024, 907, 167865. [Google Scholar] [CrossRef]
- Lai, J.Y.H.; Zhang, H.; Chiang, M.H.Y.; Lun, C.H.I.; Zhang, R.; Lau, S.C.K. The putative functions of lysogeny in mediating the survivorship of Escherichia coli in seawater and marine sediment. FEMS Microbiol. Ecol. 2018, 94, fix187. [Google Scholar] [CrossRef]
- Hassard, F.; Andrews, A.; Jones, D.L.; Parsons, L.; Jones, V.; Cox, B.A.; Daldorph, P.; Brett, H.; McDonald, J.E.; Malham, S.K. Physicochemical Factors Influence the Abundance and Culturability of Human Enteric Pathogens and Fecal Indicator Organisms in Estuarine Water and Sediment. Front. Microbiol. 2017, 8, 1996. [Google Scholar] [CrossRef]
- Norman, S.A.; Hobbs, R.C.; Wuertz, S.; Melli, A.; Beckett, L.A.; Chouicha, N.; Kundu, A.; Miller, W.A. Fecal pathogen pollution: Sources and patterns in water and sediment samples from the upper Cook Inlet, Alaska ecosystem. Environ. Sci. Process Impacts 2013, 15, 1041–1051. [Google Scholar] [CrossRef]
- Perkins, T.L.; Clements, K.; Baas, J.H.; Jago, C.F.; Jones, D.L.; Malham, S.K.; McDonald, J.E. Sediment composition influences spatial variation in the abundance of human pathogen indicator bacteria within an estuarine environment. PLoS ONE 2014, 9, e112951. [Google Scholar] [CrossRef]
- Smith, J.E.; Stocker, M.D.; Hill, R.L.; Pachepsky, Y.A. The Effect of Temperature Oscillations and Sediment Texture on Fecal Indicator Bacteria Survival in Sediments. Water Air Soil. Pollut. 2019, 230, 270. [Google Scholar] [CrossRef]
- Decho, A.W.; Gutierrez, T. Microbial Extracellular Polymeric Substances (EPSs) in Ocean Systems. Front. Microbiol. 2017, 8, 922. [Google Scholar] [CrossRef]
- Craig, D.L.; Fallowfield, H.J.; Cromar, N.J. Use of microcosms to determine persistence of Escherichia coli in recreational coastal water and sediment and validation with in situ measurements. J. Appl. Microbiol. 2004, 96, 922–930. [Google Scholar] [CrossRef]
- Pachepsky, Y.A.; Shelton, D.R. Escherichia Coliand Fecal Coliforms in Freshwater and Estuarine Sediments. Crit. Rev. Environ. Sci. Technol. 2011, 41, 1067–1110. [Google Scholar] [CrossRef]
- Stocker, M.D.; Smith, J.E.; Hernandez, C.; Macarisin, D.; Pachepsky, Y. Seasonality of E. coli and Enterococci Concentrations in Creek Water, Sediment, and Periphyton. Water Air Soil. Pollut. 2019, 230, 223. [Google Scholar] [CrossRef]
- Luna, G.M.; Quero, G.M.; Perini, L. Next generation sequencing reveals distinct fecal pollution signatures in aquatic sediments across gradients of anthropogenic influence. Adv. Oceanogr. Limnol. 2016, 7, 115–124. [Google Scholar] [CrossRef]
- Tao, K.; Liu, Y.; Ke, T.; Zhang, Y.; Xiao, L.; Li, S.; Wei, S.; Chen, L.; Hu, T. Patterns of bacterial and archaeal communities in sediments in response to dam construction and sewage discharge in Lhasa River. Ecotoxicol. Environ. Saf. 2019, 178, 195–201. [Google Scholar] [CrossRef]
- Devane, M.L.; Moriarty, E.M.; Robson, B.; Lin, S.; Wood, D.; Webster-Brown, J.; Gilpin, B.J. Relationships between chemical and microbial faecal source tracking markers in urban river water and sediments during and post-discharge of human sewage. Sci. Total Environ. 2019, 651, 1588–1604. [Google Scholar] [CrossRef]
- Vadde, K.K.; McCarthy, A.J.; Rong, R.; Sekar, R. Quantification of Microbial Source Tracking and Pathogenic Bacterial Markers in Water and Sediments of Tiaoxi River (Taihu Watershed). Front. Microbiol. 2019, 10, 699. [Google Scholar] [CrossRef]
- Sauer, E.P.; Vandewalle, J.L.; Bootsma, M.J.; McLellan, S.L. Detection of the human specific Bacteroides genetic marker provides evidence of widespread sewage contamination of stormwater in the urban environment. Water Res. 2011, 45, 4081–4091. [Google Scholar] [CrossRef]
- Ekhlas, D.; Kurisu, F.; Kasuga, I.; Cernava, T.; Berg, G.; Liu, M.; Furumai, H. Identification of new eligible indicator organisms for combined sewer overflow via 16S rRNA gene amplicon sequencing in Kanda River, Tokyo. J. Environ. Manag. 2021, 284, 112059. [Google Scholar] [CrossRef]
- Drexler, J.Z.; Johnson, H.E.; Duris, J.; Krauss, K.W. Marsh Soils as Potential Sinks for Bacteroides Fecal Indicator Bacteria, Waccamaw National Wildlife Refuge, Georgetown, SC, USA. Water Air Soil. Pollut. 2014, 225, 1861. [Google Scholar] [CrossRef]
- Boukerb, A.M.; Noel, C.; Quenot, E.; Cadiou, B.; Cheve, J.; Quintric, L.; Cormier, A.; Dantan, L.; Gourmelon, M. Comparative Analysis of Fecal Microbiomes From Wild Waterbirds to Poultry, Cattle, Pigs, and Wastewater Treatment Plants for a Microbial Source Tracking Approach. Front. Microbiol. 2021, 12, 697553. [Google Scholar] [CrossRef]
- Zan, R.; Blackburn, A.; Plaimart, J.; Acharya, K.; Walsh, C.; Stirling, R.; Kilsby, C.G.; Werner, D. Environmental DNA clarifies impacts of combined sewer overflows on the bacteriology of an urban river and resulting risks to public health. Sci. Total Environ. 2023, 889, 164282. [Google Scholar] [CrossRef]
- Manini, E.; Baldrighi, E.; Ricci, F.; Grilli, F.; Giovannelli, D.; Intoccia, M.; Casabianca, S.; Capellacci, S.; Marinchel, N.; Penna, P.; et al. Assessment of Spatio-Temporal Variability of Faecal Pollution along Coastal Waters during and after Rainfall Events. Water 2022, 14, 502. [Google Scholar] [CrossRef]
- Erb, I.K.; Suarez, C.; Frank, E.M.; Bengtsson-Palme, J.; Lindberg, E.; Paul, C.J. Escherichia coli in urban marine sediments: Interpreting virulence, biofilm formation, halotolerance, and antibiotic resistance to infer contamination or naturalization. FEMS Microbes 2024, 5, xtae024. [Google Scholar] [CrossRef]
- NSVA. Miljörapport 2019. Öresundsverket, Helsingborgs Kommun. 2020. Available online: https://www.nsva.se/vatten-och-avlopp/spillvatten/nsvas-reningsverk/oresundsverket/ (accessed on 13 October 2021).
- SMHI. SMHI Oceanographic Datacenter. Station W Landskrona. Available online: http://www.smhi.se/ (accessed on 14 June 2024).
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berg-Lyons, D.; Lozupone, C.A.; Turnbaugh, P.J.; Fierer, N.; Knight, R. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc. Natl. Acad. Sci. USA 2011, 108 (Suppl. 1), 4516–4522. [Google Scholar] [CrossRef]
- Muyzer, G.; de Waal, E.C.; Uitterlinden, A.G. Profiling of complex microbial populations by denaturing gradient gel electrophoresis analysis of polymerase chain reaction-amplified genes coding for 16S rRNA. Appl. Environ. Microbiol. 1993, 59, 695–700. [Google Scholar] [CrossRef]
- Cock, P.J.; Fields, C.J.; Goto, N.; Heuer, M.L.; Rice, P.M. The Sanger FASTQ file format for sequences with quality scores, and the Solexa/Illumina FASTQ variants. Nucleic Acids Res. 2010, 38, 1767–1771. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2024; Available online: https://www.R-project.org/ (accessed on 19 July 2024).
- Posit Team. RStudio: Integrated Development Environment for R; Posit Software; PBC: Boston, MA, USA, 2023; Available online: http://www.posit.co/ (accessed on 11 November 2023).
- Callahan, B.J.; Mcmurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. Dada2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef]
- Rausch, P.; Ruhlemann, M.; Hermes, B.M.; Doms, S.; Dagan, T.; Dierking, K.; Domin, H.; Fraune, S.; von Frieling, J.; Hentschel, U.; et al. Comparative analysis of amplicon and metagenomic sequencing methods reveals key features in the evolution of animal metaorganisms. Microbiome 2019, 7, 133. [Google Scholar] [CrossRef]
- Firke, S. janitor: Simple Tools for Examining and Cleaning Dirty Data. 2023. Available online: https://Cran.R-project.org/package=janitor (accessed on 27 January 2024).
- Alishum, A. DADA2 Formatted 16S rRNA Gene Sequences for Both Bacteria & Archaea (Version 4.3). Zenodo 2022. [Google Scholar] [CrossRef]
- Davis, N.M.; Proctor, D.M.; Holmes, S.P.; Relman, D.A.; Callahan, B.J. Simple statistical identification and removal of contaminant sequences in marker-gene and metagenomics data. Microbiome 2018, 6, 226. [Google Scholar] [CrossRef]
- Glassing, A.; Dowd, S.E.; Galandiuk, S.; Davis, B.; Chiodini, R.J. Inherent bacterial DNA contamination of extraction and sequencing reagents may affect interpretation of microbiota in low bacterial biomass samples. Gut Pathog. 2016, 8, 24. [Google Scholar] [CrossRef]
- Lathi, L.; Shetty, S. Microbiome R Package. 2012–2019. Available online: https://www.bioconductor.org/packages/release/bioc/html/microbiome.html (accessed on 27 November 2023).
- McMurdie, P.J.; Holmes, S. phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef]
- Oksanen, J.; Simpson, G.; Blanchet, F.; Kindt, R.; Legendre, P.; Minchin, P.; O’hara, R.; Solymos, P.; Stevens, M.; Szoecs, E.; et al. vegan: Community Ecology Package. 2022. Available online: https://Cran.R-project.org/package=vegan (accessed on 27 November 2023).
- Anderson, M.J. A new method for non-parametric multivariate analysisof variance. Austral Ecol. 2001, 26, 32–46. [Google Scholar]
- Martinez Arbizu, P. pairwiseAdonis: Pairwise Multilevel Comparison Using Adonis. R Package Version 0.4.1. 2017. Available online: https://github.com/pmartinezarbizu/pairwiseAdonis (accessed on 26 February 2025).
- Calle, M.L.; Pujolassos, M.; Susin, A. coda4microbiome: Compositional data analysis for microbiome cross-sectional and longitudinal studies. BMC Bioinform. 2023, 24, 82. [Google Scholar] [CrossRef]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016. [Google Scholar]
- Pfeiffer, N.; Desmarchelier, C.; Blaut, M.; Daniel, H.; Haller, D.; Clavel, T. Acetatifactor muris gen. nov., sp. nov., a novel bacterium isolated from the intestine of an obese mouse. Arch. Microbiol. 2012, 194, 901–907. [Google Scholar] [CrossRef]
- Gilroy, R.; Ravi, A.; Getino, M.; Pursley, I.; Horton, D.L.; Alikhan, N.-F.; Baker, D.; Gharbi, K.; Hall, N.; Watson, M.; et al. Extensive microbial diversity within the chicken gut microbiome revealed by metagenomics and culture. PeerJ 2021, 9, e10941. [Google Scholar] [CrossRef]
- McLellan, S.L.; Huse, S.M.; Mueller-Spitz, S.R.; Andreishcheva, E.N.; Sogin, M.L. Diversity and population structure of sewage-derived microorganisms in wastewater treatment plant influent. Environ. Microbiol. 2010, 12, 378–392. [Google Scholar] [CrossRef]
- Wu, L.; Ning, D.; Zhang, B.; Li, Y.; Zhang, P.; Shan, X.; Zhang, Q.; Brown, M.R.; Li, Z.; Van Nostrand, J.D.; et al. Global diversity and biogeography of bacterial communities in wastewater treatment plants. Nat. Microbiol. 2019, 4, 1183–1195. [Google Scholar] [CrossRef] [PubMed]
- Dueholm, M.K.D.; Nierychlo, M.; Andersen, K.S.; Rudkjøbing, V.; Knutsson, S.; Arriaga, S.; Bakke, R.; Boon, N.; Bux, F.; Christensson, M.; et al. MiDAS 4: A global catalogue of full-length 16S rRNA gene sequences and taxonomy for studies of bacterial communities in wastewater treatment plants. Nat. Commun. 2022, 13, 1908. [Google Scholar] [CrossRef]
- Li, P.; Chang, X.; Chen, X.; Wang, C.; Shang, Y.; Zheng, D.; Qi, K. Early-life antibiotic exposure increases the risk of childhood overweight and obesity in relation to dysbiosis of gut microbiota: A birth cohort study. Ann. Clin. Microbiol. Antimicrob. 2022, 21, 46. [Google Scholar] [CrossRef] [PubMed]
- Shetty, S.A.; Zuffa, S.; Bui, T.P.N.; Aalvink, S.; Smidt, H.; De Vos, W.M. Reclassification of Eubacterium hallii as Anaerobutyricum hallii gen. nov., comb. nov., and description of Anaerobutyricum soehngenii sp. nov., a butyrate and propionate-producing bacterium from infant faeces. Int. J. Syst. Evol. Microbiol. 2018, 68, 3741–3746. [Google Scholar] [CrossRef]
- Schwiertz, A.; Hold, G.L.; Duncan, S.H.; Gruhl, B.; Collins, M.D.; Lawson, P.A.; Flint, H.J.; Blaut, M. Anaerostipes caccae gen. nov., sp. nov., a New Saccharolytic, Acetate-utilising, Butyrate-producing Bacterium from Human Faeces. Syst. Appl. Microbiol. 2002, 25, 46–51. [Google Scholar] [CrossRef] [PubMed]
- Qiu, X.; Macchietto, M.G.; Liu, X.; Lu, Y.; Ma, Y.; Guo, H.; Saqui-Salces, M.; Bernlohr, D.A.; Chen, C.; Shen, S.; et al. Identification of gut microbiota and microbial metabolites regulated by an antimicrobial peptide lipocalin 2 in high fat diet-induced obesity. Int. J. Obes. 2020, 45, 143–154. [Google Scholar] [CrossRef]
- Kristensen, J.M.; Nierychlo, M.; Albertsen, M.; Nielsen, P.H. Bacteria from the Genus Arcobacter Are Abundant in Effluent from Wastewater Treatment Plants. Appl. Environ. Microbiol. 2020, 86, e03044-19. [Google Scholar] [CrossRef]
- Yatsunenko, T.; Rey, F.E.; Manary, M.J.; Trehan, I.; Dominguez-Bello, M.G.; Contreras, M.; Magris, M.; Hidalgo, G.; Baldassano, R.N.; Anokhin, A.P.; et al. Human gut microbiome viewed across age and geography. Nature 2012, 486, 222–227. [Google Scholar] [CrossRef]
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K.S.; Manichanh, C.; Nielsen, T.; Pons, N.; Levenez, F.; Yamada, T.; et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010, 464, 59–65. [Google Scholar] [CrossRef]
- Huttenhower, C.; Gevers, D.; Knight, R.; Abubucker, S.; Badger, J.H.; Chinwalla, A.T.; Creasy, H.H.; Earl, A.M.; FitzGerald, M.G.; Fulton, R.S.; et al. Structure, function and diversity of the healthy human microbiome. Nature 2012, 486, 207–214. [Google Scholar] [CrossRef]
- Ke, S.; Weiss, S.T.; Liu, Y.-Y. Dissecting the role of the human microbiome in COVID-19 via metagenome-assembled genomes. Nat. Commun. 2022, 13, 5235. [Google Scholar] [CrossRef]
- Zou, Y.; Xue, W.; Lin, X.; Lv, M.; Luo, G.; Dai, Y.; Sun, H.; Liu, S.-w.; Sun, C.-h.; Hu, T.; et al. Butyribacter intestini gen. nov., sp. nov., a butyric acid-producing bacterium of the family Lachnospiraceae isolated from human faeces, and reclassification of Acetivibrio ethanolgignens as Acetanaerobacter ethanolgignens gen. nov., comb. nov. Syst. Appl. Microbiol. 2021, 44, 126201. [Google Scholar] [CrossRef]
- McIlroy, S.J.; Albertsen, M.; Andresen, E.K.; Saunders, A.M.; Kristiansen, R.; Stokholm-Bjerregaard, M.; Nielsen, K.L.; Nielsen, P.H. ‘Candidatus Competibacter’-lineage genomes retrieved from metagenomes reveal functional metabolic diversity. ISME J. 2014, 8, 613–624. [Google Scholar] [CrossRef] [PubMed]
- Nierychlo, M.; Andersen, K.S.; Xu, Y.; Green, N.; Jiang, C.; Albertsen, M.; Dueholm, M.S.; Nielsen, P.H. MiDAS 3: An ecosystem-specific reference database, taxonomy and knowledge platform for activated sludge and anaerobic digesters reveals species-level microbiome composition of activated sludge. Water Res. 2020, 182, 115955. [Google Scholar] [CrossRef] [PubMed]
- Takada, T.; Kurakawa, T.; Tsuji, H.; Nomoto, K. Fusicatenibacter saccharivorans gen. nov., sp. nov., isolated from human faeces. Int. J. Syst. Evol. Microbiol. 2013, 63, 3691–3696. [Google Scholar] [CrossRef] [PubMed]
- Petzoldt, D.; Breves, G.; Rautenschlein, S.; Taras, D. Harryflintia acetispora gen. nov., sp. nov., isolated from chicken caecum. Int. J. Syst. Evol. Microbiol. 2016, 66, 4099–4104. [Google Scholar] [CrossRef]
- Lu, Y.; Redlinger, T.E.; Avitia, R.; Galindo, A.; Goodman, K. Isolation and genotyping of Helicobacter pylori from untreated municipal wastewater. Appl. Environ. Microbiol. 2002, 68, 1436–1439. [Google Scholar] [CrossRef]
- Naud, S.; Bellali, S.; Anani, H.; Lo, C.I.; Yacouba, A.; Tidjani Alou, M.; Armstrong, N.; Bonvalet, M.; Zitvogel, L.; Raoult, D.; et al. Luxibacter massiliensis gen. nov., sp. nov., a new bacterium isolated from the human gut microbiota. New Microbes New Infect. 2021, 40, 100850. [Google Scholar] [CrossRef]
- Albertsen, M.; McIlroy, S.J.; Stokholm-Bjerregaard, M.; Karst, S.M.; Nielsen, P.H. “Candidatus Propionivibrio aalborgensis”: A Novel Glycogen Accumulating Organism Abundant in Full-Scale Enhanced Biological Phosphorus Removal Plants. Front. Microbiol. 2016, 7, 1033. [Google Scholar] [CrossRef]
- Shkoporov, A.N.; Chaplin, A.V.; Shcherbakova, V.A.; Suzina, N.E.; Kafarskaia, L.I.; Bozhenko, V.K.; Efimov, B.A. Ruthenibacterium lactatiformans gen. nov., sp. nov., an anaerobic, lactate-producing member of the family Ruminococcaceae isolated from human faeces. Int. J. Syst. Evol. Microbiol. 2016, 66, 3041–3049. [Google Scholar] [CrossRef] [PubMed]
- Soh, M.; Miyake, S.; Lim, A.; Ding, Y.; Seedorf, H. Schaedlerella arabinosiphila gen. nov., sp. nov., a D-arabinose-utilizing bacterium isolated from faeces of C57BL/6J mice that is a close relative of Clostridium species ASF 502. Int. J. Syst. Evol. Microbiol. 2019, 69, 3616–3622. [Google Scholar] [CrossRef]
- Lozupone, C.A.; Stombaugh, J.I.; Gordon, J.I.; Jansson, J.K.; Knight, R. Diversity, stability and resilience of the human gut microbiota. Nature 2012, 489, 220–230. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.J.; Jung, D.H.; Park, C.S. Important roles of Ruminococcaceae in the human intestine for resistant starch utilization. Food Sci. Biotechnol. 2024, 33, 2009–2019. [Google Scholar] [CrossRef] [PubMed]
- Hoshino, T.; Doi, H.; Uramoto, G.I.; Wormer, L.; Adhikari, R.R.; Xiao, N.; Morono, Y.; D’Hondt, S.; Hinrichs, K.U.; Inagaki, F. Global diversity of microbial communities in marine sediment. Proc. Natl. Acad. Sci. USA 2020, 117, 27587–27597. [Google Scholar] [CrossRef]
- Anastasi, E.M.; Matthews, B.; Stratton, H.M.; Katouli, M. Pathogenic Escherichia coli found in sewage treatment plants and environmental waters. Appl. Environ. Microbiol. 2012, 78, 5536–5541. [Google Scholar] [CrossRef]
- Aslan, A.; Cole, Z.; Bhattacharya, A.; Oyibo, O. Presence of Antibiotic-Resistant Escherichia coli in Wastewater Treatment Plant Effluents Utilized as Water Reuse for Irrigation. Water 2018, 10, 805. [Google Scholar] [CrossRef]
- Raboni, M.; Gavasci, R.; Torretta, V. Assessment of the Fate of Escherichia coli in Different Stages of Wastewater Treatment Plants. Water Air Soil. Pollut. 2016, 227, 455. [Google Scholar] [CrossRef]
- Hassard, F.; Gwyther, C.L.; Farkas, K.; Andrews, A.; Jones, V.; Cox, B.; Brett, H.; Jones, D.L.; McDonald, J.E.; Malham, S.K. Abundance and Distribution of Enteric Bacteria and Viruses in Coastal and Estuarine Sediments—A Review. Front. Microbiol. 2016, 7, 1692. [Google Scholar] [CrossRef]
- Hernroth, B.; Lothigius, A.; Bolin, I. Factors influencing survival of enterotoxigenic Escherichia coli, Salmonella enterica (serovar Typhimurium) and Vibrio parahaemolyticus in marine environments. FEMS Microbiol. Ecol. 2010, 71, 272–280. [Google Scholar] [CrossRef]
- Miyagi, K.; Omura, K.; Ogawa, A.; Hanafusa, M.; Nakano, Y.; Morimatsu, S.; Sano, K. Survival of Shiga toxin-producing Escherichia coli O157 in marine water and frequent detection of the Shiga toxin gene in marine water samples from an estuary port. Epidemiol. Infect. 2001, 126, 129–133. [Google Scholar] [CrossRef]
- Saima, U.; Alam, M.; Akter, S. Survival of escherichia coli in Water Microcosm Study and Rethinking its Use as Indicator. Microbiology 2021, 90, 247–260. [Google Scholar] [CrossRef]
- Byrd, J.J.; Colwell, R.R. Long-term survival and plasmid maintenance of Escherichia coli in marine microcosms. FEMS Microbiol. Ecol. 1993, 12, 9–14. [Google Scholar] [CrossRef]
- Denamur, E.; Clermont, O.; Bonacorsi, S.; Gordon, D. The population genetics of pathogenic Escherichia coli. Nat. Rev. Microbiol. 2021, 19, 37–54. [Google Scholar] [CrossRef]
- Marsalek, J.; Rochfort, Q. Urban wet-weather flows: Sources of fecal contamination impacting on recreational waters and threatening drinking-water sources. J. Toxicol. Environ. Health A 2004, 67, 1765–1777. [Google Scholar] [CrossRef] [PubMed]
- Newton, R.J.; Bootsma, M.J.; Morrison, H.G.; Sogin, M.L.; McLellan, S.L. A microbial signature approach to identify fecal pollution in the waters off an urbanized coast of Lake Michigan. Microb. Ecol. 2013, 65, 1011–1023. [Google Scholar] [CrossRef]
- Berthe, T.; Ratajczak, M.; Clermont, O.; Denamur, E.; Petit, F. Evidence for coexistence of distinct Escherichia coli populations in various aquatic environments and their survival in estuary water. Appl. Environ. Microbiol. 2013, 79, 4684–4693. [Google Scholar] [CrossRef]
- Rumball, N.A.; Mayer, H.C.; McLellan, S.L. Selective survival of Escherichia coli phylotypes in freshwater beach sand. Appl. Environ. Microbiol. 2021, 87, e02473-20. [Google Scholar] [CrossRef]
- Williams, A.P.; Avery, L.M.; Killham, K.; Jones, D.L. Persistence, dissipation, and activity of Escherichia coli O157:H7 within sand and seawater environments. FEMS Microbiol. Ecol. 2007, 60, 24–32. [Google Scholar] [CrossRef]
- Byappanahalli, M.N.; Whitman, R.L.; Shively, D.A.; Sadowsky, M.J.; Ishii, S. Population structure, persistence, and seasonality of autochthonous Escherichia coli in temperate, coastal forest soil from a Great Lakes watershed. Environ. Microbiol. 2006, 8, 504–513. [Google Scholar] [CrossRef]
- Davies, C.M.; Long, J.A.H.; Donald, M.; Ashbolt, N.J. Survival of Fecal Microorganisms in Marine and Freshwater Sediments. Appl. Environ. Microbiol. 1995, 61, 1888–1896. [Google Scholar] [CrossRef] [PubMed]
- Sciarrillo, R.; Zuzolo, D.; Cicchella, D.; Iannone, F.; Cammino, G.; Guarino, C. Contamination and ecological risk assessment of the seaport of Naples (Italy): Insights from marine sediments. J. Geochem. Explor. 2020, 210, 106449. [Google Scholar] [CrossRef]
- Devane, M.L.; Moriarty, E.; Weaver, L.; Cookson, A.; Gilpin, B. Fecal indicator bacteria from environmental sources; strategies for identification to improve water quality monitoring. Water Res. 2020, 185, 116204. [Google Scholar] [CrossRef] [PubMed]
- McLellan, S.L.; Newton, R.J.; Vandewalle, J.L.; Shanks, O.C.; Huse, S.M.; Eren, A.M.; Sogin, M.L. Sewage reflects the distribution of human faecal Lachnospiraceae. Environ. Microbiol. 2013, 15, 2213–2227. [Google Scholar] [CrossRef]
- Newton, R.J.; McLellan, S.L.; Dila, D.K.; Vineis, J.H.; Morrison, H.G.; Eren, A.M.; Sogin, M.L. Sewage reflects the microbiomes of human populations. mBio 2015, 6, e02574. [Google Scholar] [CrossRef]
- Jephson, T. Diel Vertical Migration in Marine Dinoflagellates. Ph.D. Thesis, Department of Biology, Lund University, Lund, Sweden, 2012. [Google Scholar]
- NSVA. Miljörapport 2021. Öresundsverket, Helsingborgs Kommun. 2022. Available online: https://nsva.se/wp-content/uploads/2024/06/miljorapport-oresundsverket-2021.pdf (accessed on 4 April 2022).
- Al Aukidy, M.; Verlicchi, P. Contributions of combined sewer overflows and treated effluents to the bacterial load released into a coastal area. Sci. Total Environ. 2017, 607–608, 483–496. [Google Scholar] [CrossRef]
- Koboević, Ž.; Mišković, D.; Capor Hrošik, R.; Koboević, N. Analysis of Sea Pollution by Sewage from Vessels. Sustainability 2021, 14, 263. [Google Scholar] [CrossRef]
- Araujo, S.; Henriques, I.S.; Leandro, S.M.; Alves, A.; Pereira, A.; Correia, A. Gulls identified as major source of fecal pollution in coastal waters: A microbial source tracking study. Sci. Total Environ. 2014, 470–471, 84–91. [Google Scholar] [CrossRef]
- Tarek, M.H.; Hubbart, J.; Garner, E. Microbial source tracking to elucidate the impact of land-use and physiochemical water quality on fecal contamination in a mixed land-use watershed. Sci. Total Environ. 2023, 872, 162181. [Google Scholar] [CrossRef]
- Henry, R.; Schang, C.; Coutts, S.; Kolotelo, P.; Prosser, T.; Crosbie, N.; Grant, T.; Cottam, D.; O’Brien, P.; Deletic, A.; et al. Into the deep: Evaluation of SourceTracker for assessment of faecal contamination of coastal waters. Water Res. 2016, 93, 242–253. [Google Scholar] [CrossRef]
- Carney, R.L.; Brown, M.V.; Siboni, N.; Raina, J.B.; Kahlke, T.; Mitrovic, S.M.; Seymour, J.R. Highly heterogeneous temporal dynamics in the abundance and diversity of the emerging pathogens Arcobacter at an urban beach. Water Res. 2020, 171, 115405. [Google Scholar] [CrossRef] [PubMed]
- Fraser, A.N.; Preheim, S.P. Bacterial community composition and functional potential associated with a variety of urban stormwater sources. Urban. Ecosyst. 2021, 24, 1379–1390. [Google Scholar] [CrossRef]
- Patel, G.B.; Sprott, G.D. Methanosaeta concilii gen. nov. sp. nov. (“Methanothrix concilii”) and Methanosaeta thermoacetophila nom. rev., comb. nov. Int. J. Syst. Bacteriol. 1990, 40, 79–82. [Google Scholar] [CrossRef]
- Satoh, H.; Miura, Y.; Tsushima, I.; Okabe, S. Layered structure of bacterial and archaeal communities and their in situ activities in anaerobic granules. Appl. Environ. Microbiol. 2007, 73, 7300–7307. [Google Scholar] [CrossRef]
- Mino, S.; Kudo, H.; Arai, T.; Sawabe, T.; Takai, K.; Nakagawa, S. Sulfurovum aggregans sp. nov., a hydrogen-oxidizing, thiosulfate-reducing chemolithoautotroph within the Epsilonproteobacteria isolated from a deep-sea hydrothermal vent chimney, and an emended description of the genus Sulfurovum. Int. J. Syst. Evol. Microbiol. 2014, 64, 3195–3201. [Google Scholar] [CrossRef]
- Frigaard, N.U.; Dahl, C. Sulfur metabolism in phototrophic sulfur bacteria. Adv. Microb. Physiol. 2009, 54, 103–200. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, M.; Higashioka, Y.; Kojima, H.; Fukui, M. Desulfosarcina widdelii sp. nov. and Desulfosarcina alkanivorans sp. nov., hydrocarbon-degrading sulfate-reducing bacteria isolated from marine sediment and emended description of the genus Desulfosarcina. Int. J. Syst. Evol. Microbiol. 2017, 67, 2994–2997. [Google Scholar] [CrossRef]
- Chieffi, D.; Fanelli, F.; Fusco, V. Arcobacter butzleri: Up-to-date taxonomy, ecology, and pathogenicity of an emerging pathogen. Compr. Rev. Food Sci. Food Saf. 2020, 19, 2071–2109. [Google Scholar] [CrossRef] [PubMed]
- European Union. Country Profiles on Urban Waste Water Treatment: Denmark. Available online: https://water.europa.eu/freshwater/countries/uwwt/denmark (accessed on 11 June 2024).
- European Union. Country Profiles on Urban Waste Water Treatment: Sweden. Available online: https://water.europa.eu/freshwater/countries/uwwt/sweden (accessed on 11 June 2024).
- Treusch, A.H.; Vergin, K.L.; Finlay, L.A.; Donatz, M.G.; Burton, R.M.; Carlson, C.A.; Giovannoni, S.J. Seasonality and vertical structure of microbial communities in an ocean gyre. ISME J. 2009, 3, 1148–1163. [Google Scholar] [CrossRef]
- Roth, W.G.; Leckie, M.P.; Dietzler, D.N. Restoration of Colony-Forming Activity in Osmotically Stressed Escherichia coli by Betaine. Appl. Environ. Microbiol. 1988, 54, 3142–3146. [Google Scholar] [CrossRef]
- Tolu, J.; Rydberg, J.; Meyer-Jacob, C.; Gerber, L.; Bindler, R. Spatial variability of organic matter molecular composition and elemental geochemistry in surface sediments of a small boreal Swedish lake. Biogeosciences 2017, 14, 1773–1792. [Google Scholar] [CrossRef]
- Haller, L.; Tonolla, M.; Zopfi, J.; Peduzzi, R.; Wildi, W.; Pote, J. Composition of bacterial and archaeal communities in freshwater sediments with different contamination levels (Lake Geneva, Switzerland). Water Res. 2011, 45, 1213–1228. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Frank, E.M.; Suarez, C.; Erb, I.K.; Jephson, T.; Lindberg, E.; Paul, C.J. Microbial Contamination in Urban Marine Sediments: Source Identification Using Microbial Community Analysis and Fecal Indicator Bacteria. Microorganisms 2025, 13, 983. https://doi.org/10.3390/microorganisms13050983
Frank EM, Suarez C, Erb IK, Jephson T, Lindberg E, Paul CJ. Microbial Contamination in Urban Marine Sediments: Source Identification Using Microbial Community Analysis and Fecal Indicator Bacteria. Microorganisms. 2025; 13(5):983. https://doi.org/10.3390/microorganisms13050983
Chicago/Turabian StyleFrank, Ellinor M., Carolina Suarez, Isabel K. Erb, Therese Jephson, Elisabet Lindberg, and Catherine J. Paul. 2025. "Microbial Contamination in Urban Marine Sediments: Source Identification Using Microbial Community Analysis and Fecal Indicator Bacteria" Microorganisms 13, no. 5: 983. https://doi.org/10.3390/microorganisms13050983
APA StyleFrank, E. M., Suarez, C., Erb, I. K., Jephson, T., Lindberg, E., & Paul, C. J. (2025). Microbial Contamination in Urban Marine Sediments: Source Identification Using Microbial Community Analysis and Fecal Indicator Bacteria. Microorganisms, 13(5), 983. https://doi.org/10.3390/microorganisms13050983