

Genetic Evolution of Antibiotic Resistance and Virulence Genes in Escherichia coli Isolates from a Chinese Hospital over a 12-Year Period

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Clinical Isolates
2.2. Antibiotic Susceptibilities
2.3. Whole-Genome Sequencing
2.4. Bioinformatics Analysis
3. Results
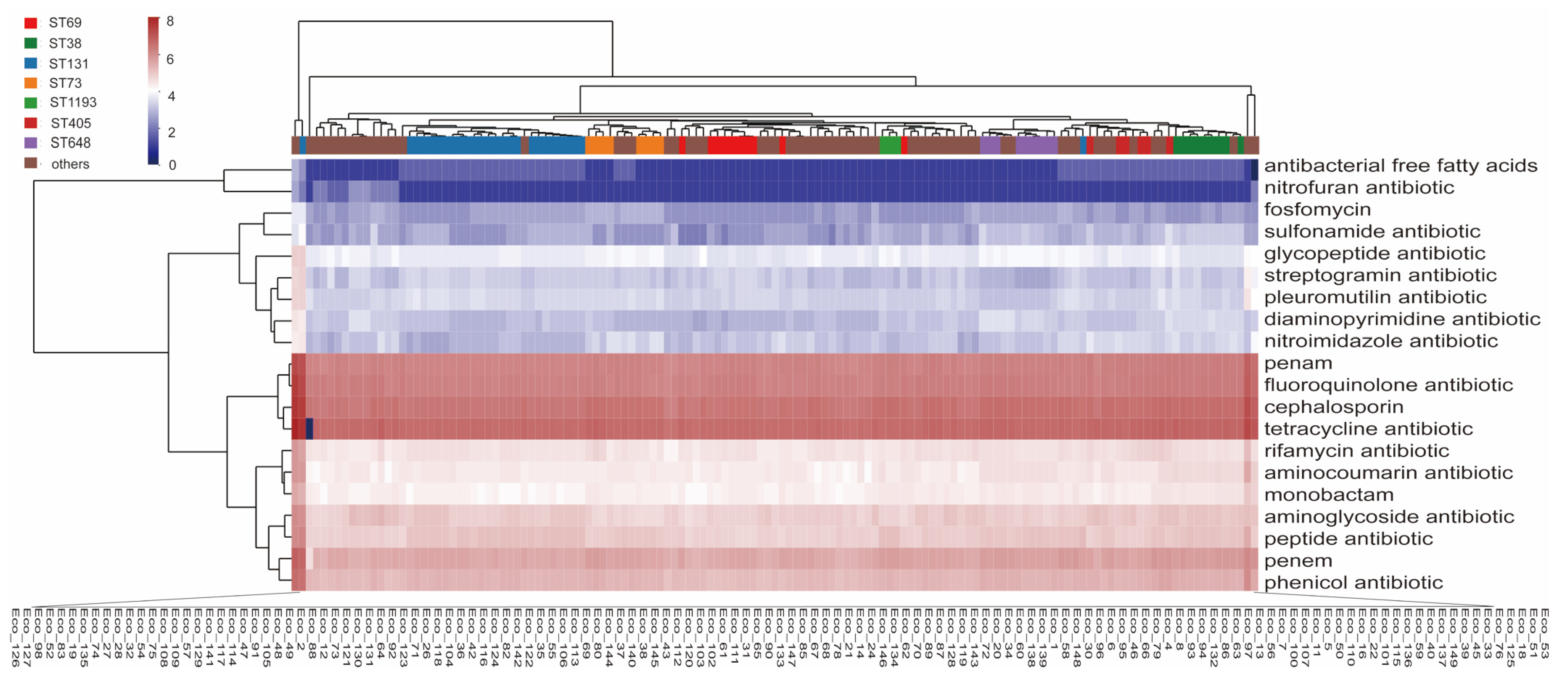
3.1. Antibiotic Resistance of the E. coli Isolates
3.2. Multilocus Sequence Types and Phylogenetic Relationships of the Isolates
3.3. Antibiotic Resistance Genes in the Isolates
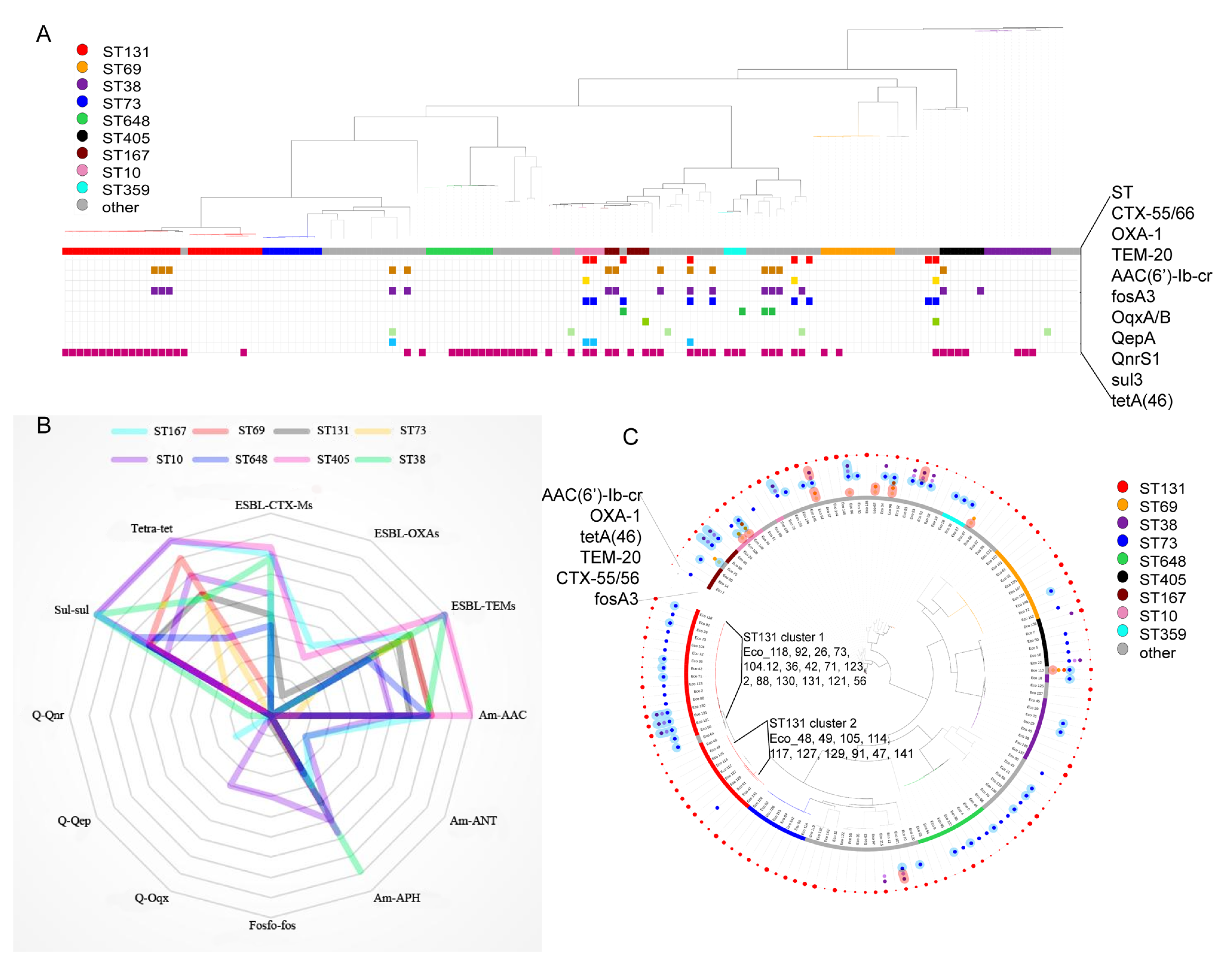
3.4. Comparison of Antibiotic Resistance Genes Among Different STs
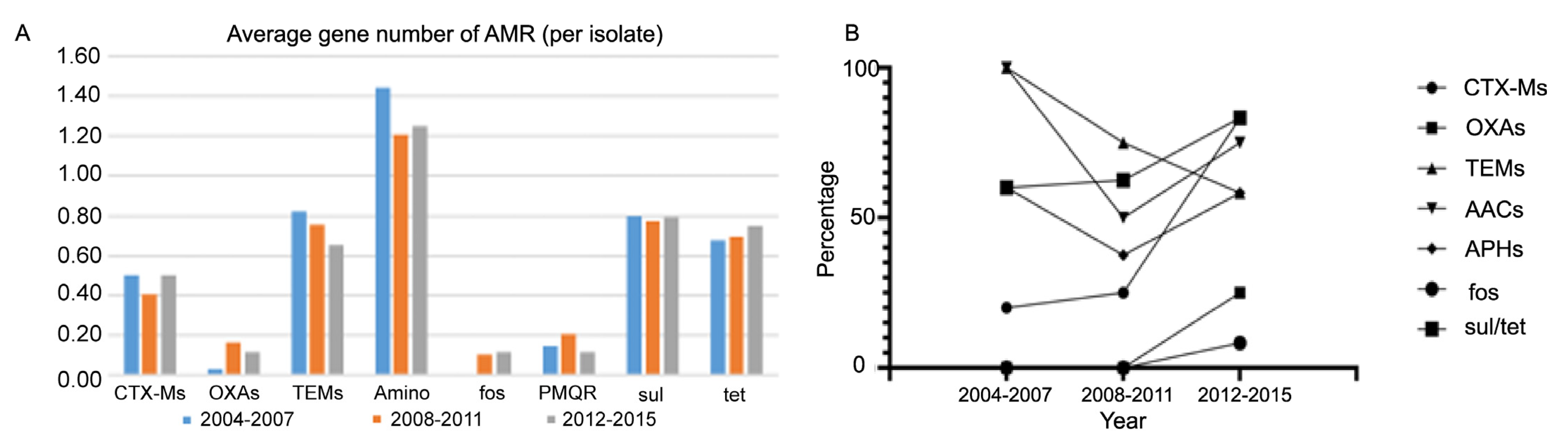
3.5. The Changing Pattern of Antibiotic-Resistant Gene Content over Time
3.6. Plasmid-Mediated Antibiotic-Resistant Genes
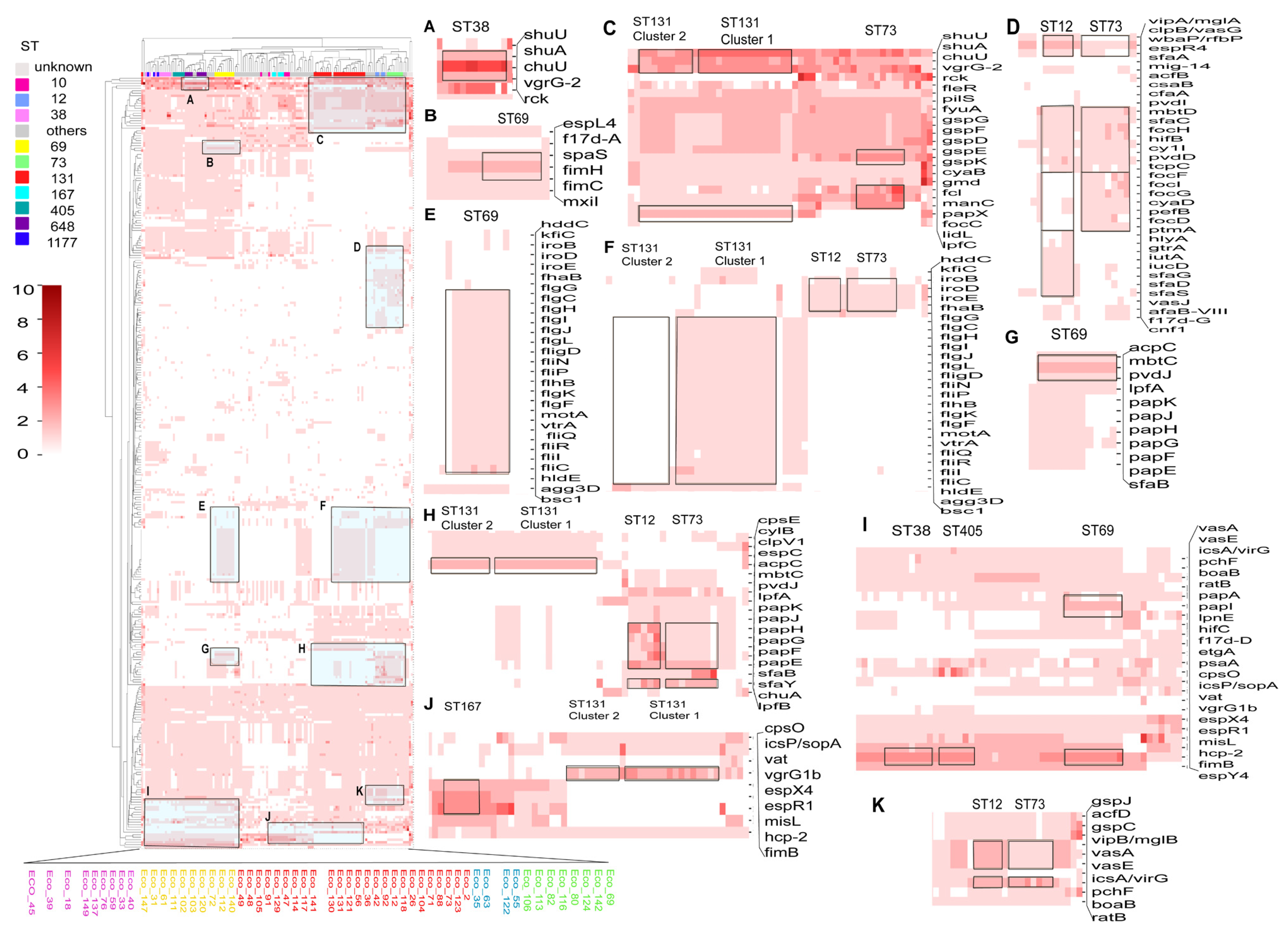
3.7. Comparison of Virulence Genes Among Different STs
3.8. Comparison of Secretion Systems and Genomic Islands Among Different STs
4. Discussion
4.1. Evolution of Antibiotic Resistance and Virulence
4.2. Convergence of Resistance and Virulence
4.3. Implications of Horizontal Gene Transfer
4.4. Clinical and Public Health Implications
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Xiao, S.; Tang, C.; Zeng, Q.; Xue, Y.; Chen, Q.; Chen, E.; Han, L. Antimicrobial Resistance and Molecular Epidemiology of Escherichia coli from Bloodstream Infection in Shanghai, China, 2016–2019. Front. Med. 2021, 8, 803837. [Google Scholar] [CrossRef] [PubMed]
- Bai, C.; Zhang, X.; Yang, D.; Li, D.; Feng, H.; Li, Y. Clinical Analysis of Bloodstream Infection of Escherichia coli in Patients with Pancreatic Cancer from 2011 to 2019. Can. J. Infect. Dis. Med. Microbiol. 2022, 2022, 1338188. [Google Scholar] [CrossRef]
- Blandy, O.; Honeyford, K.; Gharbi, M.; Thomas, A.; Ramzan, F.; Ellington, M.J.; Hope, R.; Holmes, A.H.; Johnson, A.P.; Aylin, P.; et al. Factors that impact on the burden of Escherichia coli bacteraemia: Multivariable regression analysis of 2011–2015 data from West London. J. Hosp. Infect. 2019, 101, 120–128. [Google Scholar] [CrossRef]
- CHINET. 2021. Available online: http://www.chinets.com (accessed on 15 August 2024).
- Zhao, S.Y.; Wang, Y.C.; Xiao, S.Z.; Jiang, X.F.; Guo, X.K.; Ni, Y.X.; Han, L.Z. Drug susceptibility and molecular epidemiology of Escherichia coli in bloodstream infections in Shanghai, China, 2011–2013. Infect. Dis. 2015, 47, 310–318. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Li, P.; Yu, X.; Zhang, X.; Guo, Q.; Xu, X.; Wang, M.; Wang, M. Molecular Characteristics of Escherichia coli Causing Bloodstream Infections During 2010–2015 in a Tertiary Hospital, Shanghai, China. Infect. Drug Resist. 2021, 14, 2079–2086. [Google Scholar] [CrossRef] [PubMed]
- Puvača, N.; de Llanos Frutos, R. Antimicrobial Resistance in Escherichia coli Strains Isolated from Humans and Pet Animals. Antibiotics 2021, 10, 69. [Google Scholar] [CrossRef]
- Paitan, Y. Current Trends in Antimicrobial Resistance of Escherichia coli. Curr. Top. Microbiol. Immunol. 2018, 416, 181–211. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Jaimes, T.; Monroy-Pérez, E.; Garzón, J.; Morales-Espinosa, R.; Navarro-Ocaña, A.; García-Cortés, L.R.; Nolasco-Alonso, N.; Gaytán-Núñez, F.K.; Moreno-Noguez, M.; Vaca-Paniagua, F.; et al. High Virulence and Multidrug Resistance of Escherichia coli Isolated in Periodontal Disease. Microorganisms 2022, 11, 45. [Google Scholar] [CrossRef]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. Horizontal gene transfer and the evolution of transcriptional regulation in Escherichia coli. Genome Biol. 2008, 9, R4. [Google Scholar] [CrossRef]
- Frazão, N.; Sousa, A.; Lässig, M.; Gordo, I. Horizontal gene transfer overrides mutation in Escherichia coli colonizing the mammalian gut. Proc. Natl. Acad. Sci. USA 2019, 116, 17906–17915. [Google Scholar] [CrossRef]
- Pakbin, B.; Brück, W.M.; Rossen, J.W.A. Virulence Factors of Enteric Pathogenic Escherichia coli: A Review. Int. J. Mol. Sci. 2021, 22, 9922. [Google Scholar] [CrossRef] [PubMed]
- Geslain, G.; Cointe, A.; Bidet, P.; Courroux, C.; Abasse, S.; Mariani, P.; Bonacorsi, S. Fatal Meningitis from Shiga Toxin-Producing Escherichia coli in 2 Full-Term Neonates, France. Emerg. Infect. Dis. 2023, 29, 1703–1705. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.R. Virulence factors in Escherichia coli urinary tract infection. Clin. Microbiol. Rev. 1991, 4, 80–128. [Google Scholar] [CrossRef] [PubMed]
- Asadi Karam, M.R.; Habibi, M.; Bouzari, S. Urinary tract infection: Pathogenicity, antibiotic resistance and development of effective vaccines against Uropathogenic Escherichia coli. Mol. Immunol. 2019, 108, 56–67. [Google Scholar] [CrossRef]
- Roshani, M.; Taheri, M.; Goodarzi, A.; Yosefimashouf, R.; Shokoohizadeh, L. Evaluation of antibiotic resistance, toxin-antitoxin systems, virulence factors, biofilm-forming strength and genetic linkage of Escherichia coli strains isolated from bloodstream infections of leukemia patients. BMC Microbiol. 2023, 23, 327. [Google Scholar] [CrossRef]
- Kim, B.; Kim, J.H.; Lee, Y. Virulence Factors Associated with Escherichia coli Bacteremia and Urinary Tract Infection. Ann. Lab. Med. 2022, 42, 203–212. [Google Scholar] [CrossRef]
- Wang, M.H.; Kim, K.S. Cytotoxic necrotizing factor 1 contributes to Escherichia coli meningitis. Toxins 2013, 5, 2270–2280. [Google Scholar] [CrossRef]
- Sarowska, J.; Futoma-Koloch, B.; Jama-Kmiecik, A.; Frej-Madrzak, M.; Ksiazczyk, M.; Bugla-Ploskonska, G.; Choroszy-Krol, I. Virulence factors, prevalence and potential transmission of extraintestinal pathogenic Escherichia coli isolated from different sources: Recent reports. Gut Pathog. 2019, 11, 10. [Google Scholar] [CrossRef]
- Ben Zakour, N.L.; Alsheikh-Hussain, A.S.; Ashcroft, M.M.; Khanh Nhu, N.T.; Roberts, L.W.; Stanton-Cook, M.; Schembri, M.A.; Beatson, S.A. Sequential Acquisition of Virulence and Fluoroquinolone Resistance Has Shaped the Evolution of Escherichia coli ST131. mBio 2016, 7, e00347-16. [Google Scholar] [CrossRef]
- Mora-Rillo, M.; Fernandez-Romero, N.; Navarro-San Francisco, C.; Diez-Sebastian, J.; Romero-Gomez, M.P.; Fernandez, F.A.; Lopez, J.R.A.; Mingorance, J. Impact of virulence genes on sepsis severity and survival in Escherichia coli bacteremia. Virulence 2015, 6, 93–100. [Google Scholar] [CrossRef]
- Nicolas-Chanoine, M.H.; Bertrand, X.; Madec, J.Y. Escherichia coli ST131, an intriguing clonal group. Clin. Microbiol. Rev. 2014, 27, 543–574. [Google Scholar] [CrossRef] [PubMed]
- Rogers, B.A.; Sidjabat, H.E.; Paterson, D.L. Escherichia coli O25b-ST131: A pandemic, multiresistant, community-associated strain. J. Antimicrob. Chemother. 2011, 66, 1–14. [Google Scholar] [CrossRef]
- Taati Moghadam, M.; Mirzaei, M.; Fazel Tehrani Moghaddam, M.; Babakhani, S.; Yeganeh, O.; Asgharzadeh, S.; Farahani, H.E.; Shahbazi, S. The Challenge of Global Emergence of Novel Colistin-Resistant Escherichia coli ST131. Microb. Drug Resist. 2021, 27, 1513–1524. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Xiao, R.; Feng, J.; Wang, X.; Lai, J.; Kang, W.; Li, Y.; Zhu, X.; Ji, T.; Huang, X.; et al. Distribution of virulence genes and antimicrobial resistance of Escherichia coli isolated from hospitalized neonates: A multi-center study across China. Heliyon 2024, 10, e35991. [Google Scholar] [CrossRef]
- Wang, S.; Zhao, S.; Zhou, Y.; Jin, S.; Ye, T.; Pan, X. Antibiotic resistance spectrum of E. coli strains from different samples and age-grouped patients: A 10-year retrospective study. BMJ Open 2023, 13, e067490. [Google Scholar] [CrossRef]
- Huang, L.; Huang, C.; Yan, Y.; Sun, L.; Li, H. Urinary Tract Infection Etiological Profiles and Antibiotic Resistance Patterns Varied Among Different Age Categories: A Retrospective Study from a Tertiary General Hospital During a 12-Year Period. Front. Microbiol. 2021, 12, 813145. [Google Scholar] [CrossRef]
- Magiorakos, A.P.; Srinivasan, A.; Carey, R.B.; Carmeli, Y.; Falagas, M.E.; Giske, C.G.; Harbarth, S.; Hindler, J.F.; Kahlmeter, G.; Olsson-Liljequist, B.; et al. Multidrug-resistant, extensively drug-resistant and pandrug-resistant bacteria: An international expert proposal for interim standard definitions for acquired resistance. Clin. Microbiol. Infect. 2012, 18, 268–281. [Google Scholar] [CrossRef] [PubMed]
- Kallonen, T.; Brodrick, H.J.; Harris, S.R.; Corander, J.; Brown, N.M.; Martin, V.; Peacock, S.J.; Parkhill, J. Systematic longitudinal survey of invasive Escherichia coli in England demonstrates a stable population structure only transiently disturbed by the emergence of ST131. Genome Res. 2017, 27, 1437–1449. [Google Scholar] [CrossRef]
- Rodriguez, I.; Figueiredo, A.S.; Sousa, M.; Aracil-Gisbert, S.; Fernandez-de-Bobadilla, M.D.; Lanza, V.F.; Rodriguez, C.; Zamora, J.; Loza, E.; Mingo, P.; et al. A 21-Year Survey of Escherichia coli from Bloodstream Infections (BSI) in a Tertiary Hospital Reveals How Community-Hospital Dynamics of B2 Phylogroup Clones Influence Local BSI Rates. mSphere 2021, 6, e0086821. [Google Scholar] [CrossRef]
- Royer, G.; Darty, M.M.; Clermont, O.; Condamine, B.; Laouenan, C.; Decousser, J.W.; Vallenet, D.; Lefort, A.; de Lastours, V.; Denamur, E.; et al. Phylogroup stability contrasts with high within sequence type complex dynamics of Escherichia coli bloodstream infection isolates over a 12-year period. Genome Med. 2021, 13, 77. [Google Scholar] [CrossRef]
- Bogema, D.R.; McKinnon, J.; Liu, M.; Hitchick, N.; Miller, N.; Venturini, C.; Iredell, J.; Darling, A.E.; Roy Chowdury, P.; Djordjevic, S.P. Whole-genome analysis of extraintestinal Escherichia coli sequence type 73 from a single hospital over a 2 year period identified different circulating clonal groups. Microb. Genom. 2020, 6, e000255. [Google Scholar] [CrossRef] [PubMed]
- Bialek-Davenet, S.; Criscuolo, A.; Ailloud, F.; Passet, V.; Jones, L.; Delannoy-Vieillard, A.S.; Garin, B.; Le Hello, S.; Arlet, G.; Nicolas-Chanoine, M.H.; et al. Genomic definition of hypervirulent and multidrug-resistant Klebsiella pneumoniae clonal groups. Emerg. Infect. Dis. 2014, 20, 1812–1820. [Google Scholar] [CrossRef] [PubMed]
- Piazza, A.; Corbella, M.; Marchetti, V.M.; Merla, C.; Mileto, I.; Kuka, A.; Petazzoni, G.; Gaiarsa, S.; Migliavacca, R.; Baldanti, F.; et al. Clinical isolates of ST131 blaOXA-244-positive Escherichia coli, Italy, December 2022 to July 2023. Euro Surveill. 2024, 29, 2400073. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Feng, C.; Jia, H.; Yang, Q.; Zou, Q. Genetic Evolution of Antibiotic Resistance and Virulence Genes in Escherichia coli Isolates from a Chinese Hospital over a 12-Year Period. Microorganisms 2025, 13, 954. https://doi.org/10.3390/microorganisms13040954
Feng C, Jia H, Yang Q, Zou Q. Genetic Evolution of Antibiotic Resistance and Virulence Genes in Escherichia coli Isolates from a Chinese Hospital over a 12-Year Period. Microorganisms. 2025; 13(4):954. https://doi.org/10.3390/microorganisms13040954
Chicago/Turabian StyleFeng, Chengjie, Hongbing Jia, Qian Yang, and Qinghua Zou. 2025. "Genetic Evolution of Antibiotic Resistance and Virulence Genes in Escherichia coli Isolates from a Chinese Hospital over a 12-Year Period" Microorganisms 13, no. 4: 954. https://doi.org/10.3390/microorganisms13040954
APA StyleFeng, C., Jia, H., Yang, Q., & Zou, Q. (2025). Genetic Evolution of Antibiotic Resistance and Virulence Genes in Escherichia coli Isolates from a Chinese Hospital over a 12-Year Period. Microorganisms, 13(4), 954. https://doi.org/10.3390/microorganisms13040954