
The Subgingival Microbial Composition in Health and Periodontitis with Different Probing Depths

 , and
, and
Abstract
1. Introduction
2. Materials and Methods
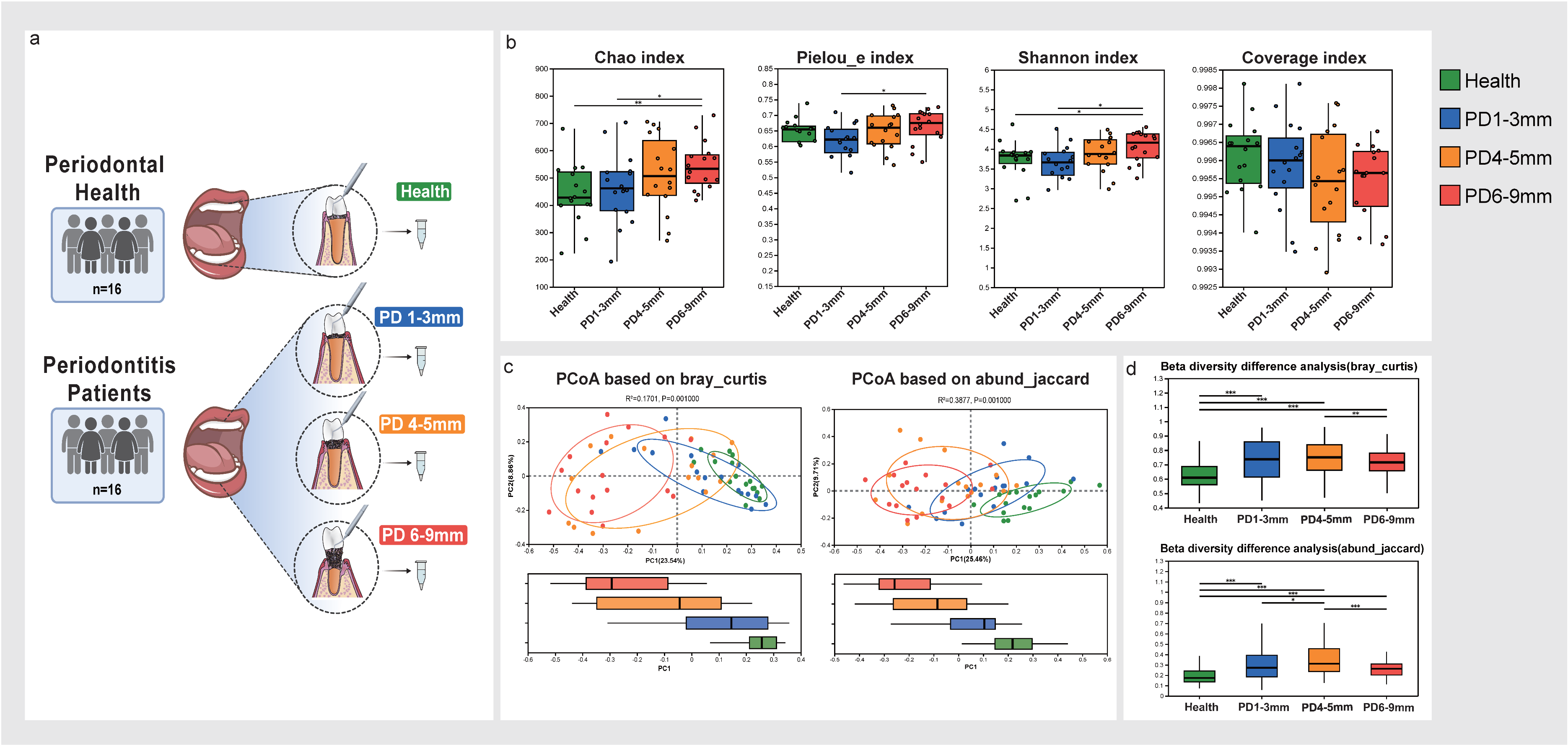
2.1. Study Population and Clinical Examination
2.2. Sample Collection and Processing
2.3. DNA Extraction, PCR Amplification, and Sequencing
2.4. Bioinformatic Analysis, Statistical Analysis, and Visualization
3. Results
3.1. Microbial Diversity Analysis in Health and Periodontitis Across Varying Probing Depths
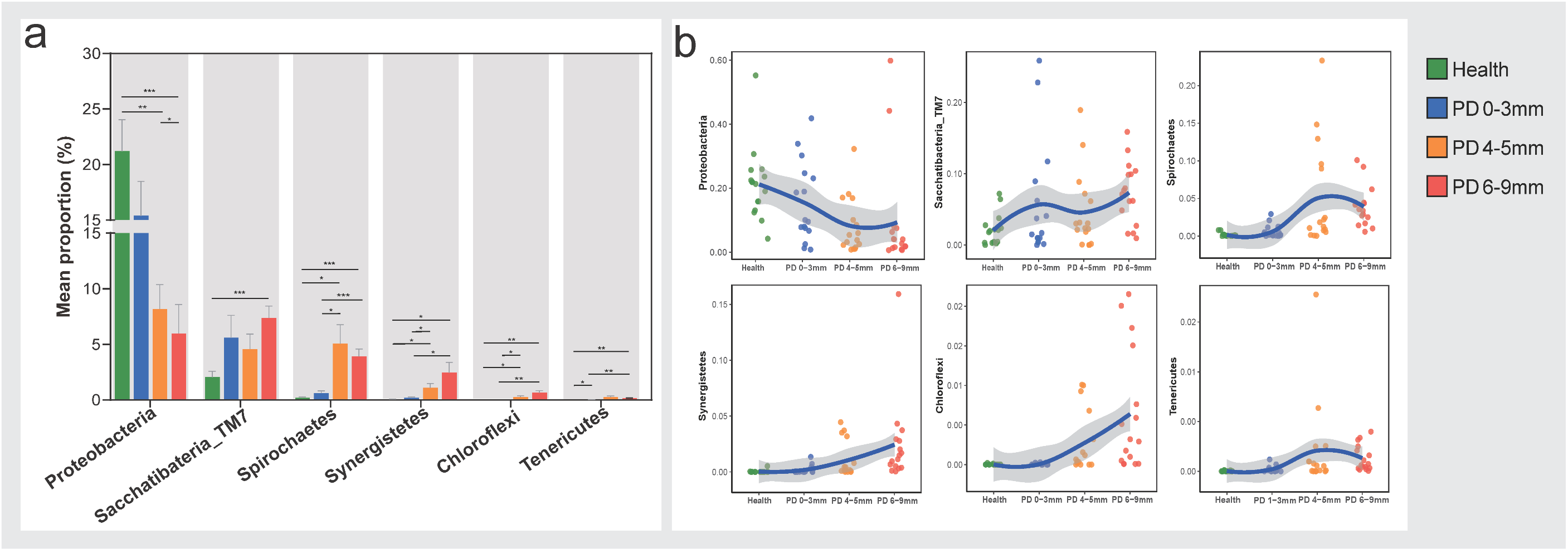
3.2. Compositions of Subgingival Microbiota at the Phylum Level
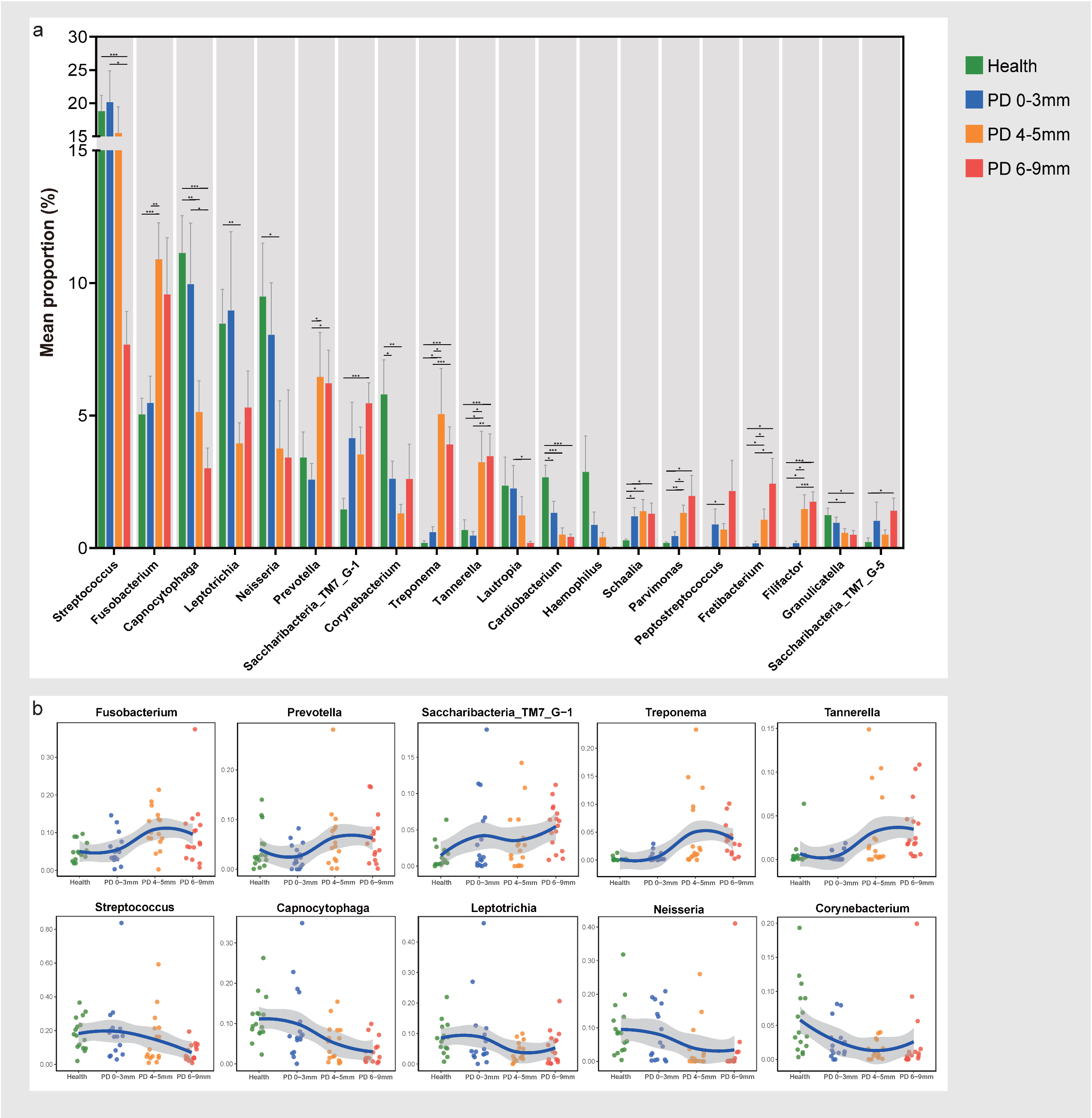
3.3. Compositions of Subgingival Microbiota at the Genus Level
3.4. Co-Occurrence Network Based on Species Level Revealed Different Interaction Patterns in Each Group
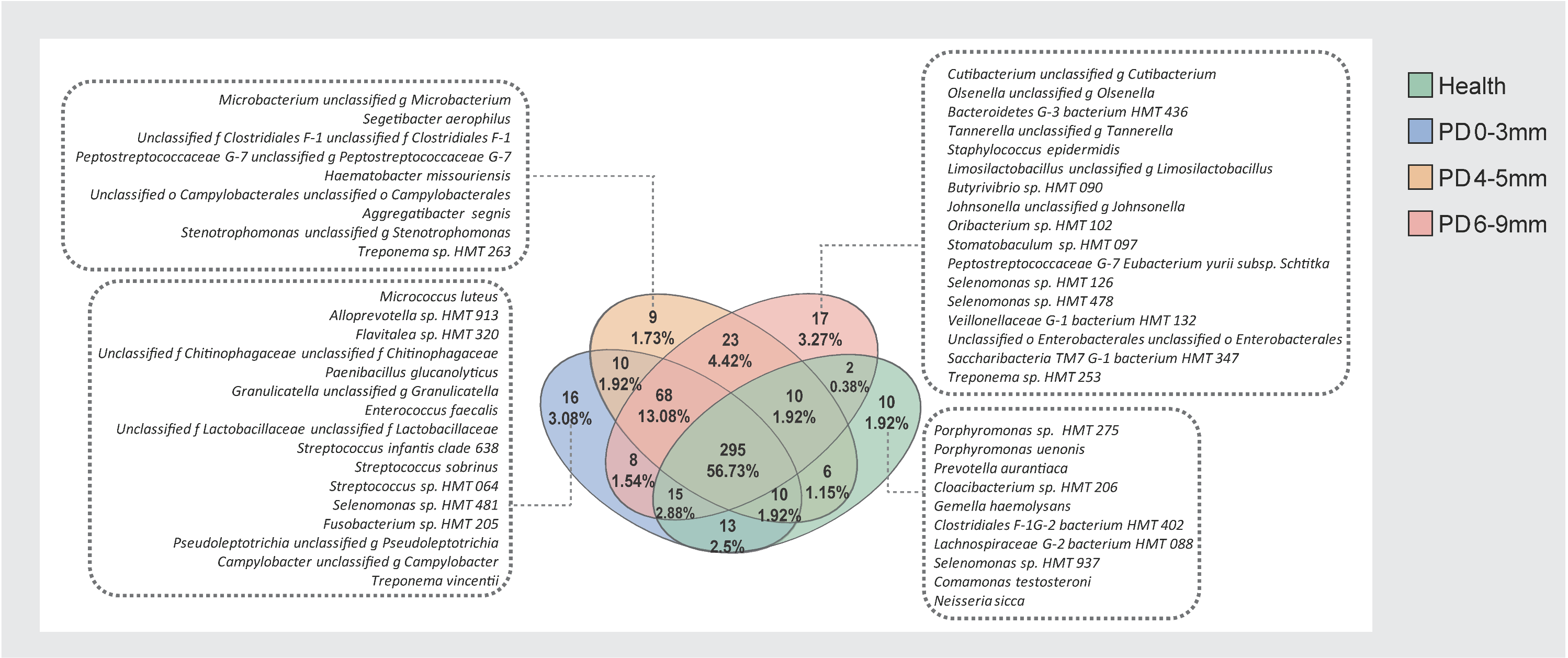
3.5. Core and Unique Microbial Species Across Health and Periodontitis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mombelli, A. Microbial colonization of the periodontal pocket and its significance for periodontal therapy. Periodontology 2000 2018, 76, 85–96. [Google Scholar] [CrossRef] [PubMed]
- Slots, J. Periodontitis: Facts, fallacies and the future. Periodontology 2000 2017, 75, 7–23. [Google Scholar] [CrossRef] [PubMed]
- Papapanou, P.N.; Sanz, M.; Buduneli, N.; Dietrich, T.; Feres, M.; Fine, D.H.; Flemmig, T.F.; Garcia, R.; Giannobile, W.V.; Graziani, F.; et al. Periodontitis: Consensus report of workgroup 2 of the 2017 World Workshop on the Classification of Periodontal and Peri-Implant Diseases and Conditions. J. Clin. Periodontol. 2018, 89, S162–S170. [Google Scholar]
- Van der Velden, U. What exactly distinguishes aggressive from chronic periodontitis: Is it mainly a difference in the degree of bacterial invasiveness? Periodontology 2000 2017, 75, 24–44. [Google Scholar] [CrossRef]
- Berezow, A.B.; Darveau, R.P. Microbial shift and periodontitis. Periodontology 2000 2011, 55, 36–47. [Google Scholar] [CrossRef]
- Haffajee, A.D.; Socransky, S.S. Microbiology of periodontal diseases: Introduction. Periodontology 2000 2005, 38, 9–12. [Google Scholar] [CrossRef]
- Abusleme, L.; Dupuy, A.K.; Dutzan, N.; Silva, N.; Burleson, J.A.; Strausbaugh, L.D.; Gamonal, J.; Diaz, P.I. The subgingival microbiome in health and periodontitis and its relationship with community biomass and inflammation. ISME J. 2013, 7, 1016–1025. [Google Scholar] [CrossRef]
- Ng, E.; Tay, J.R.H.; Balan, P.; Ong, M.M.A.; Bostanci, N.; Belibasakis, G.N.; Seneviratne, C.J. Metagenomic sequencing provides new insights into the subgingival bacteriome and aetiopathology of periodontitis. J. Periodontal Res. 2021, 56, 205–218. [Google Scholar] [CrossRef]
- Iniesta, M.; Chamorro, C.; Ambrosio, N.; Marín, M.J.; Sanz, M.; Herrera, D. Subgingival microbiome in periodontal health, gingivitis and different stages of periodontitis. J. Clin. Periodontol. 2023, 50, 905–920. [Google Scholar] [CrossRef]
- Antezack, A.; Etchecopar-Etchart, D.; La Scola, B.; Monnet-Corti, V. New putative periodontopathogens and periodontal health-associated species: A systematic review and meta-analysis. J. Periodontal. Res. 2023, 58, 893–906. [Google Scholar] [CrossRef]
- Socransky, S.S.; Haffajee, A.D.; Cugini, M.A.; Smith, C.; Kent, R.L., Jr. Microbial complexes in subgingival plaque. J. Clin. Periodontol. 1998, 25, 134–144. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, G.V.O.; Mosley, G.A.; Ross, W.; Dagher, A.; Martins, B.; Fernandes, J.C.H. Revisiting Socransky’s Complexes: A Review Suggesting Updated New Bacterial Clusters (GF-MoR Complexes) for Periodontal and Peri-Implant Diseases and Conditions. Microorganisms 2024, 12, 2214. [Google Scholar] [CrossRef] [PubMed]
- Loesche, W.J.; Gusberti, F.; Mettraux, G.; Higgins, T.; Syed, S. Relationship between oxygen tension and subgingival bacterial flora in untreated human periodontal pockets. Infect. Immun. 1983, 42, 659–667. [Google Scholar] [CrossRef]
- Haffajee, A.D.; Socransky, S.S.; Goodson, J.M. Subgingival temperature (I). Relation to baseline clinical parameters. J. Clin. Periodontol. 1992, 19, 401–408. [Google Scholar] [CrossRef]
- Galgut, P.N. The relevance of pH to gingivitis and periodontitis. J. Int. Acad. Periodontol. 2001, 3, 61–67. [Google Scholar]
- Ge, X.; Rodriguez, R.; Trinh, M.; Gunsolley, J.; Xu, P. Oral microbiome of deep and shallow dental pockets in chronic periodontitis. PLoS ONE 2013, 8, e65520. [Google Scholar] [CrossRef]
- Cai, Z.; Lin, S.; Hu, S.; Zhao, L. Structure and Function of Oral Microbial Community in Periodontitis Based on Integrated Data. Front. Cell. Infect. Microbiol. 2021, 11, 663756. [Google Scholar] [CrossRef]
- Shi, M.; Wei, Y.; Hu, W.; Nie, Y.; Wu, X.; Lu, R. The Subgingival Microbiome of Periodontal Pockets with Different Probing Depths in Chronic and Aggressive Periodontitis: A Pilot Study. Front. Cell. Infect. Microbiol. 2018, 8, 124. [Google Scholar] [CrossRef]
- Pérez-Chaparro, P.J.; McCulloch, J.A.; Mamizuka, E.M.; Moraes, A.; Faveri, M.; Figueiredo, L.C.; Duarte, P.M.; Feres, M. Do different probing depths exhibit striking differences in microbial profiles? J. Clin. Periodontol. 2018, 45, 26–37. [Google Scholar] [CrossRef]
- He, J.; Liu, Y.; Xu, H.; Wei, X.; Chen, M. Insights into the variations in microbial community structure during the development of periodontitis and its pathogenesis. Clin. Oral. Investig. 2024, 28, 675. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Chen, F.; Cai, Y.; Chen, Z.; Luan, Q.; Yu, X. Measuring the subgingival microbiota in periodontitis patients: Comparison of the surface layer and the underlying layers. Microbiol. Immunol. 2020, 64, 99–112. [Google Scholar] [CrossRef] [PubMed]
- Caton, J.G.; Armitage, G.; Berglundh, T.; Chapple, I.L.C.; Jepsen, S.; Kornman, K.S.; Mealey, B.L.; Papapanou, P.N.; Sanz, M.; Tonetti, M.S. A new classification scheme for periodontal and peri-implant diseases and conditions—Introduction and key changes from the 1999 classification. J. Clin. Periodontol. 2018, 89, S1–S8. [Google Scholar]
- Feng, X.; Zhu, L.; Xu, L.; Meng, H.; Zhang, L.; Ren, X.; Lu, R.; Tian, Y.; Shi, D.; Wang, X. Distribution of 8 periodontal microorganisms in family members of Chinese patients with aggressive periodontitis. Arch. Oral Biol. 2015, 60, 400–407. [Google Scholar] [CrossRef]
- Liu, C.; Zhao, D.; Ma, W.; Guo, Y.; Wang, A.; Wang, Q.; Lee, D.J. Denitrifying sulfide removal process on high-salinity wastewaters in the presence of Halomonas sp. Appl. Microbiol. Biotechnol. 2016, 100, 1421–1426. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Magoč, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef]
- Edgar, R.C. UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 2013, 10, 996–998. [Google Scholar] [CrossRef]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef]
- Carr, A.; Diener, C.; Baliga, N.S.; Gibbons, S.M. Use and abuse of correlation analyses in microbial ecology. ISME J. 2019, 13, 2647–2655. [Google Scholar] [CrossRef]
- Aja, E.; Mangar, M.; Fletcher, H.M.; Mishra, A. Filifactor alocis: Recent Insights and Advances. J. Dent. Res. 2021, 100, 790–797. [Google Scholar] [CrossRef]
- Khemwong, T.; Kobayashi, H.; Ikeda, Y.; Matsuura, T.; Sudo, T.; Kano, C.; Mikami, R.; Izumi, Y. Fretibacterium sp. human oral taxon 360 is a novel biomarker for periodontitis screening in the Japanese population. PLoS ONE 2019, 14, e0218266. [Google Scholar] [CrossRef] [PubMed]
- Mark Welch, J.L.; Rossetti, B.J.; Rieken, C.W.; Dewhirst, F.E.; Borisy, G.G. Biogeography of a human oral microbiome at the micron scale. Proc. Natl. Acad. Sci. USA 2016, 113, E791–E800. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Chu, Y.; Liu, J.R.; Liu, W.Y.; Ouyang, X.Y. Subgingival Microbial Profiles of Young Chinese Adults with Stage I/II Periodontitis, Gingivitis and Periodontal Health Status. Chin. J. Dent. Res. 2021, 24, 167–175. [Google Scholar] [PubMed]
- Esparbès, P.; Legrand, A.; Bandiaky, O.N.; Chéraud-Carpentier, M.; Martin, H.; Montassier, E.; Soueidan, A. Subgingival Microbiota and Cytokines Profile Changes in Patients with Periodontitis: A Pilot Study Comparing Healthy and Diseased Sites in the Same Oral Cavities. Microorganisms 2021, 9, 2364. [Google Scholar] [CrossRef]
- He, X.; McLean, J.S.; Edlund, A.; Yooseph, S.; Hall, A.P.; Liu, S.Y.; Dorrestein, P.C.; Esquenazi, E.; Hunter, R.C.; Cheng, G.; et al. Cultivation of a human-associated TM7 phylotype reveals a reduced genome and epibiotic parasitic lifestyle. Proc. Natl. Acad. Sci. USA 2015, 112, 244–249. [Google Scholar] [CrossRef]
- Zhong, Q.; Liao, B.; Liu, J.; Shen, W.; Wang, J.; Wei, L.; Ma, Y.; Dong, P.T.; Bor, B.; McLean, J.S.; et al. Episymbiotic Saccharibacteria TM7x modulates the susceptibility of its host bacteria to phage infection and promotes their coexistence. Proc. Natl. Acad. Sci. USA 2024, 121, e2319790121. [Google Scholar] [CrossRef]
- Tian, J.; Utter, D.R.; Cen, L.; Dong, P.-T.; Shi, W.; Bor, B.; Qin, M.; McLean, J.S.; He, X. Acquisition of the arginine deiminase system benefits epiparasitic Saccharibacteria and their host bacteria in a mammalian niche environment. Proc. Natl. Acad. Sci. USA 2022, 119, e2114909119. [Google Scholar] [CrossRef]
- Lu, H.; Zou, P.; Zhang, Y.; Zhang, Q.; Chen, Z.; Chen, F. The sampling strategy of oral microbiome. iMeta 2022, 1, e23. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Health | Periodontitis | ||||
|---|---|---|---|---|---|
| Number | 16 | 16 | |||
| Gender | Male | 8 | 9 | ||
| Female | 8 | 7 | |||
| Age | 29.73 ± 5.51 | 41.69 ± 12.68 | |||
| Number of teeth | 28 | 26.94 ± 1.18 | |||
| Probing depth (PD) | ≤3 mm | 4.02 ± 1.87 | |||
| Percentage of PD | PD 0–3 mm | 100% | PD 0–3 mm | 55% | |
| PD 4–5 mm | 25% | ||||
| PD 6–9 mm | 19% | ||||
| PD ≥ 10 mm | 1% | ||||
| Bleeding on probing | 4.53 ± 2.77% | 83.42 ± 0.5% | |||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, J.; Geng, Y.; Guo, J.; Peng, J.; Xu, H.; Zhao, B.; Huang, S.; Qin, M.; Du, W.; Tian, J. The Subgingival Microbial Composition in Health and Periodontitis with Different Probing Depths. Microorganisms 2025, 13, 930. https://doi.org/10.3390/microorganisms13040930
Wang J, Geng Y, Guo J, Peng J, Xu H, Zhao B, Huang S, Qin M, Du W, Tian J. The Subgingival Microbial Composition in Health and Periodontitis with Different Probing Depths. Microorganisms. 2025; 13(4):930. https://doi.org/10.3390/microorganisms13040930
Chicago/Turabian StyleWang, Jingyan, Yiran Geng, Jing Guo, Jiahan Peng, He Xu, Bingqian Zhao, Shiyan Huang, Man Qin, Wenbin Du, and Jing Tian. 2025. "The Subgingival Microbial Composition in Health and Periodontitis with Different Probing Depths" Microorganisms 13, no. 4: 930. https://doi.org/10.3390/microorganisms13040930
APA StyleWang, J., Geng, Y., Guo, J., Peng, J., Xu, H., Zhao, B., Huang, S., Qin, M., Du, W., & Tian, J. (2025). The Subgingival Microbial Composition in Health and Periodontitis with Different Probing Depths. Microorganisms, 13(4), 930. https://doi.org/10.3390/microorganisms13040930