
Quorum Sensing in Chromobacterium subtsugae ATCC 31532 (Formerly Chromobacterium violaceum ATCC 31532): Transcriptomic and Genomic Analyses

 ,
,  ,
,  ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Bacterial Strain and Growth Conditions
2.2. RNA Extraction and Quality Control
2.3. cDNA Library Preparation
2.4. cDNA Library Sequencing
2.5. Bioinformatic Analysis of RNA-Seq Dataset
2.6. Bioinformatic Analysis of C. subtsugae ATCC 31532 Genome
3. Results
3.1. Differential Expression of Genes Predicted to Be Under QS Regulation in C. subtsugae ATCC 31532 Strain
3.2. Putative CsuR Receptor Binding Sites in C. subtsugae ATCC 31532 Genome
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Adeolu, M.; Gupta, R.S. Phylogenomics and molecular signatures for the order Neisseriales: Proposal for division of the order Neisseriales into the emended family Neisseriaceae and Chromobacteriaceae fam. nov. Antonie van Leeuwenhoek 2013, 104, 1–24. [Google Scholar] [CrossRef]
- Bergonzini, C. Sopra un nuovo bacterio colorato. Annu. Soc. Nat. Modena 1880, 14, 149–158. [Google Scholar]
- ATCC. Available online: https://www.atcc.org/ (accessed on 24 February 2025).
- Miller, M.B.; Bassler, B.L. Quorum sensing in bacteria. Annu. Rev. Microbiol. 2001, 55, 165–199. [Google Scholar] [CrossRef] [PubMed]
- McClean, K.H.; Winson, M.K.; Fish, L.; Taylor, A.; Chhabra, S.R.; Camara, M.; Daykin, M.; Lamb, J.H.; Swift, S.; Bycroft, B.W.; et al. Quorum sensing and Chromobacterium violaceum: Exploitation of violacein production and inhibition for the detection of N-acylhomoserine lactones. Microbiology 1997, 143, 3703–3711. [Google Scholar] [CrossRef]
- Morohoshi, T.; Kato, M.; Fukamachi, K.; Kato, N.; Ikeda, T. N-acylhomoserine lactone regulates violacein production in Chromobacterium violaceum type strain ATCC 12472. FEMS Microbiol. Lett. 2008, 279, 124–130. [Google Scholar] [CrossRef]
- Chen, G.; Swem, L.R.; Swem, D.L.; Stauff, D.L.; O’Loughlin, C.T.; Jeffrey, P.D.; Bassler, B.L.; Hughson, F.M. A strategy for antagonizing quorum sensing. Mol. Cell 2011, 42, 199–209. [Google Scholar] [CrossRef]
- Hoshino, T. Violacein and related tryptophan metabolites produced by Chromobacterium violaceum: Biosynthetic mechanism and pathway for construction of violacein core. Appl. Microbiol. Biotechnol. 2011, 91, 1463–1475. [Google Scholar] [CrossRef]
- Balibar, C.J.; Walsh, C.T. In vitro biosynthesis of violacein from L-tryptophan by the enzymes VioA-E from Chromobacterium violaceum. Biochemistry 2006, 45, 15444–15457. [Google Scholar] [CrossRef] [PubMed]
- Kothari, V.; Sharma, S.; Padia, D. Recent research advances on Chromobacterium violaceum. Asian Pac. J. Trop. Med. 2017, 10, 744–752. [Google Scholar] [CrossRef]
- Deryabin, D.G.; Galadzhieva, A.A.; Duskaev, G.K. Screening of N-hexanamide and 2H-1,3-benzodioxol derivatives for quorum sensing modulation in Chromobacterium violaceum. Microbiology 2020, 89, 733–739. [Google Scholar] [CrossRef]
- Stauff, D.L.; Bassler, B.L. Quorum sensing in Chromobacterium violaceum: DNA recognition and gene regulation by the CviR receptor. J. Bacteriol. 2011, 193, 3871–3878. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Hinshaw, K.C.; Macdonald, S.J.; Chandler, J.R. Draft genome sequence of Chromobacterium violaceum strain CV017. Genome Announc. 2016, 4, e00080-16. [Google Scholar] [CrossRef]
- Harrison, A.M.; Soby, S.D. Reclassification of Chromobacterium violaceum ATCC 31532 and its quorum biosensor mutant CV026 to Chromobacterium subtsugae. AMB Express 2020, 10, 202. [Google Scholar] [CrossRef]
- Batista, J.H.; da Silva Neto, J.F. Chromobacterium violaceum pathogenicity: Updates and insights from genome sequencing of novel Chromobacterium species. Front. Microbiol. 2017, 8, 2213. [Google Scholar] [CrossRef] [PubMed]
- Martin, P.A.W.; Gundersen-Rindal, D.; Blackburn, M.; Buyer, J. Chromobacterium subtsugae sp. nov., a betaproteobacterium toxic to Colorado potato beetle and other insect pests. Int. J. Syst. Evol. Microbiol. 2007, 57, 993–999. [Google Scholar] [CrossRef]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Burrows-Wheeler Aligner. Available online: http://bio-bwa.sourceforge.net (accessed on 24 February 2025).
- NCBI. Chromobacterium Subtsugae Strain ATCC 31532 Chromosome, Complete Genome. Available online: https://www.ncbi.nlm.nih.gov/nuccore/NZ_CP142381.1 (accessed on 24 February 2025).
- NCBI. Genome Annotation. Available online: https://www.ncbi.nlm.nih.gov/datasets/gene/GCF_035919665.1/ (accessed on 25 February 2025).
- featureCounts: A Ultrafast and Accurate Read Summarization Program. Available online: https://subread.sourceforge.net/featureCounts.html (accessed on 24 February 2025).
- Analyzing RNA-Seq Data with DESeq2. Available online: https://www.bioconductor.org/packages/release/bioc/vignettes/DESeq2/inst/doc/DESeq2.html (accessed on 25 February 2025).
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef]
- Rice, P.; Longden, I.; Bleasby, A. EMBOSS: The European molecular biology open software suite. Trends Genet. 2000, 16, 276–277. [Google Scholar] [CrossRef]
- Park, H.; Park, S.; Yang, Y.H.; Choi, K.Y. Microbial synthesis of violacein pigment and its potential applications. Crit. Rev. Biotechnol. 2021, 41, 879–901. [Google Scholar] [CrossRef]
- Koirala, P.; Doody, C.; Blackwell, H.; Chandler, J.R. Regulation of an antibiotic resistance efflux pump by quorum sensing and a TetR-family repressor in Chromobacterium subtsugae. bioRxiv 2023, 3. 2023.09.02.556004. [Google Scholar]
- Horinouchi, S.; Beppu, T. A-factor as a microbial hormone that controls cellular differentiation and secondary metabolism in Streptomyces griseus. Mol. Microbiol. 1994, 12, 859–864. [Google Scholar] [CrossRef]
- InterPro. Classification of Protein Families. Available online: https://www.ebi.ac.uk/interpro/entry/InterPro/IPR006385/ (accessed on 24 February 2025).
- Kato, J.-y.; Funa, N.; Watanabe, H.; Ohnishi, Y.; Horinouchi, S. Biosynthesis of γ-butyrolactone autoregulators that switch on secondary metabolism and morphological development in Streptomyces. Proc. Natl. Acad. Sci. USA 2007, 104, 2378–2383. [Google Scholar] [CrossRef] [PubMed]
- NCBI. Predicted Small Integral Membrane Protein (DUF2165). Available online: https://www.ncbi.nlm.nih.gov/Structure/cdd/cl02290 (accessed on 24 February 2025).
- Batista, B.B.; de Lima, V.M.; Picinato, B.A.; Koide, T.; da Silva Neto, J.F. A quorum-sensing regulatory cascade for siderophore-mediated iron homeostasis in Chromobacterium violaceum. mSystems 2024, 9, e0139723. [Google Scholar] [CrossRef] [PubMed]
- Ahn, S.K.; Cuthbertson, L.; Nodwell, J.R. Genome context as a predictive tool for identifying regulatory targets of the TetR family transcriptional regulators. PLoS ONE 2012, 7, e50562. [Google Scholar] [CrossRef]
- J. Craig Venter Institute. Genome Property Definition Page. Available online: https://genome-properties.jcvi.org/cgi-bin/GenomePropDefinition.cgi?prop_acc=GenProp1090 (accessed on 24 February 2025).
- Schaefer, A.L.; Val, D.L.; Hanzelka, B.L.; Cronan, J.E., Jr.; Greenberg, E.P. Generation of cell-to-cell signals in quorum sensing: Acyl homoserine lactone synthase activity of a purified Vibrio fischeri LuxI protein. Proc. Natl. Acad. Sci. USA 1996, 93, 9505–9509. [Google Scholar] [CrossRef]
- Talfournier, F.; Stines-Chaumeil, C.; Branlant, G. Methylmalonate-semialdehyde dehydrogenase from Bacillus subtilis: Substrate specificity and coenzyme A binding. J. Biol. Chem. 2011, 286, 21971–21981. [Google Scholar] [CrossRef] [PubMed]
- Park, S.C.; Kim, P.-H.; Lee, G.-S.; Kang, S.G.; Ko, H.-J.; Yoon, S.-I. Structural and biochemical characterization of the Bacillus cereus 3-hydroxyisobutyrate dehydrogenase. Biochem. Biophys. Res. Commun. 2016, 474, 522–527. [Google Scholar] [CrossRef]
- Stojkova, P.; Spidlova, P.; Stulik, J. Nucleoid-associated protein HU: A lilliputian in gene regulation of bacterial virulence. Front. Cell. Infect. Microbiol. 2019, 9, 159. [Google Scholar] [CrossRef]
- Aravind, L.; Anantharaman, V.; Balaji, S.; Babu, M.M.; Iyer, L.M. The many faces of the helix-turn-helix domain: Transcription regulation and beyond. FEMS Microbiol. Rev. 2005, 29, 231–262. [Google Scholar] [CrossRef]
- Nesmelova, I.V.; Hackett, P.B. DDE transposases: Structural similarity and diversity. Adv. Drug Deliv. Rev. 2010, 62, 1187–1195. [Google Scholar] [CrossRef]
- Koonin, E.V.; Aravind, L.; Leipe, D.D.; Iyer, L.M. Evolutionary history and higher order classification of AAA ATPases. J. Struct. Biol. 2004, 146, 11–31. [Google Scholar]
- de Almeida, R.; Trevilato, P.B.; Bartoleti, L.A.; Proença-Módena, J.L.; Hanna, E.S.; Gregoracci, G.B.; Brocchi, M. Bacteriophages and insertion sequences of Chromobacterium violaceum ATCC 12472. Genet. Mol. Res. 2004, 3, 76–84. [Google Scholar]
- Igarashi, K.; Kashiwagi, K. Polyamine transport in bacteria and yeast. Biochem. J. 1999, 344, 633–642. [Google Scholar] [CrossRef] [PubMed]
- Prentice, J.A.; Bridges, A.A.; Bassler, B.L. Synergy between c-di-GMP and quorum-sensing signaling in Vibrio cholerae biofilm morphogenesis. J. Bacteriol. 2022, 204, e0024922. [Google Scholar] [CrossRef]
- Eberhardt, R.Y.; Chang, Y.; Bateman, A.; Murzin, A.G.; Axelrod, H.L.; Hwang, W.C.; Aravind, L. Filling out the structural map of the NTF2-like superfamily. BMC Bioinform. 2013, 14, 327. [Google Scholar] [CrossRef]
- Pages, J.-M.; James, C.E.; Winterhalter, M. The porin and the permeating antibiotic: A selective diffusion barrier in Gram-negative bacteria. Nat. Rev. Microbiol. 2008, 6, 893–903. [Google Scholar] [CrossRef]
- Kostlanova, N.; Mitchell, E.P.; Lortat-Jacob, H.; Oscarson, S.; Lahmann, M.; Gilboa-Garber, N.; Chambat, G.; Wimmerova, M.; Imberty, A. The fucose-binding lectin from Ralstonia solanacearum. A new type of β-propeller architecture formed by oligomerization and interacting with fucoside, fucosyllactose, and plant xyloglucan. J. Biol. Chem. 2005, 280, 27839–27849. [Google Scholar] [PubMed]
- Dimitrova, P.D.; Damyanova, T.; Paunova-Krasteva, T. Chromobacterium violaceum: A model for evaluating the anti-quorum sensing activities of plant substances. Sci. Pharm. 2023, 91, 33. [Google Scholar] [CrossRef]
- Gogoleva, N.E.; Shlykova, L.V.; Gorshkov, V.Y.; Daminova, A.G.; Gogolev, Y.V. Effect of topology of quorum sensing-related genes in Pectobacterium atrosepticum on their expression. Mol. Biol. 2014, 48, 583–589. [Google Scholar] [CrossRef]
- Mendoza-Vargas, A.; Olvera, L.; Olvera, M.; Grande, R.; Vega-Alvarado, L.; Taboada, B.; Jimenez-Jacinto, V.; Salgado, H.; Juarez, K.; Contreras-Moreira, B.; et al. Genome-wide identification of transcription start sites, promoters and transcription factor binding sites in E. coli. PLoS ONE 2009, 4, e7526. [Google Scholar] [CrossRef]
- Murakami, K.S. Structural biology of bacterial RNA polymerase. Biomolecules 2015, 5, 848–864. [Google Scholar] [CrossRef] [PubMed]
- Egland, K.A.; Greenberg, E.P. Quorum sensing in Vibrio fischeri: Elements of the luxl promoter. Mol. Microbiol. 1999, 31, 1197–1204. [Google Scholar] [CrossRef]
- Verma, S.C.; Miyashiro, T. Quorum sensing in the squid-Vibrio symbiosis. Int. J. Mol. Sci. 2013, 14, 16386–16401. [Google Scholar] [CrossRef]
- Leal, A.M.; de Queiroz, J.D.; Medeiros, S.R.; Lima, T.K.; Agnez-Lima, L.F. Violacein induces cell death by triggering mitochondrial membrane hyperpolarization in vitro. BMC Microbiol. 2015, 15, 115. [Google Scholar] [CrossRef] [PubMed]
- Dennis, G.; Sherman, B.T.; Hosack, D.A.; Yang, J.; Gao, W.; Lane, H.C.; Lempicki, R.A. DAVID: Database for annotation, visualization, and integrated discovery. Genome Biol. 2003, 4, R60. [Google Scholar] [CrossRef]
- Favero, F.; Tolentino, T.A.; Fernandes, V.; Treptow, W.; Pereira, A.L.; Machado, A.H.L. α-Alkylidene δ-lactones inhibit quorum sensing phenotypes in Chromobacterium strain CV026 showing interaction with the CviR receptor. RSC Adv. 2023, 26, 18045–18057. [Google Scholar] [CrossRef]
- Nivetha, R.; Meenakumari, M.; Peroor, M.D.A.; Janarthanan, S. Fucose-binding lectins: Purification, characterization and potential biomedical applications. Mol. Biol. Rep. 2023, 50, 10589–10603. [Google Scholar] [CrossRef] [PubMed]
- Chaparian, R.R.; Ball, A.S.; van Kessel, J.C. Hierarchical transcriptional control of the LuxR quorum-sensing regulon of Vibrio harveyi. J. Bacteriol. 2020, 202, e00047-20. [Google Scholar] [CrossRef]
- Guo, L.; Ruan, Q.; Ma, D.; Wen, J. Revealing quorum-sensing networks in Pseudomonas aeruginosa infections through internal and external signals to prevent new resistance trends. Microbiol. Res. 2024, 289, 127915. [Google Scholar] [CrossRef]
- Deochand, D.K.; Grove, A. MarR family transcription factors: Dynamic variations on a common scaffold. Crit. Rev. Biochem. Mol. Biol. 2017, 52, 595–613. [Google Scholar] [CrossRef]
- Colclough, A.L.; Scadden, J.; Blair, J.M.A. TetR-family transcription factors in Gram-negative bacteria: Conservation, variation and implications for efflux-mediated antimicrobial resistance. BMC Genom. 2019, 20, 731. [Google Scholar] [CrossRef] [PubMed]
- Creamer, K.E.; Kudo, Y.; Moore, B.S.; Jensen, P.R. Phylogenetic analysis of the salinipostin γ-butyrolactone gene cluster uncovers new potential for bacterial signalling-molecule diversity. Microb. Genom. 2021, 7, 000568. [Google Scholar] [CrossRef] [PubMed]
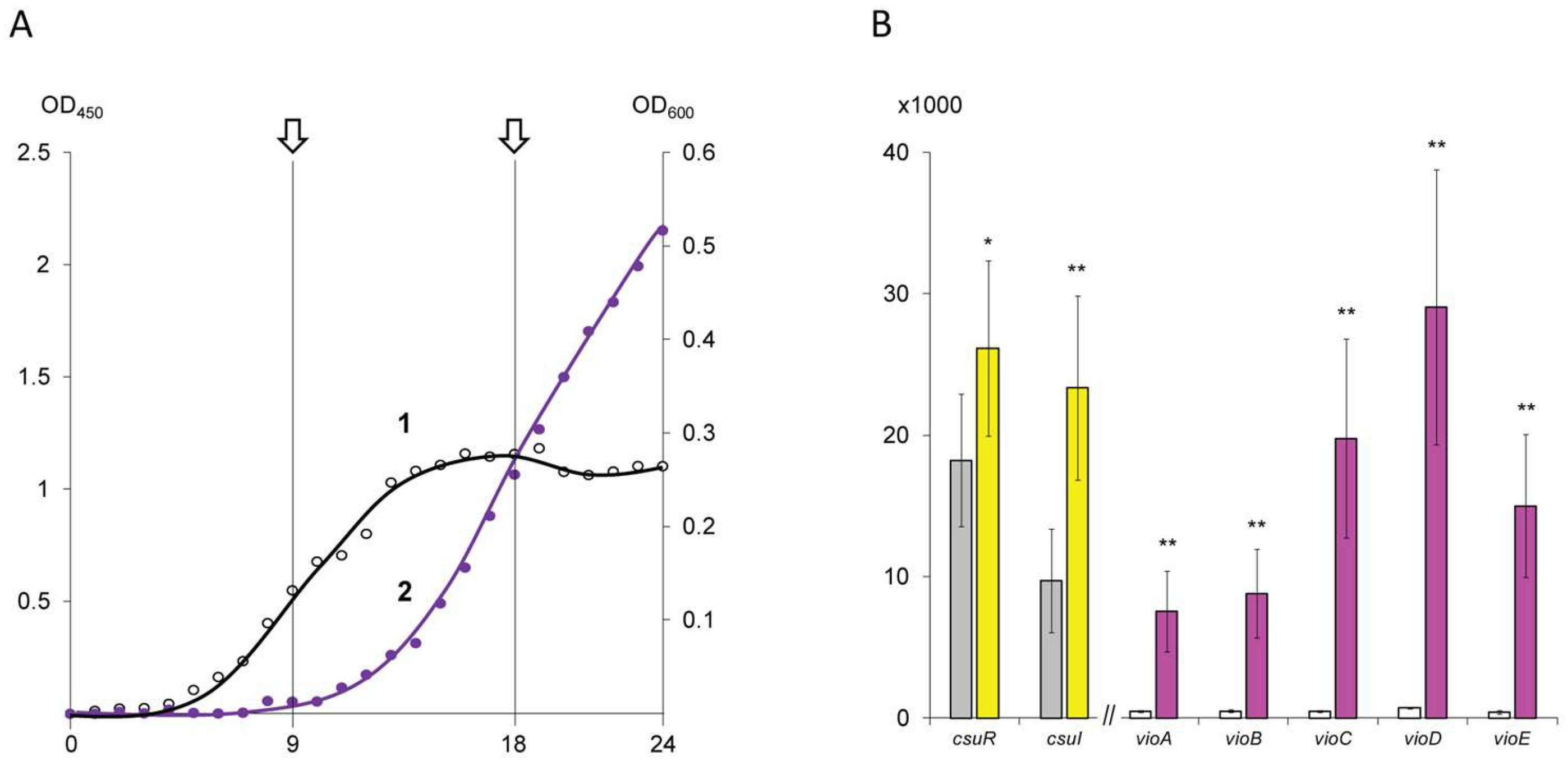
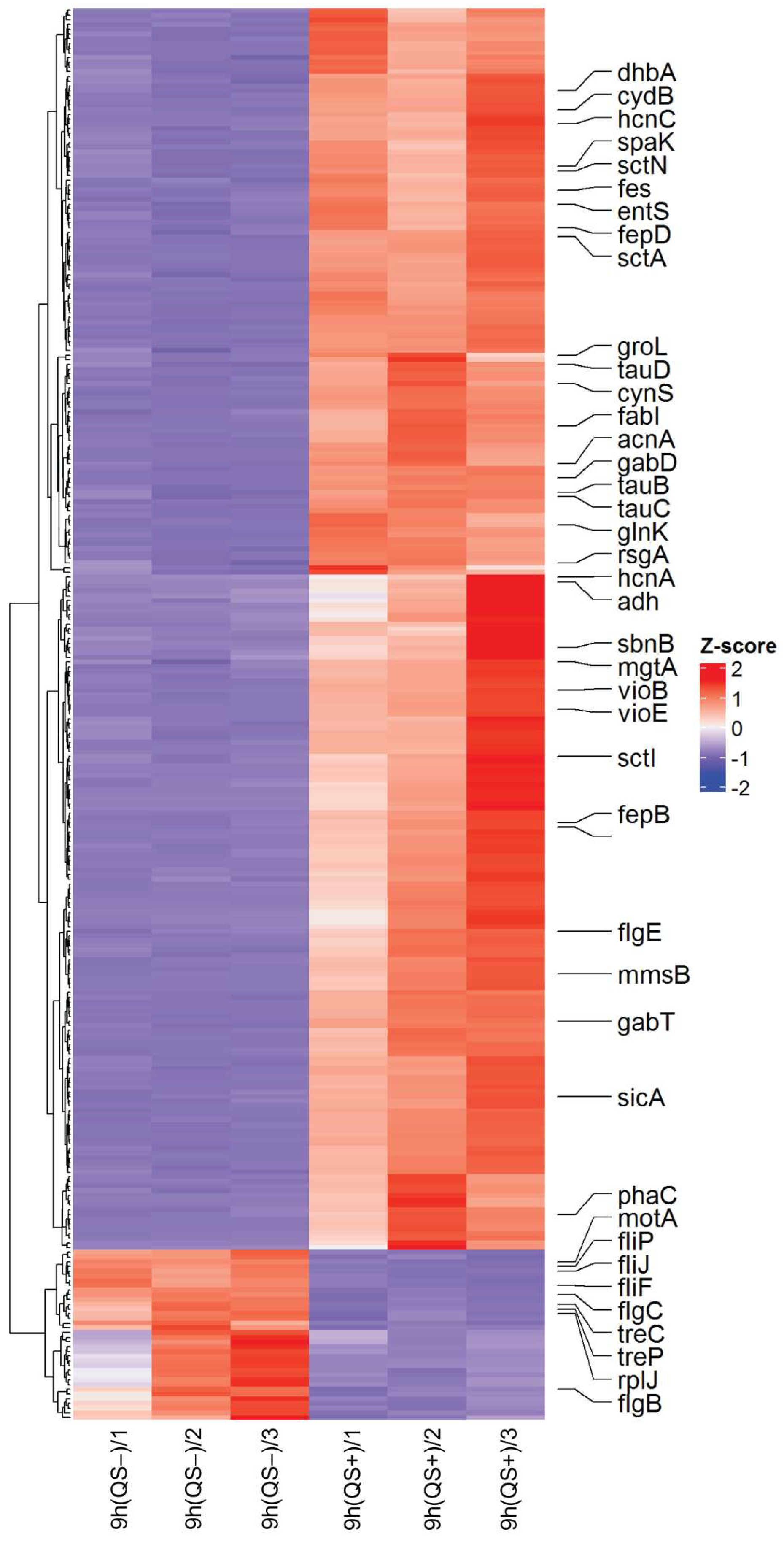
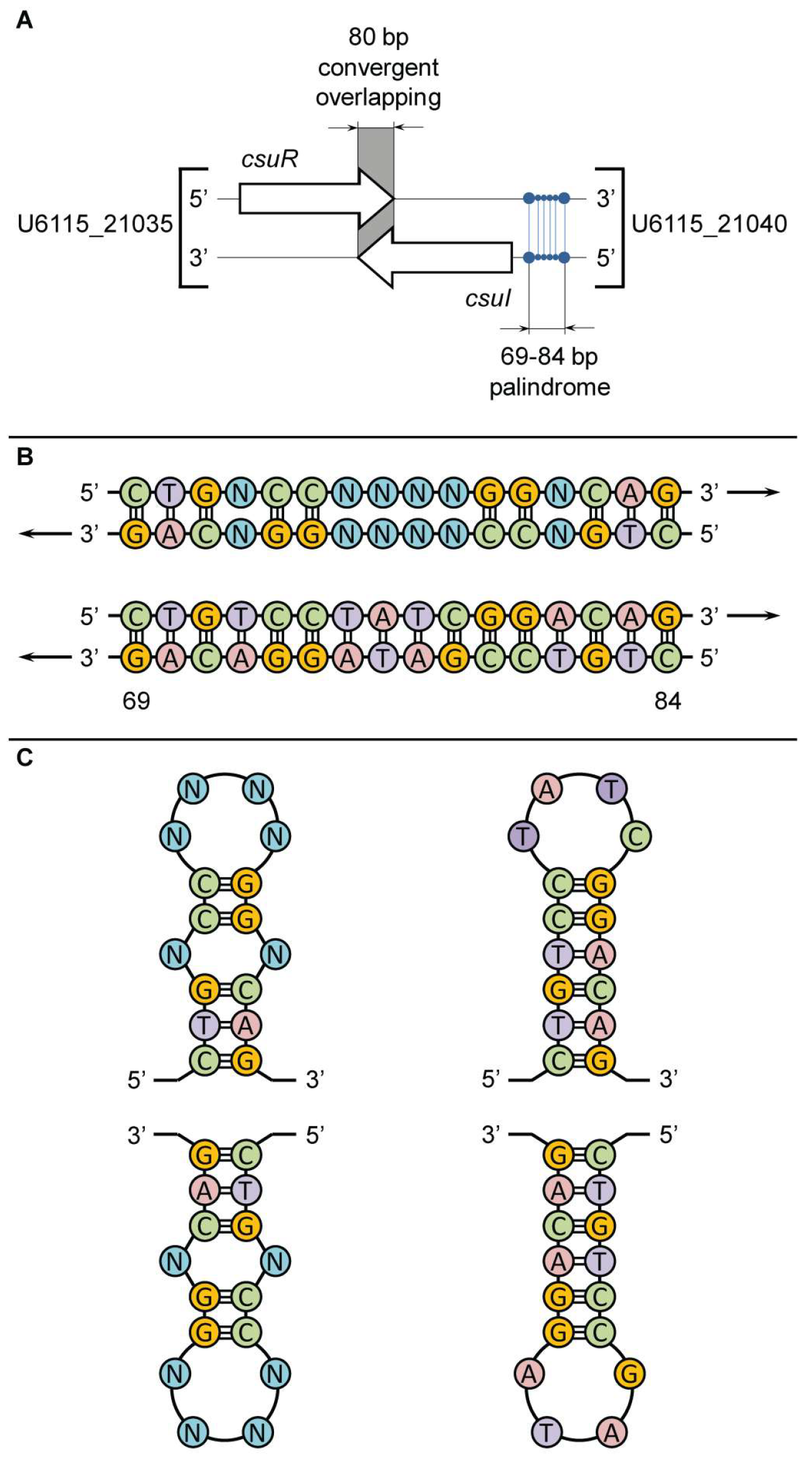
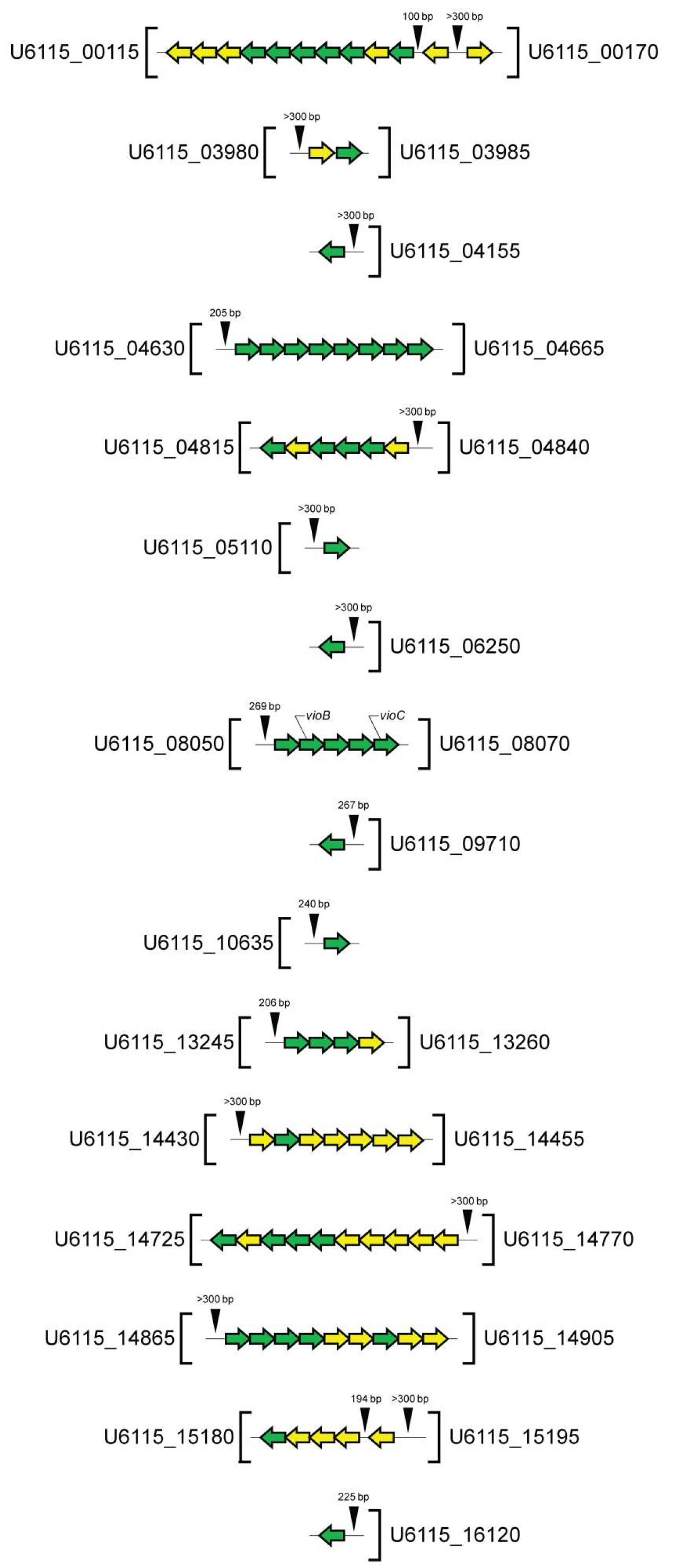
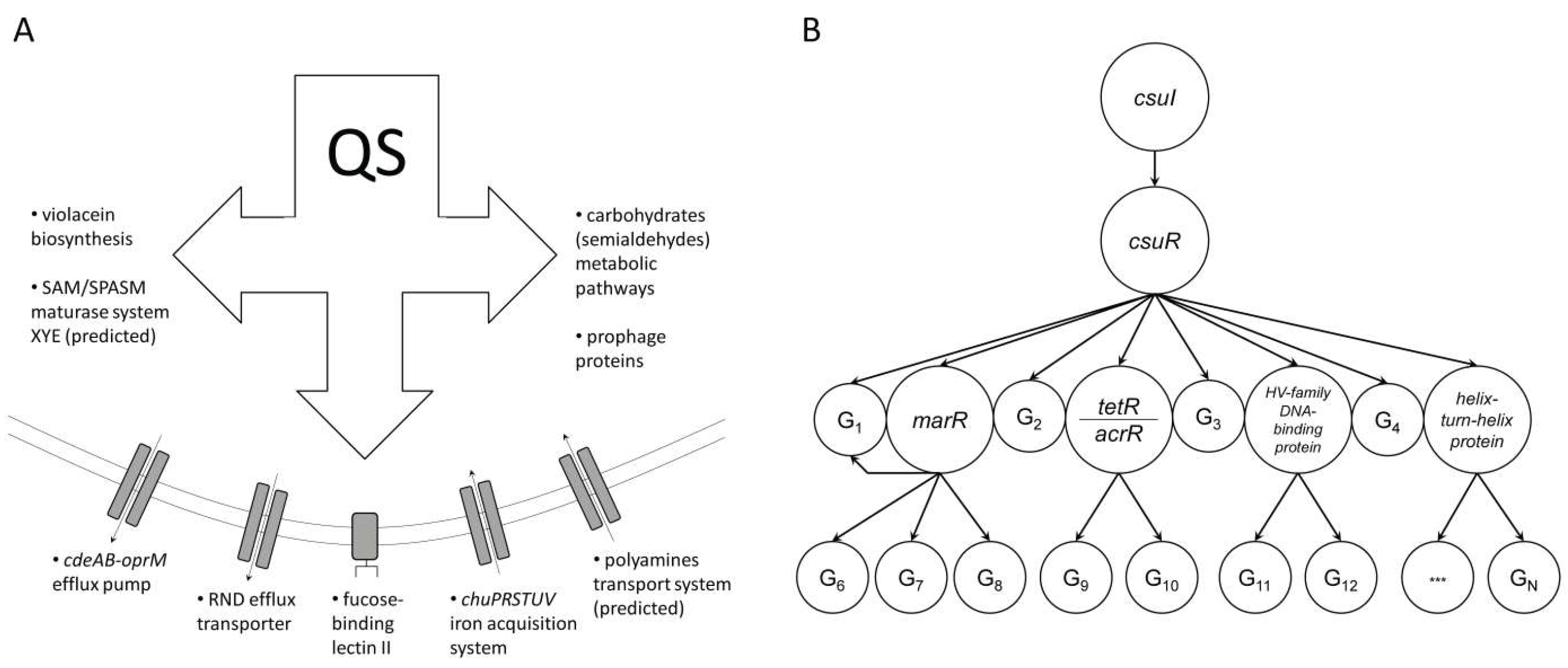

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ORF No. | Product Description | 9 h (QS−) Sample Expression | 18 h (QS+) Sample Expression | Log2 (Fold Change) |
|---|---|---|---|---|
| … | … | |||
| U6115_00130 | acyltransferase | 265.58 | 6104.15 | 4.5223 |
| U6115_00135 | flavin reductase family protein | 222.29 | 3654.83 | 4.038 |
| U6115_00140 | SDR family oxidoreductase | 308.96 | 14,269.74 | 5.5261 |
| U6115_00145 | MFS transporter | 352.10 | 12,816.07 | 5.1849 |
| U6115_00150 | cupin domain-containing protein | 168.27 | 5722.99 | 5.0879 |
| … | … | |||
| U6115_00160 | FAD-binding oxidoreductase | 239.46 | 9032.96 | 5.2337 |
| … | … | |||
| … | … | |||
| U6115_03985 | nuclear transport factor 2 family protein | 426.80 | 10,029.22 | 4.5549 |
| U6115_04155 | DUF1842 domain-containing protein | 3578.32 | 76,174.05 | 4.4121 |
| U6115_04630 | hypothetical protein | 463.24 | 90,275.91 | 7.6056 |
| U6115_04635 | AfsA-related hotdog domain-containing protein | 227.75 | 111,013.08 | 8.9269 |
| U6115_04640 | HAD-IB family hydrolase | 80.45 | 46,715.15 | 9.1777 |
| U6115_04645 | DUF2165 family protein | 81.60 | 29,104.70 | 8.4833 |
| U6115_04650 | MarR family transcriptional regulator | 82.29 | 27,891.04 | 8.3998 |
| U6115_04655 | efflux transporter outer membrane subunit | 169.53 | 51,325.66 | 8.2401 |
| U6115_04660 | HlyD family efflux transporter periplasmic adaptor subunit | 241.02 | 78,278.93 | 8.344 |
| U6115_04665 | DHA2 family efflux MFS transporter permease subunit | 505.29 | 75,629.69 | 7.2257 |
| U6115_04815 | ABC transporter ATP-binding protein | 739.58 | 17,230.47 | 4.5431 |
| … | … | |||
| U6115_04825 | ABC transporter substrate-binding protein | 732.78 | 12,679.79 | 4.1139 |
| U6115_04830 | hemin-degrading factor | 2186.39 | 40,476.44 | 4.211 |
| U6115_04835 | TonB-dependent hemoglobin/transferrin/lactoferrin family receptor | 4640.67 | 75,458.91 | 4.0235 |
| … | … | |||
| U6115_05110 | porin | 5085.22 | 86,187.67 | 4.0832 |
| U6115_06250 | hypothetical protein | 4504.26 | 84,702.20 | 4.2331 |
| U6115_08050 | FAD-dependent oxidoreductase | 463.89 | 7559.13 | 4.0287 |
| U6115_08055 | iminophenyl-pyruvate dimer synthase VioB | 522.94 | 8812.35 | 4.0771 |
| U6115_08060 | NAD(P)/FAD-dependent oxidoreductase | 505.23 | 19,786.24 | 5.293 |
| U6115_08065 | tryptophan hydroxylase | 739.89 | 29,065.83 | 5.2973 |
| U6115_08070 | violacein biosynthesis enzyme VioE | 424.25 | 15,023.60 | 5.1455 |
| U6115_09710 | hypothetical protein | 1044.89 | 17,971.71 | 4.1047 |
| U6115_10635 | hypothetical protein | 1021.42 | 19,171.38 | 4.2317 |
| U6115_13245 | DUF1843 domain-containing protein | 78.56 | 2425.74 | 4.9472 |
| U6115_13250 | DUF1842 domain-containing protein | 318.76 | 11,303.45 | 5.148 |
| U6115_13255 | DUF1842 domain-containing protein | 247.45 | 9744.09 | 5.2992 |
| … | … | |||
| … | … | |||
| U6115_14435 | CoA-acylating methylmalonate-semialdehyde dehydrogenase | 3465.35 | 63,003.03 | 4.1846 |
| … | … | |||
| U6115_14725 | HU family DNA-binding protein | 199.18 | 3597.91 | 4.1771 |
| … | … | |||
| U6115_14735 | hypothetical protein | 77.15 | 1750.40 | 4.5112 |
| U6115_14740 | hypothetical protein | 63.99 | 1474.44 | 4.5138 |
| U6115_14745 | hypothetical protein | 35.72 | 1204.11 | 5.0607 |
| … | … | |||
| U6115_14865 | peptidase | 264.97 | 4716.73 | 4.1546 |
| U6115_14870 | hypothetical protein | 193.55 | 6526.27 | 5.0729 |
| U6115_14875 | hypothetical protein | 23.64 | 717.12 | 4.9079 |
| U6115_14880 | capsid cement protein | 21.49 | 503.90 | 4.5344 |
| … | … | |||
| U6115_14895 | hypothetical protein | 62.70 | 1760.16 | 4.8166 |
| … | … | |||
| U6115_15180 | DUF3138 family protein | 638.68 | 26,856.19 | 5.3953 |
| … | … | |||
| U6115_16120 | fucose-binding lectin II | 1206.95 | 64,464.05 | 5.7393 |
| ORF Promoter | DNA Strand | Sequence Variant for Alignment | 16-Base-Pair Sequence Found (Matching Nucleotides Are Underlined) | Position in Relation to the Transcription Start Site (−bp) | Ideal Sequence Similarity (%) |
|---|---|---|---|---|---|
| U6115_RS00160 | reverse | inverted reverse motif | TGATCCGCTACGGCGG | 79–94 | 56.25 |
| U6115_RS00165 | reverse | reverse motif | GAGAGGGCAGAGGGTG | 135–150 | 56.25 |
| U6115_RS00170 | forward | inverted forward motif | GCTAGACAAGCATGGC | 107–122 | 56.25 |
| reverse motif | CGGACGAAAGCCGGCC | 126–141 | 56.25 | ||
| U6115_RS03980 | forward | forward motif | CAGTCCGATCATAAAT | 88–103 | 62.50 |
| reverse motif | GACAGGCTGCCCTGTC | 146–161 | 62.50 | ||
| U6115_RS04155 | reverse | inverted forward motif | GGCGGTAAATCCGGTC | 127–142 | 62.50 |
| U6115_RS04630 | forward | reverse motif | GAGAAAATATCCTATG | 101–116 | 62.50 |
| inverted reverse motif | CTAAATGATAGCCGAG | 114–129 | 56.25 | ||
| U6115_RS04840 | reverse | inverted forward motif | GCCAGGGTCCACCGTT | 81–96 | 56.25 |
| U6115_RS05110 | forward | reverse motif | GGCGGAATATACCGTC | 77–92 | 62.50 |
| U6115_RS06250 | reverse | reverse motif | GCTATTTTAGCTTTTC | 120–135 | 56.25 |
| U6115_RS08050 | forward | reverse motif | AAGAGCTGAGCCATTC | 73–88 | 56.25 |
| inverted reverse motif | CCGCCCGCCGGGAAAC | 135–150 | 56.25 | ||
| U6115_RS09710 | reverse | inverted reverse motif | CTGACCGTTAAGAACG | 91–106 | 68.75 |
| U6115_RS10635 | forward | inverted reverse motif | CATTCCATTTCGTCAG | 135–150 | 56.25 |
| inverted forward motif | GTCATTCCATTTCGTC | 137–152 | 56.25 | ||
| forward motif | CTGGGCTGCGGGCGAG | 169–184 | 56.25 | ||
| reverse motif | GTTTGGTTTGCCTGGG | 180–195 | 56.25 | ||
| U6115_RS13245 | forward | reverse motif | GAAAGCACTGCCCGGC | 73–88 | 62.50 |
| forward motif | CTGTACTTAAGGGCAG | 116–131 | 68.75 | ||
| inverted forward motif | CCCAGCCCAAGCTGAC | 177–192 | 56.25 | ||
| U6115_RS14430 | forward | not found | |||
| U6115_RS14770 | reverse | not found | |||
| U6115_RS14865 | forward | forward motif | CACTCATCTCTTACAT | 121–136 | 56.25 |
| U6115_RS15190 | reverse | reverse motif | GCCGGGCCCGCCTGTT | 80–95 | 62.50 |
| inverted reverse motif | CAGGCCGGCAGGGCCG | 108–123 | 62.50 | ||
| forward motif | CTGTTCTGGCCTGCCG | 158–173 | 56.25 | ||
| U6115_RS15195 | reverse | reverse motif | CGCGCGTTAGCCCGTC | 177–192 | 62.50 |
| U6115_RS16120 | reverse | inverted reverse motif | CCGTCCGTTTGGAGCC | 170–185 | 62.50 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Deryabin, D.G.; Inchagova, K.S.; Nikonorova, E.R.; Karimov, I.F.; Duskaev, G.K. Quorum Sensing in Chromobacterium subtsugae ATCC 31532 (Formerly Chromobacterium violaceum ATCC 31532): Transcriptomic and Genomic Analyses. Microorganisms 2025, 13, 1021. https://doi.org/10.3390/microorganisms13051021
Deryabin DG, Inchagova KS, Nikonorova ER, Karimov IF, Duskaev GK. Quorum Sensing in Chromobacterium subtsugae ATCC 31532 (Formerly Chromobacterium violaceum ATCC 31532): Transcriptomic and Genomic Analyses. Microorganisms. 2025; 13(5):1021. https://doi.org/10.3390/microorganisms13051021
Chicago/Turabian StyleDeryabin, Dmitry G., Ksenia S. Inchagova, Eugenia R. Nikonorova, Ilshat F. Karimov, and Galimzhan K. Duskaev. 2025. "Quorum Sensing in Chromobacterium subtsugae ATCC 31532 (Formerly Chromobacterium violaceum ATCC 31532): Transcriptomic and Genomic Analyses" Microorganisms 13, no. 5: 1021. https://doi.org/10.3390/microorganisms13051021
APA StyleDeryabin, D. G., Inchagova, K. S., Nikonorova, E. R., Karimov, I. F., & Duskaev, G. K. (2025). Quorum Sensing in Chromobacterium subtsugae ATCC 31532 (Formerly Chromobacterium violaceum ATCC 31532): Transcriptomic and Genomic Analyses. Microorganisms, 13(5), 1021. https://doi.org/10.3390/microorganisms13051021