
Hirudo verbana Microbiota Dynamics: A Key Factor in Hirudotherapy-Related Infections?

 ,
,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Leeches
2.2. Collection of Samples from the Leeches
2.3. DNA Extraction and Pools
2.4. PCR Amplification and Sequencing (Next-Generation Sequencing (NGS))
2.5. Bioinformatics Analyses
2.6. Statistical Analysis
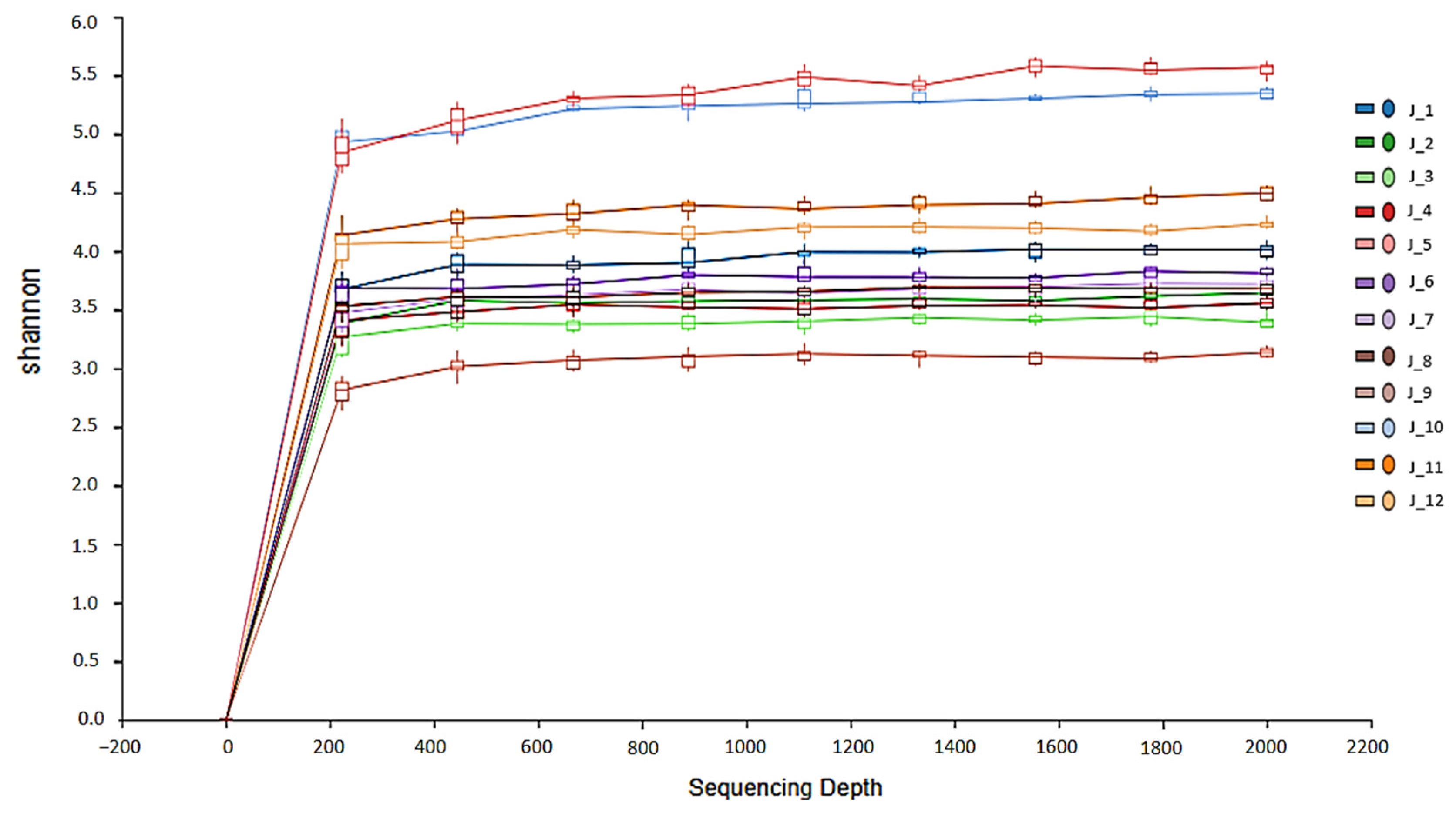
3. Results
3.1. Microbiota
3.2. Relative Abundance of Bacterial Taxa at Phylum Level
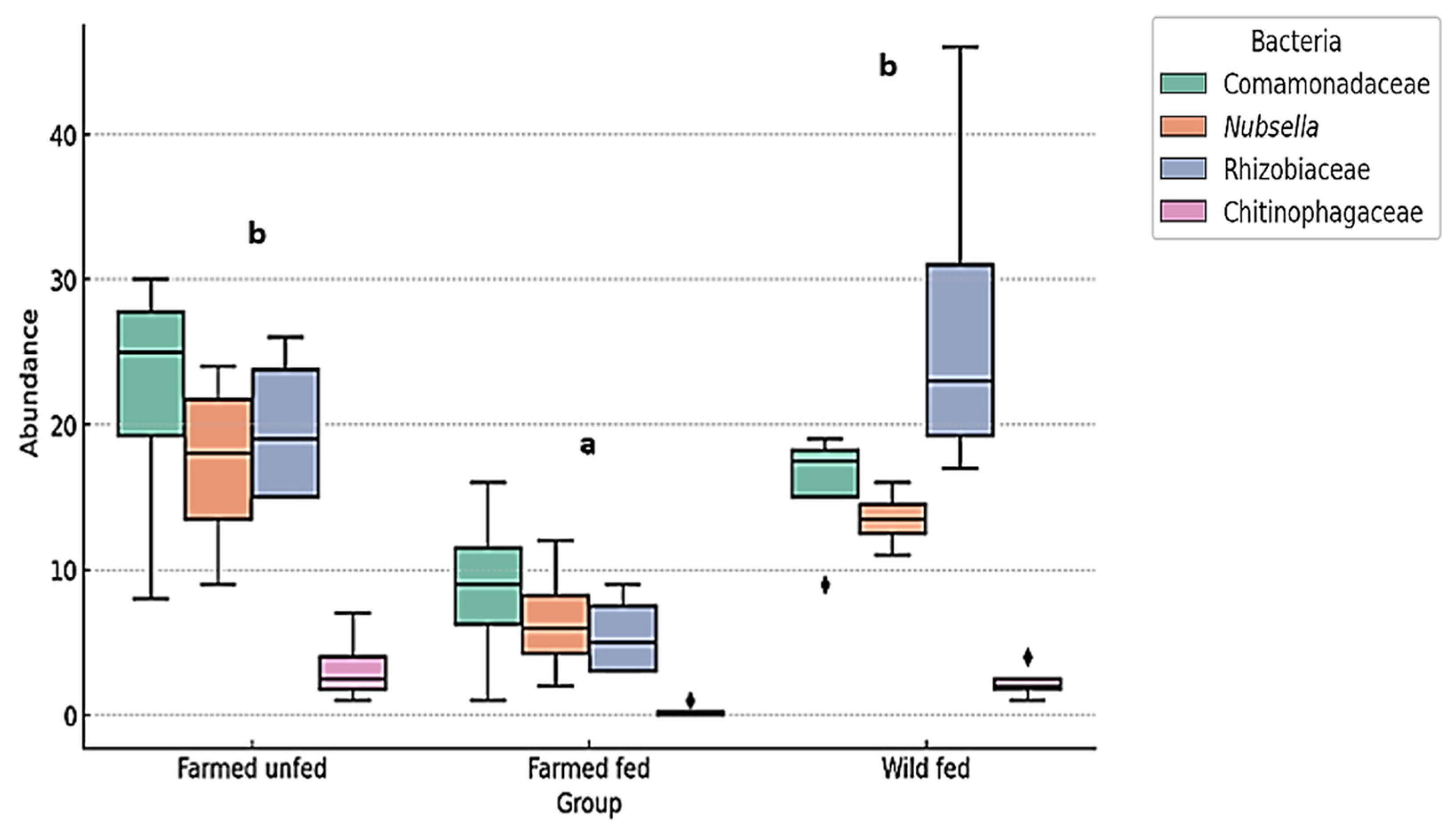
3.3. Relative Abundance (%) of Bacteria at Family and Genus Level
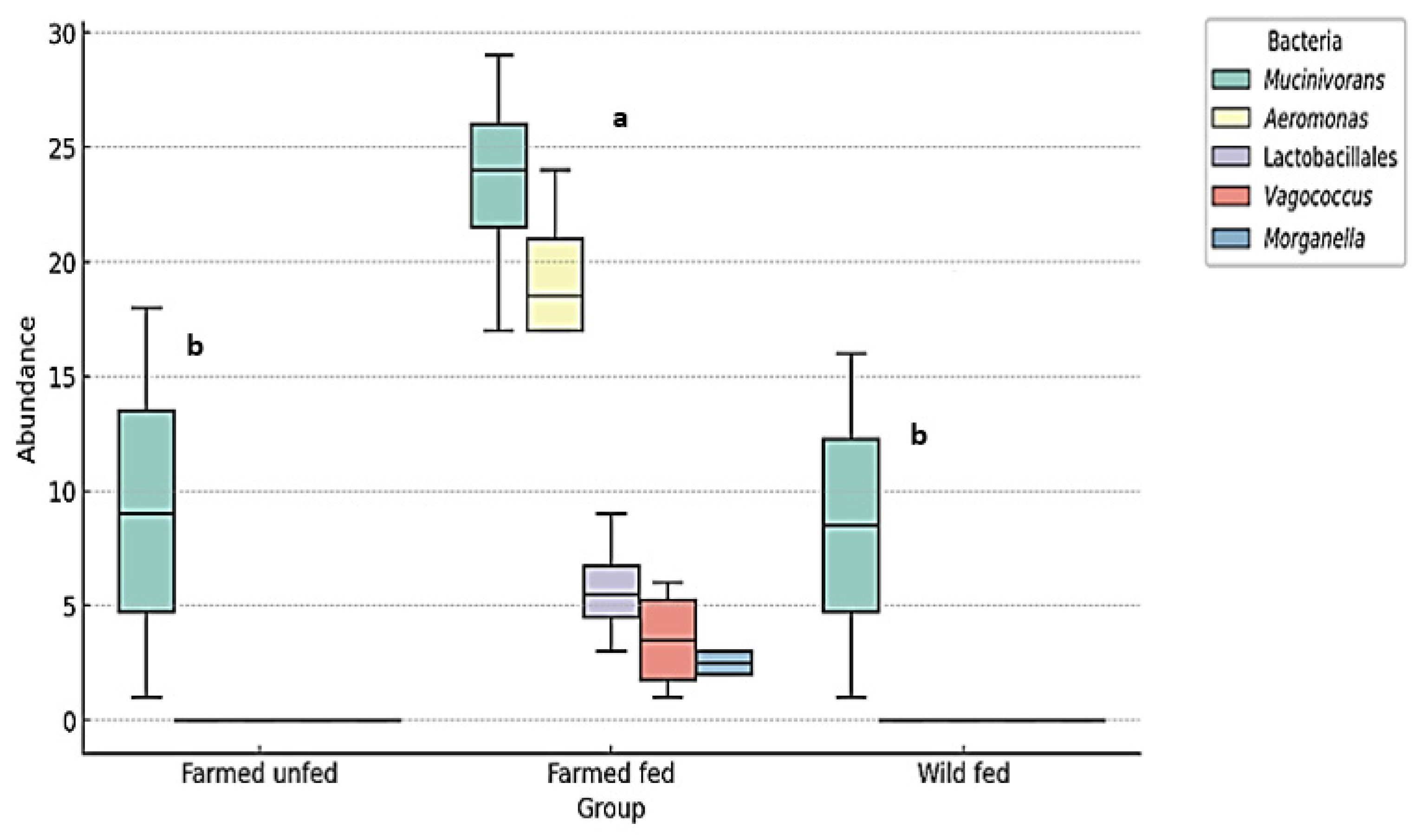
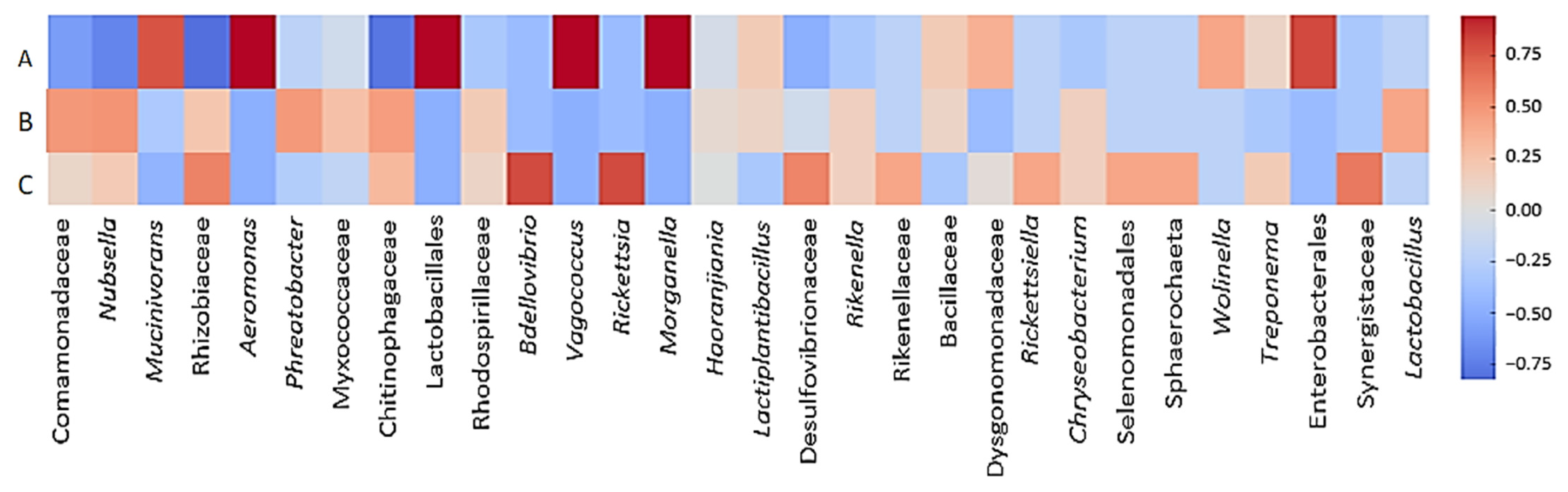
3.4. Opportunistic Pathogenic Bacteria That May Cause Infection During Hirudotherapy
4. Discussion
4.1. H. verbana Microbiota Diversity
4.2. Aeromonas and Mucinivorans Abundance
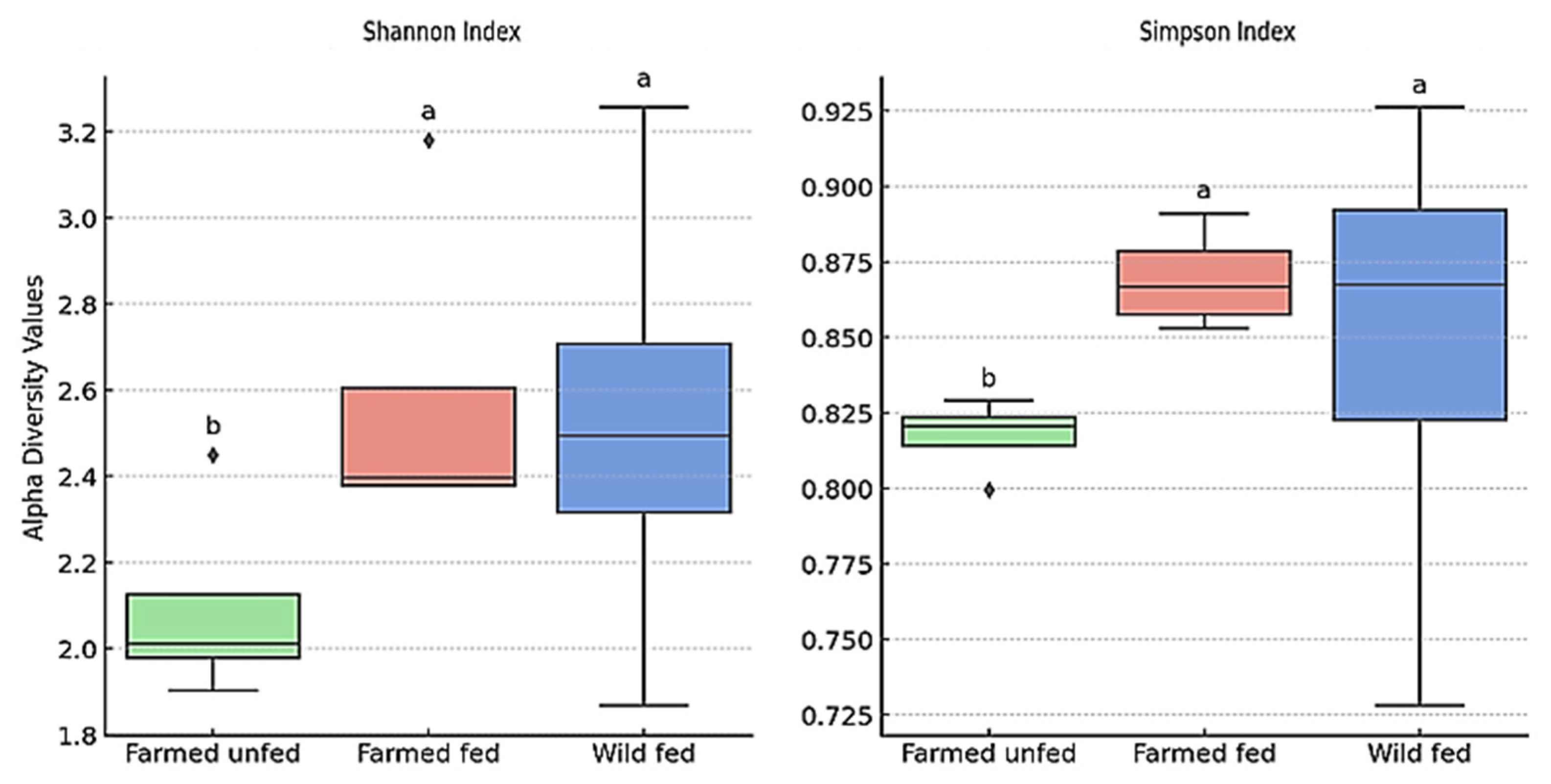
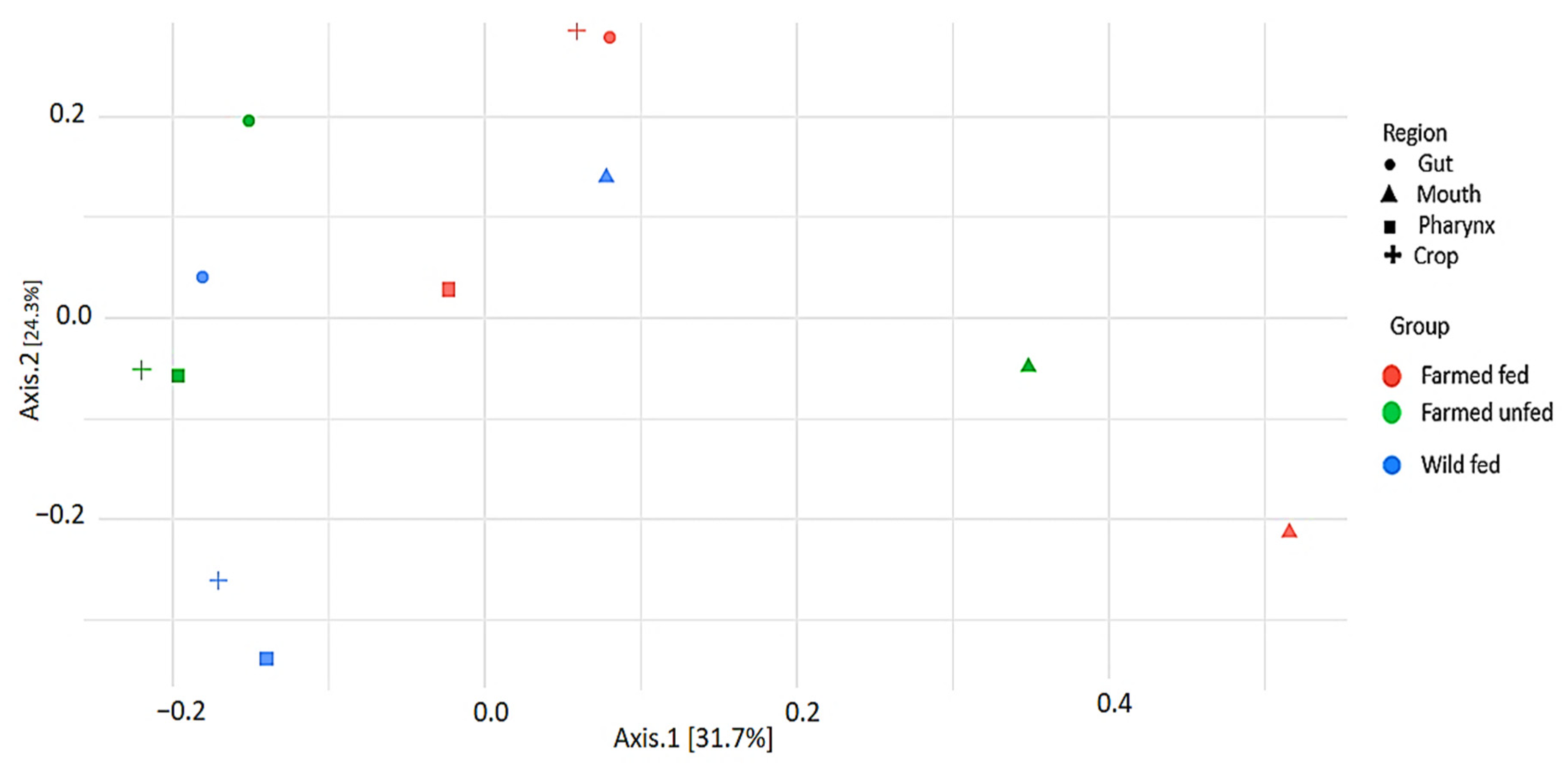
4.3. Beta and Alpha Diversity
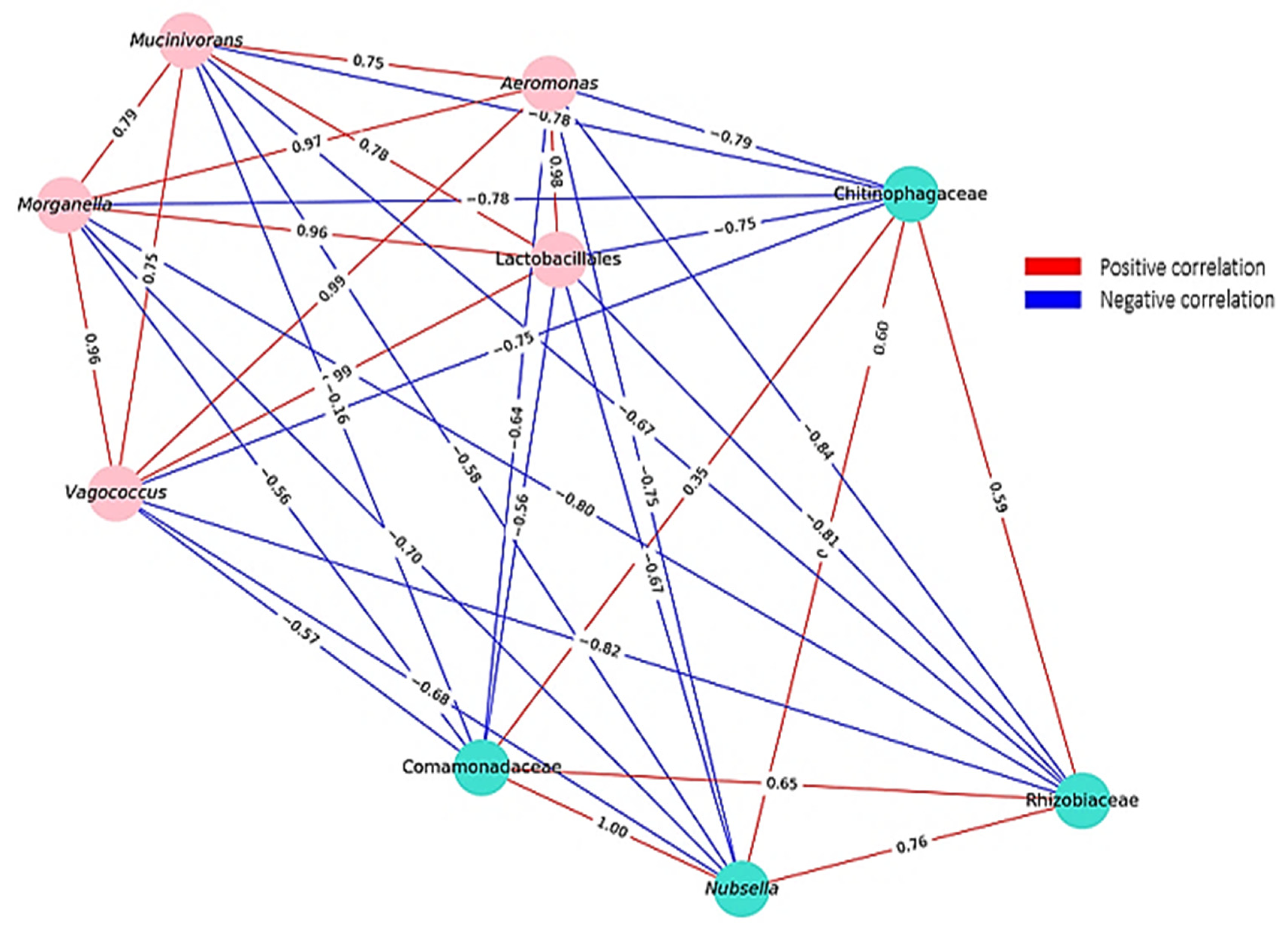
4.4. Correlation Dynamics
4.5. Infection Risk Assessment of Hirudotherapy
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Suhov, K. Clinical Hirudotherapy: A Practical Guide; Neformat: Moscow, Russia, 2018. [Google Scholar]
- Savinov, B.A. Hirudotherapy. Guidebook for Doctors; Medicine: Moscow, Russia, 2004. [Google Scholar]
- Hackenberger, P.N.; Janis, J.E. A comprehensive review of medicinal leeches in plastic and reconstructive surgery. Plast. Reconstr. Surg. Glob. Open 2019, 7, e2555. [Google Scholar] [CrossRef] [PubMed]
- Mumcuoglu, K.Y.; Pidhorz, C.; Cohen, R.; Ofek, A.; Lipton, H.A. The use of the medicinal leech, Hirudo medicinalis, in reconstructive plastic surgery. Internet J. Plast. Surg. 2007, 4, 2. Available online: https://ispub.com/IJPS/4/2/3993 (accessed on 13 April 2025).
- Utevsky, S.; Zagmajster, M.; Atemasov, A.; Zinenko, O.; Utevska, O.; Utevsky, A.; Trontelj, P. Distribution and status of medicinal leeches (genus Hirudo) in the Western Palaearctic: Anthropogenic, ecological, or historical effects? Aquat. Conserv. Mar. Freshwat. Ecosyst. 2010, 20, 198–210. [Google Scholar] [CrossRef]
- Sağlam, N. Internal and external morphological characteristics of the medicinal leech species Hirudo sulukii and Hirudo verbana. Turkiye Parazitol. Derg. 2019, 43, 204–209. [Google Scholar] [CrossRef] [PubMed]
- Faleh, N.; Hassan, Z.N.; Shafeeq, M.A.A. Leeches review: Biology, ecology, and medical importance. Indian J. Public Health Res. Dev. 2019, 10, 2587–2590. [Google Scholar] [CrossRef]
- Marden, J.N.; McClure, E.A.; Beka, L.; Graf, J. Host matters: Medicinal leech digestive-tract symbionts and their pathogenic potential. Front. Microbiol. 2016, 7, 1569. [Google Scholar] [CrossRef]
- Worthen, P.L.; Gode, C.J.; Graf, J. Culture-independent characterization of the digestive-tract microbiota of the medicinal leech reveals a tripartite symbiosis. Appl. Environ. Microbiol. 2006, 72, 4775–4781. [Google Scholar] [CrossRef]
- Mumcuoglu, K.Y.; Huberman, L.; Cohen, R.; Temper, V.; Adler, A.; Galun, R.; Block, C. Elimination of symbiotic Aeromonas spp. from the intestinal tract of the medicinal leech, Hirudo medicinalis, using ciprofloxacin feeding. Clin. Microbiol. Infect. 2010, 16, 563–567. [Google Scholar] [CrossRef]
- Maltz, M.A.; Bomer, L.; Lapierre, P.; Morrison, H.G.; McClure, E.A.; Sogin, M.L.; Graf, J. Metagenomic analysis of the medicinal leech gut microbiota. Front. Microbiol. 2014, 5, 151. [Google Scholar] [CrossRef]
- Tasiemski, A.; Massol, F.; Cuvillier-Hot, V.; Boidin-Wichlacz, C.; Roger, E.; Rodet, F.; Fournier, I.; Thomas, F.; Salzet, M. Reciprocal immune benefit based on complementary production of antibiotics by the leech Hirudo verbana and its gut symbiont Aeromonas veronii. Sci. Rep. 2015, 5, 17498. [Google Scholar] [CrossRef]
- Neupane, S.; Modry, D.; Pafčo, B.; Zurek, L. Bacterial community of the digestive tract of the European medicinal leech (Hirudo verbana) from the Danube River. Microb. Ecol. 2019, 77, 1082–1090. [Google Scholar] [CrossRef]
- Bomar, L.; Stephens, W.Z.; Nelson, M.C.; Velle, K.; Guillemin, K.; Graf, J. Draft genome sequence of Aeromonas veronii Hm21, an asymbiotic isolate from the medicinal leech digestive tract. Genome Announc. 2013, 1, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Nelson, M.C.; Bomar, L.; Maltz, M.; Graf, J. Mucinivorans hirudinis gen. nov., sp. nov., an anaerobic, mucin-degrading bacterium isolated from the digestive tract of the medicinal leech, Hirudo verbana. Int. J. Syst. Evol. Microbiol. 2015, 65, 990–995. [Google Scholar] [CrossRef]
- Bauters, T.G.M.; Buyle, F.M.A.; Verschraegen, G.; Vermis, K.; Vogelaers, D.; Claeys, G.; Robays, H. Infection risk related to the use of medicinal leeches. Pharm. World Sci. 2007, 29, 122–125. [Google Scholar] [CrossRef]
- Whitaker, I.S.; Josty, I.C.; Hawkins, S.; Azzopardi, E.; Naderi, N.; Graf, J.; Pharm, L.D.B. Medicinal leeches and the microsurgeon: A four-year study, clinical series and risk-benefit review. Microsurgery 2011, 31, 281–287. [Google Scholar] [CrossRef]
- Brauer, P.R.; Saadah, M.; Fritz, M.A.; Wu, S.S.; Lamarre, E.D. Analysis of antibiotic-resistant infections associated with hirudotherapy. Am. J. Otolaryngol. 2024, 45, 104500. [Google Scholar] [CrossRef]
- Sağlam, N. Identification Key to the Freshwater and Marine Leeches; Fırat Üniversitesi Basım Evi: Elazığ, Turkey, 2004. [Google Scholar]
- Sağlam, N. Leech Biology and Breeding Techniques; Fırat Üniversitesi Basım Evi: Elazığ, Turkey, 2004. [Google Scholar]
- Sawyer, R.T. Leech Biology and Behavior; Oxford University Press/Clarendon Press: Oxford, UK, 1986. [Google Scholar]
- Davies, R.W. Annelida: Leeches, Polychaetes, and Acanthobdellids. In Ecology and Classification of North American Freshwater Invertebrates; Thorp, J.H., Covich, A.P., Eds.; Academic Press: San Diego, CA, USA, 1991. [Google Scholar]
- Barnes, R.D. Invertebrate Zoology; W.B. Saunders Company: Philadelphia, PA, USA, 1974. [Google Scholar]
- Klindworth, A.; Pruesse, E.; Schweer, T.; Peplies, C.; Quast, C.; Horn, M.; Glöckner, F.O. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 2013, 41, e1. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. Cambridge: Babraham Bioinformatics, Babraham Institute. 2010. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 24 January 2025).
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [PubMed]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J. Reproducible, interactive, scalable, and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Bokulich, N.A.; Kaehler, B.D.; Rideout, J.R.; Dillon, M.; Bolyen, E.; Knight, R.; Huttley, G.A.; Caporaso, J.G. Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin. Microbiome 2018, 6, 90. [Google Scholar] [CrossRef]
- Robeson, M.S., II; O’Rourke, D.R.; Kaehler, B.D.; Ziemski, M.; Dillon, M.R.; Foster, J.T.; Bokulich, N.A. RESCRIPt: Reproducible sequence taxonomy reference database management for the masses. bioRxiv 2020. [CrossRef]
- Graf, J.; Kikuchi, Y.; Rio, R.V. Leeches and their microbiota: Naturally simple symbiosis models. Trends Microbiol. 2006, 14, 365–371. [Google Scholar] [CrossRef]
- Kikuchi, Y.; Graf, J. Spatial and temporal population dynamics of a naturally occurring two-species microbial community inside the digestive tract of the medicinal leech. Appl. Environ. Microbiol. 2007, 73, 1984–1991. [Google Scholar] [CrossRef]
- McClure, E.A.; Nelson, M.C.; Lin, A.; Graf, J. Macrobdella decora: Old World leech gut microbial community structure conserved in a New World leech. Appl. Environ. Microbiol. 2021, 87, e02082-20. [Google Scholar] [CrossRef] [PubMed]
- Nelson, M.C.; Graf, J. Bacterial symbioses of the medicinal leech Hirudo verbana. Gut Microbes 2012, 3, 322–331. [Google Scholar] [CrossRef] [PubMed]
- Whitaker, I.S.; Maltz, M.; Siddall, M.E.; Graf, J. Characterization of the digestive tract microbiota of Hirudo orientalis (medicinal leech) and antibiotic resistance profile. Plast. Reconstr. Surg. 2014, 133, 408e–418e. [Google Scholar] [CrossRef]
- Asker, D.; Beppu, T.; Ueda, K. Nubsella zeaxanthinifaciens gen. nov., sp. nov., a zeaxanthin-producing bacterium of the family Sphingobacteriaceae isolated from freshwater. Int. J. Syst. Evol. Microbiol. 2008, 58 Pt 3, 601. [Google Scholar] [CrossRef]
- Willems, A. The family Comamonadaceae. In The Prokaryotes, 4th ed.; Rosenberg, E., DeLong, E.F., Lory, S., Stackebrandt, E., Thompson, F., Eds.; Springer Press: Berlin/Heidelberg, Germany, 2013. [Google Scholar]
- Alves, L.M.; de Souza, J.A.; Varani, A.M.; de Macedo Lemos, E.G. The family Rhizobiaceae. In The Prokaryotes, 4th ed.; Rosenberg, E., DeLong, E.F., Lory, S., Stackebrandt, E., Thompson, F., Eds.; Springer Press: Berlin/Heidelberg, Germany, 2013. [Google Scholar]
- Baldani, J.I.; Videira, S.S.; Teixeira, K.R.S.; Reis, V.M.; de Oliveira, A.L.M.; de Souza, E.S.M.; Pedraza, R.; Baldani, V.L.D. The family Rhodospirillaceae. In The Prokaryotes, 4th ed.; Rosenberg, E., DeLong, E.F., Lory, S., Stackebrandt, E., Thompson, F., Eds.; Springer Press: Berlin/Heidelberg, Germany, 2013. [Google Scholar]
- Baek, K.; Choi, A. Complete genome sequence of Phreatobacter sp. strain NMCR1094, a formate-utilizing bacterium isolated from a freshwater stream. Microbiol. Resour. Announc. 2019, 8, e00860-19. [Google Scholar] [CrossRef] [PubMed]
- Garcia, R.; Müller, R. The family Myxococcaceae. In The Prokaryotes, 4th ed.; Rosenberg, E., DeLong, E.F., Lory, S., Stackebrandt, E., Thompson, F., Eds.; Springer Press: Berlin/Heidelberg, Germany, 2013. [Google Scholar]
- Graf, J. Symbiosis of Aeromonas veronii biovar sobria and Hirudo medicinalis, the medicinal leech: A novel model for digestive tract associations. Infect. Immun. 1999, 67, 1–7. [Google Scholar] [CrossRef]
- Mumcuoglu, K.Y. Recommendations for the use of leeches in reconstructive plastic surgery. Evid. Based Complement. Altern. Med. 2014, 2014, 205929. [Google Scholar] [CrossRef]
- Tian, Q.; Cheng, B.; Wu, Z.; Ji, H.; Shao, G.; Gao, T.; Zhang, X.; Liu, F. Microbial-feeding interactions reveal the effects of feeding blood on the gut microbiota of the aquaculture leech (Hirudo nipponica). Isr. J. Aquacult—Bamidgeh 2023, 75, 1838433. [Google Scholar] [CrossRef]
- Bomar, L.; Graf, J. Investigation into the physiologies of Aeromonas veronii in vitro and inside the digestive tract of the medicinal leech using RNA-seq. Biol. Bull. 2012, 223, 155–166. [Google Scholar] [CrossRef] [PubMed]
- Lent, C.M.; Fliegner, K.H.; Freedman, E.; Dickinson, M.H. Ingestive behaviour and physiology of the medicinal leech. J. Exp. Biol. 1988, 137, 513–527. [Google Scholar] [CrossRef]
- Joslin, J.; Biondich, A.; Walker, K.; Zanghi, N. A comprehensive review of hirudiniasis: From historic uses of leeches to modern treatments of their bites. Wilderness Environ. Med. 2017, 28, 355–361. [Google Scholar] [CrossRef]
- Perotti, M.A.; Clarke, H.K.; Turner, B.D.; Braig, H.R. Rickettsia as obligate and mycetomic bacteria. FASEB J. 2006, 20, 2372–2374. [Google Scholar] [CrossRef] [PubMed]
- Waso, M.; Reyneke, B.; Havenga, B.; Khan, S.; Khan, W. Insights into Bdellovibrio spp. mechanisms of action and potential applications. World J. Microbiol. Biotechnol. 2021, 37, 85. [Google Scholar] [CrossRef]
- Cao, H.; He, S.; Wang, H.; Hou, S.; Lu, L.; Yang, X. Bdellovibrio, potential biocontrol bacteria against pathogenic Aeromonas hydrophila. Vet. Microbiol. 2012, 154, 413–418. [Google Scholar] [CrossRef]
- Kuever, J. The family Desulfovibrionaceae. In The Prokaryotes, 4th ed.; Rosenberg, E., DeLong, E.F., Lory, S., Stackebrandt, E., Thompson, F., Eds.; Springer Press: Berlin/Heidelberg, Germany, 2013. [Google Scholar]
- Yörük, G.N.; Güner, A. Classification of lactic acid bacteria and the importance of Weissella species in food microbiology. Atatürk Üniversitesi Vet. Bil. Derg. 2011, 6, 163–176. (In Turkish) [Google Scholar]
- Inoue, R.; Tsuruta, T.; Nojima, İ.; Nakayama, K.; Tsukahara, T.; Yajima, T. Postnatal changes in the expression of genes for cryptdins 1–6 and the role of luminal bacteria in cryptdin gene expression in mouse small intestine. FEMS Immunol. Med. Microbiol. 2008, 52, 407–416. [Google Scholar] [CrossRef]
- Ryu, J.H.; Kim, S.H.; Lee, H.Y.; Bai, J.Y.; Nam, Y.; Bae, J.W.; Lee, D.G.; Shin, S.C.; Ha, E.M.; Lee, W.J. Innate immune homeostasis by the homeobox gene caudal and commensal-gut mutualism in Drosophila. Science 2008, 319, 777–782. [Google Scholar] [CrossRef]
- Riley, M.A.; Gordon, D.M. The ecological role of bacteriocins in bacterial competition. Trends Microbiol. 1999, 7, 129–133. [Google Scholar] [CrossRef] [PubMed]
- Braschler, T.R.; Merino, S.; Tomas, J.M.; Graf, J. Complement resistance is essential for colonization of the digestive tract of Hirudo medicinalis by Aeromonas strains. Appl. Environ. Microbiol. 2003, 69, 4268–4271. [Google Scholar] [CrossRef] [PubMed]
- Slesak, G.; Inthalath, S.; Dittrich, S.; Paris, D.H.; Newton, P.N. Leeches as further potential vectors for rickettsial infections. Proc. Natl. Acad. Sci. USA 2015, 112, E6593–E6599. [Google Scholar] [CrossRef]
- Kikuchi, Y.; Fukatsu, T. Rickettsia infection in natural leech population. Microb. Ecol. 2005, 49, 265–271. [Google Scholar] [CrossRef] [PubMed]
- Zinicola, M.; Lima, F.; Lima, S.; Machado, V.; Gomez, M.; Döpfer, D.; Guard, C.; Bicalho, R. Altered microbiomes in bovine digital dermatitis lesions, and the gut as a pathogen reservoir. PLoS ONE 2015, 10, e0120504. [Google Scholar] [CrossRef]
- Angelakis, E.; Bachar, D.; Yasir, M.; Musso, D.; Djossou, F.; Gaborit, B.; Brah, S. Treponema species enrich the gut microbiota of traditional living populations but are absent from urban individuals. New Microbes New Infect. 2018, 27, 14–21. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pool No. | Main Pool | Type of Sample |
|---|---|---|
| J1 | Farmed unfed mouth | Tissue |
| J2 | Farmed unfed pharynx | Tissue |
| J3 | Farmed unfed crop | Tissue |
| J4 | Farmed unfed intestine | Tissue |
| J5 | Farmed fed mouth | Tissue |
| J6 | Farmed fed pharynx | ILF + Tissue |
| J7 | Farmed fed crop | ILF |
| J8 | Farmed fed intestine | ILF |
| J9 | Wild fed mouth | Tissue |
| J10 | Wild fed pharynx | ILF + Tissue |
| J11 | Wild fed crop | ILF |
| J12 | Wild fed intestine | ILF |
| Group | Region | Shannon Index (Mean ± SD) | Simpson Index (Mean ± SD) |
|---|---|---|---|
| Farmed Unfed | Mouth | 2.45 ± 0.12 a | 0.829 ± 0.017 a |
| Pharynx | 2.02 ± 0.10 ab | 0.819 ± 0.016 ab | |
| Crop | 1.90 ± 0.10 b | 0.800 ± 0.016 b | |
| Intestine | 2.00 ± 0.10 ab | 0.822 ± 0.016 ab | |
| Farmed Fed | Mouth | 3.18 ± 0.16 c | 0.891 ± 0.018 c |
| Pharynx | 2.41 ± 0.12 abc | 0.853 ± 0.017 abc | |
| Crop | 2.38 ± 0.12 abc | 0.859 ± 0.017 abc | |
| Intestine | 2.38 ± 0.12 abc | 0.874 ± 0.017 abc | |
| Wild Fed | Mouth | 1.87 ± 0.09 d | 0.728 ± 0.015 d |
| Pharynx | 3.26 ± 0.16 cd | 0.926 ± 0.019 cd | |
| Crop | 2.52 ± 0.13 bc | 0.854 ± 0.017 bc | |
| Intestine | 2.46 ± 0.12 bc | 0.881 ± 0.018 bc |
| Pathogens | Farmed Unfed | Farmed Fed | Wild Fed |
|---|---|---|---|
| Aeromonas | 0.08 (M) | 17.30 (M); 17.11 (F); 24.10 (C); 20.11 (I) | 0,42 (M); 0.02 (F) 0.03 (C) |
| Pseudomonas | 0.42 (M); | 0.41 (M); 0.14 (F); 0.64 (C); 0.33 (I) | 0.09 (M) |
| Acinetobacter | 0.07 (M) | 0.35 (M); 0.42 (F); 0.90 (C); 0.60 (I) | - |
| Staphylococcus | 0.13 (M) | 0.24 (M) | - |
| Morganella | - | 3 (M); 2.11 (F); 3.02 (C); 2.14 (I) | - |
| Enterococcus | - | 0.53 (M) | - |
| Streptococcus | - | 0.32 (M) | - |
| Rickettsia | - | 0.03 (M) | 9.38 (F); 1.27 (C); 1.12 (I) |
| Anaplasma | - | 0.01 (M); 0.04 (F); 0.07 (C); 0.05 (I) | 0.01 (C); 0.01 (I) |
| Serratia | - | - | 0.04 (F); 0.07 (C); 0.02 (I) |
| Clostridia | - | - | 1.20 (F); 1.12 (C) |
| Porphyromonas | - | - | 0.96 (F) |
| Bacteroides | 0.75 (F) | ||
| Fuzobacterium | 0.05 (M) | 0.53 (M) | 0.02 (M) |
| Bacillaceae | 0.57 (M) | 3.85 (M) | |
| Treponema | 0.12 (F); 0.33 (C); | 1.10 (F) | 1.95 (F); 0.53 (I) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karasartova, D.; Arslan-Akveran, G.; Sensoz, S.; Mumcuoglu, K.Y.; Taylan-Ozkan, A. Hirudo verbana Microbiota Dynamics: A Key Factor in Hirudotherapy-Related Infections? Microorganisms 2025, 13, 918. https://doi.org/10.3390/microorganisms13040918
Karasartova D, Arslan-Akveran G, Sensoz S, Mumcuoglu KY, Taylan-Ozkan A. Hirudo verbana Microbiota Dynamics: A Key Factor in Hirudotherapy-Related Infections? Microorganisms. 2025; 13(4):918. https://doi.org/10.3390/microorganisms13040918
Chicago/Turabian StyleKarasartova, Djursun, Gonul Arslan-Akveran, Sabiha Sensoz, Kosta Y. Mumcuoglu, and Aysegul Taylan-Ozkan. 2025. "Hirudo verbana Microbiota Dynamics: A Key Factor in Hirudotherapy-Related Infections?" Microorganisms 13, no. 4: 918. https://doi.org/10.3390/microorganisms13040918
APA StyleKarasartova, D., Arslan-Akveran, G., Sensoz, S., Mumcuoglu, K. Y., & Taylan-Ozkan, A. (2025). Hirudo verbana Microbiota Dynamics: A Key Factor in Hirudotherapy-Related Infections? Microorganisms, 13(4), 918. https://doi.org/10.3390/microorganisms13040918