1. Introduction
Watermelon is an important economic crop in China. According to the National Bureau of Statistics, between 2018 and 2022, China’s watermelon-cultivation area reached 1.5 million hectares, and the average yield of watermelons was about 62.35 million tons (
https://data.stats.gov.cn/easyquery.htm?cn=C01 (accessed on 10 February 2025)). China is the largest producer of watermelons in the world [
1]. Based on the climate, geographical conditions, and cultivation practices, China’s watermelon-production areas can be divided into three major regions: the northern semi-arid area, northwestern arid area, and southern humid area. The northern semi-arid area experiences cold winters and warm, dry summers, with low annual rainfall, but has fertile soil, resulting in high watermelon yields. The northwestern arid area is characterized by dry conditions with little rainfall and large temperature differences between day and night, leading to high-quality watermelons with high sugar contents. The southern humid area has a humid and rainy climate, supporting multi-season cultivation, with watermelons coming to market early and having a long supply period.
Watermelon is threatened by various diseases throughout its growth period [
2]. Among them,
Fusarium wilt of watermelon, caused by
Fusarium oxysporum f. sp.
niveum (FoN), is present in watermelon-production areas around the world [
3]. FoN invades the plant through root hairs or wounds in the roots. It multiplies and spreads within the vascular vessels, producing toxins that damage cells and block the vessels, disrupting water transport. This leads to wilting or death of the plant, causing significant losses in watermelon production [
4]. In recent years,
Fusarium wilt of watermelon has become increasingly severe in China due to continuous cropping obstacles, climate change, and the lack of disease-resistant varieties. Conventional control strategies for FoN primarily rely on resistant cultivars and fungicide applications, yet these approaches face limitations in long-term efficacy and environmental safety [
5,
6]. Although breeding resistant varieties is considered one of the most effective strategies for controlling
Fusarium wilt of watermelon, the development of commercially viable and highly resistant cultivars remains limited, primarily due to challenges, such as prolonged breeding cycles and inefficient selection processes. Chemical control methods for
Fusarium wilt of watermelon include the use of prothioconazole and thiophanate-methyl. [
6]. But it has been difficult to solve defects, and the long-term use of chemical agents can lead to drug resistance, environmental pollution, and harm to non-target organisms and results in pesticide residues. Therefore, people are increasingly interested in exploring effective and environmentally friendly alternative methods for controlling the disease. Biological control has the advantages of low pollution, high specificity, and environmental friendliness and has broad application prospects [
7]. Therefore, environmentally friendly biological control, including the use of hypovirulence-related mycoviruses for control, is considered a feasible alternative method [
8].
Mycoviruses are viruses that infect fungi and can replicate within fungal cells [
9]. Studies have shown that most fungal viruses exhibit asymptomatic infection of the host, but some fungi may exhibit phenotypic abnormalities, such as an abnormal colony morphology, reduced spore production, slower mycelial growth, decreased pathogenicity, or increased pathogenicity after being infected [
10,
11]. These abnormalities can reduce the host’s virulence, thus providing an environmentally friendly and sustainable means of disease control. Attenuated mycoviruses have been reported in a variety of fungal species, including
Fusarium graminearum [
12],
Colletotrichum spp. [
13], and
Penicillium digitatum [
14]. In practical applications, certain mycoviruses known to induce hypovirulence in fungi have been developed as biocontrol agents against plant fungal diseases. For instance,
Cryphonectria hypovirus 1 (CHV1), which infects the chestnut blight fungus
Cryphonectria parasitica, has been successfully used for the biological control of chestnut blight in Europe [
15]. Similarly,
Pestalotiopsis theae chrysovirus-1 (PtCV1) [
16] in the tea gray blight pathogen and
Leptosphaeria biglobosa quadrivirus 1 (LbQV1) [
17] in the blackleg pathogen of oilseed rape have been found to enhance plant resistance to their respective fungal hosts. Additionally, it has been reported that spraying
Brassica napus at the early flowering stage with a strain of
Sclerotinia sclerotiorum hypovirulence-associated DNA virus 1 (SsHADV-1)-infected DT-8 can reduce the severity of stem rot by 67.6% and increase the yield by 14.9% [
18]. The large-scale application of existing mycoviral formulations is limited by the low transmission efficiency, narrow host range, and insufficient stability, making them less effective in complex field disease systems. Therefore, it is crucial to identify novel mycoviruses with high transmission efficiency, broad-spectrum antifungal activity, and genetic stability to enhance biocontrol effectiveness.
Despite growing interest in mycoviruses as potential biocontrol agents, their areal distribution, diversity, and ecological roles in FoN remain largely unexplored. In this study, we employed high-throughput sequencing (HTS) to systematically analyze the virome of 150 FoN strains isolated from the three major watermelon-producing regions in China. Our objectives were threefold: (1) to characterize the diversity and composition of mycoviruses associated with FoN populations, (2) to identify novel mycoviruses with potential hypovirulence-inducing properties, and (3) to elucidate areal patterns in mycovirus distribution and their implications for biocontrol. This study represents the first large-scale areal analysis of FoN mycoviruses in China, contributing to sustainable disease management and improved watermelon production.
2. Materials and Methods
2.1. Fungus Isolation and Purification
Watermelon samples infected with FoN were collected from three key watermelon-producing areas in China: Hebei Province (36°01′ N–42°37′ N, 113°04′ E–119°53′ E) in the northern semi-arid area, Fujian Province (23°30′ N–28°22′ N, 115°50′ E–124°07′ E) in the southern humid area, and Xinjiang Uygur Autonomous Region (34°22′ N–49°15′ N, 73°40′ E–96°18′ E) in the northwestern arid area. The northern semi-arid area is characterized by a mid-latitude steppe climate, with an annual average temperature of 16.38 °C, which is 1.76% higher than the national average in China. Its annual precipitation is typically around 20.33 mm, and the humidity level is 47.18%. (
https://weatherandclimate.com/ (accessed on 16 July 2024)). In the northern semi-arid area, watermelon cultivation is primarily based on a one-crop-per-year system. The southern humid area has a humid subtropical climate with no dry season. Its annual average temperature is 19.79 °C, 5.17% higher than the national average in China. The annual precipitation is typically around 87.27 mm, and the humidity level is 82.02%. In the southern humid area, watermelon cultivation is primarily based on a two-crop-per-year system. In some areas, through the application of facility cultivation and early-maturing varieties, an efficient three-crop-per-year planting model can be achieved. The northwestern arid area is classified as having a mid-latitude desert climate. Its annual average temperature is 3.13 °C, which is 11.49% lower than the national average in China. The annual precipitation is typically around 14.75 mm, and the humidity level is 45.83%. The northwestern arid area has a relatively cold climate and a short frost-free period, so watermelon cultivation is typically based on a one-crop-per-year system. We collected 200, 223 and 178 watermelon
Fusarium wilt samples from the northern semi-arid area, northwestern arid area, and southern humid area, respectively, with a total of 601 samples. Through pathogen isolation, purification, morphological identification, and molecular characterization, we isolated 140, 162, and 113 strains of 415 FoN from the northern semi-arid area, the southern humid area, and the northwestern arid area, respectively, and purified and identified them using established methods [
19].
Fifty strains were randomly selected from each producing area for biological characterization and sequencing, and the selected strains were cultured on PDA at 25 °C. The colony diameter was measured daily for 5 days using the cross method, with three replicates per day. Average values were calculated, and the mycelial growth rate was determined. After 5 days of culture, the morphology of the cultured strain was observed and photographed. The selected strains were inoculated respectively into 100 mL of Potato Dextrose Broth (PDB). The cultures were incubated in a shaking incubator at 25 °C and 180 rpm for 3 days. Post-cultivation, mycelia were removed via filtration through a sterilized filter cloth. The spore yield was estimated using a five-point sampling method combined with hemocytometer counting. To ensure reliability, all experiments were conducted in triplicate for each strain.
2.2. Extraction of Total RNA
The selected strains were cultured respectively on cellophane-overlaid PDA media (PDA-CF) at 25 °C in complete darkness for 5 days. After incubation, the mycelia of 50 strains from the same area were mixed together, ground into a fine powder using a mortar and pestle with liquid nitrogen, and subjected to total RNA extraction using the TransZolUpPlusRNA kit (TransGenBiotech, Beijing, China) following the manufacturer’s protocol. The quality and quantity of the extracted RNA were evaluated using two complementary methods: spectrophotometric analysis with a Nanodrop 2000 system (Thermo Scientific, Waltham, MA, USA) and electrophoretic separation on a 1.0% agarose gel. To ensure consistency in downstream applications, RNA samples were normalized to a uniform concentration of 200 ng/μL.
2.3. Library Preparation and Sequencing
RNA purification, reverse transcription, library preparation, and high-throughput sequencing were conducted by Shanghai Majorbio Bio-pharm Biotechnology Co., Ltd. (MajorBio, Shanghai, China). For each sample, 1 μg of total RNA was processed to construct strand-specific sequencing libraries after ribosomal RNA (rRNA) depletion. Libraries were prepared using the Illumina Stranded Total RNA Prep, with ligation using the Ribo-Zero Plus kit, strictly adhering to the manufacturer’s protocols. The pooled libraries were then subjected to paired-end sequencing on an Illumina NovaSeq X Plus platform (Illumina, San Diego, CA, USA). Raw sequencing data often contain adapter sequences, low-quality bases, uncertain bases (denoted as N), and short sequences, which can compromise the accuracy of downstream analyses. To address this, we first used fastp (v0.19.6, available at
https://github.com/OpenGene/fastp (accessed on 1 February 2024)) [
20] to perform quality control. This involved trimming adapter sequences from the 3′ and 5′ ends of the reads. Reads shorter than 50 nt after trimming, with an average quality score below 20, or containing N bases were discarded, ensuring that only high-quality paired-end reads were retained. Next, we performed de novo assembly on the cleaned reads using MEGAHIT (v1.3.0, Dinghua Li, Shenzhen, China), generating contigs for each sample. Contigs with a length of ≥1 kb, and those identified as circular were selected for further analysis.
2.4. Virus-Related Sequence Assembly and Annotation
For viral contig identification, assembled sequences were screened against the NCBI nr protein database through DIAMOND BLASTX (v0.9.10, Benjamin Buchfink, Tübingen, Baden-Württemberg, Germany) [
21] with a stringent e-value cutoff (1 × 10
−5) to balance sensitivity and specificity. Contigs aligned to identical reference sequences were consolidated into extended contigs using DNAMAN (v8.0, Lynnon Biosoft, Vaudreuil, QC, Canada). Final sequence annotation, ORF prediction, and amino acid analyses were performed using DNAMAN. The raw sequencing data from the metavirome libraries have been deposited in the NCBI Sequence Read Archive (SRA) database under BioProject accession number PRJNA1158555.
2.5. Phylogenetic Analysis of Mycoviruses
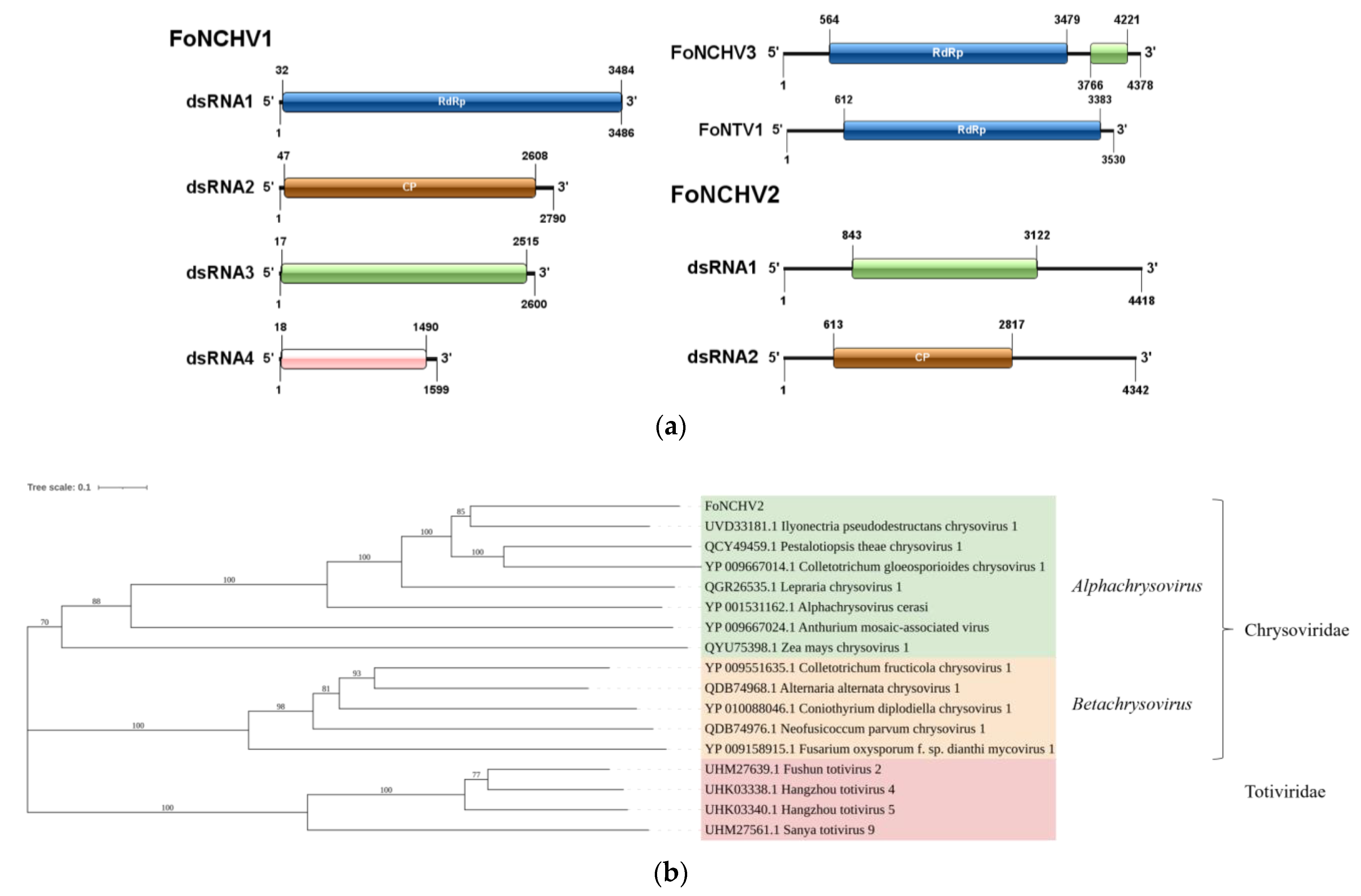
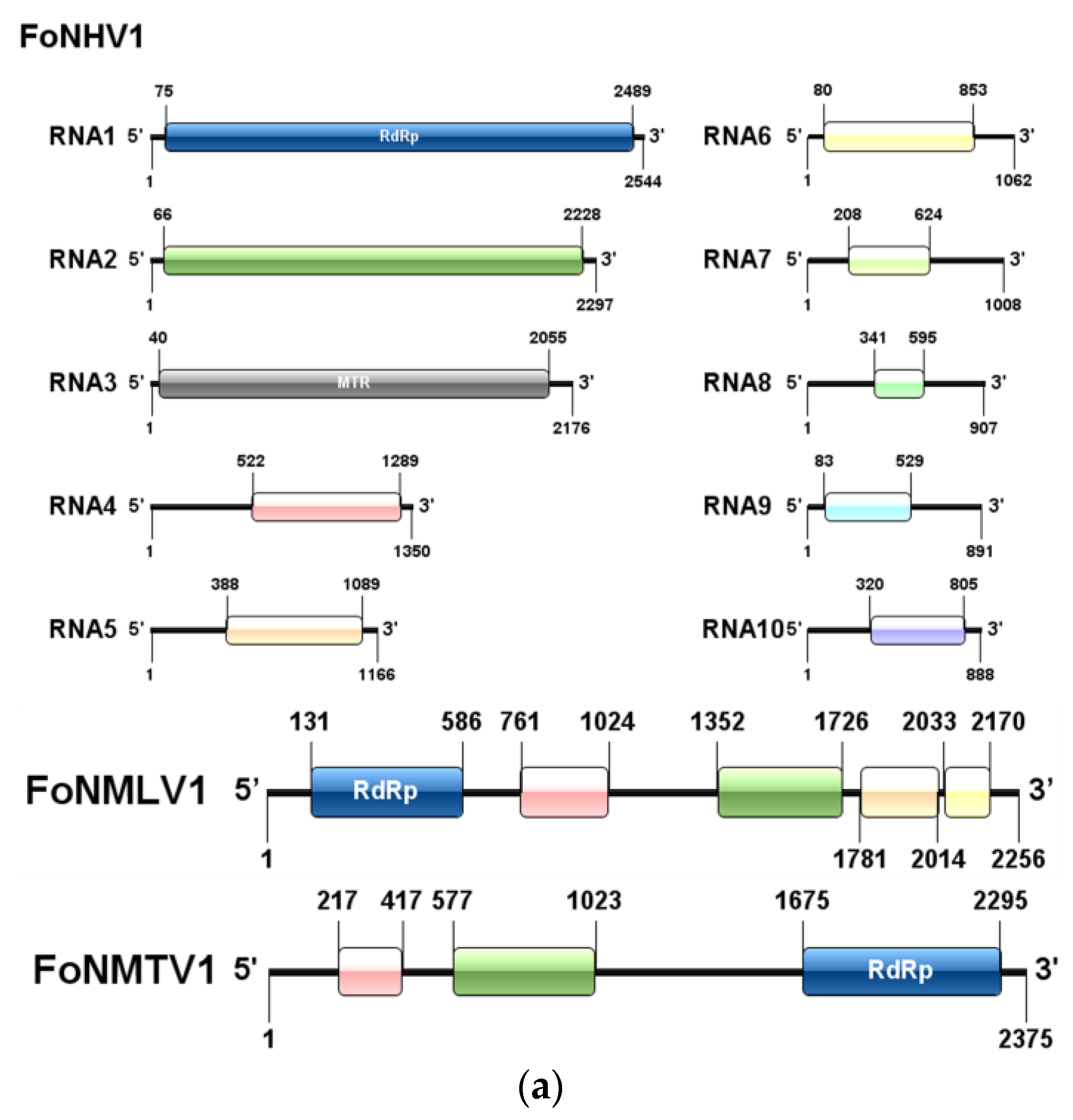
To analyze the evolutionary relationships of the newly identified viruses, the RNA-dependent RNA polymerase protein (RdRp) or Coat Protein (CP) sequences encoded by the putative mycoviruses were compared using BLASTp against the NCBI-nr database. The core conserved domains of RdRp were aligned with MAFFT (v7.427, Kazutaka Katoh, Mishima, Japan) using the E-INS-i algorithm. The alignments were then refined with trimAl (v1.4, Salvador Capella-Gutiérrez, Madrid, Spain) to remove low-quality regions, applying the automated heuristic method based on similarity statistics.
The phylogenetic tree was constructed using MEGA7 software (Molecular Evolutionary Genetics Analysis v7.0, Sudhir Kumar, Tempe, AZ, USA). First, the target sequences were aligned using the ClustalW algorithm to ensure accurate sequence alignment. After alignment, the Neighbor-Joining (NJ) method in MEGA7 was employed to construct the phylogenetic tree. The NJ tree was built based on the tp-distance model, which is suitable for calculating evolutionary distances for nucleotide sequences. Bootstrap testing with 1000 replicates was performed to assess the node support of the phylogenetic tree, and nodes with bootstrap values greater than 70% were considered statistically well-supported. The final phylogenetic tree was visualized and edited using MEGA7.
2.6. Putative Mycovirus Sequence Confirmation
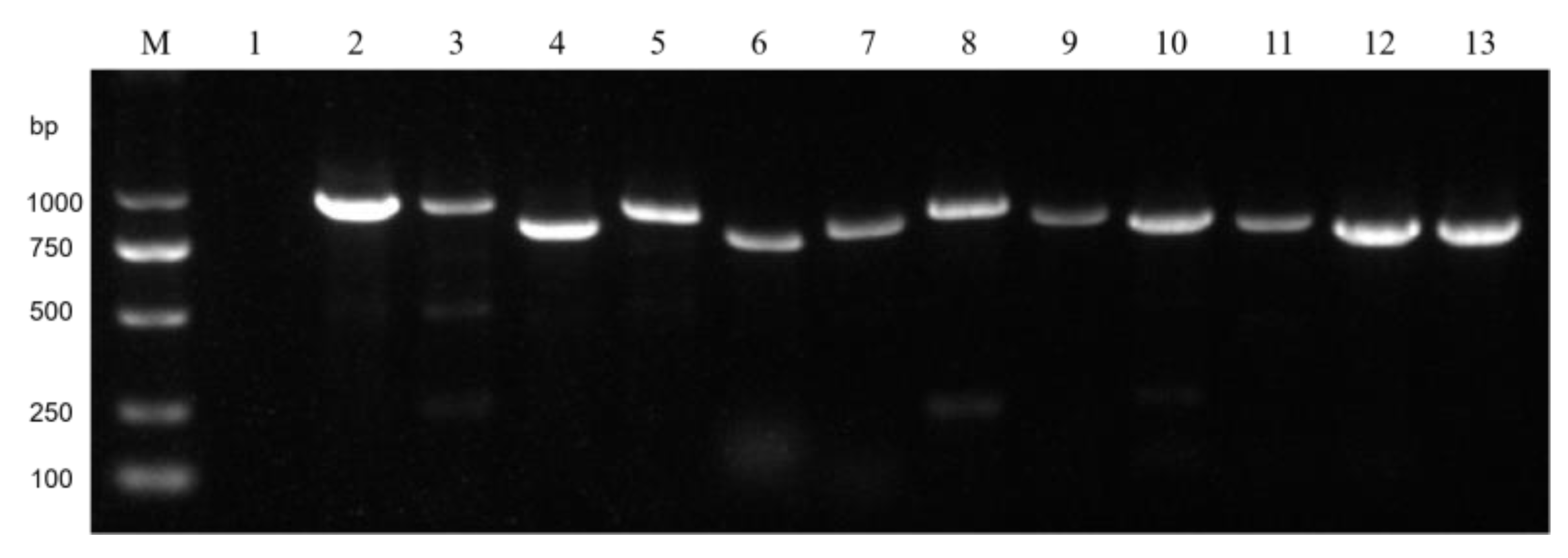
Total RNA of the 150 selected strains was extracted separately with TransZol Up (TransGen Biotech, Beijing, China) and quantified using a NanoDrop spectrophotometer (Thermo Scientific, Waltham, MA, USA). cDNA synthesis was performed using the Fastking gDNA Dispelling RT SuperMix kit (TIANGEN, Beijing, China) according to the manufacturer’s protocol. Two viruses from each of the three main nucleic acid types (dsRNA, +ssRNA, and −ssRNA) were detected via RT-PCR. Specific primers were designed with prime 5 software (
Table S1). PCR amplification was conducted with the M5 HiPer plus Taq HiFi PCR mix (2×, with blue dye) (Mei5bio, Beijing, China). The PCR conditions included an initial denaturation at 95 °C for 5 min, followed by 35 cycles of 95 °C for 45 s, 58 °C for 45 s, and 72 °C for 45 s, with a final extension at 72 °C for 5 min. RT-PCR products were separated through 1% agarose gel electrophoresis and visualized using a gel imaging system. The presence of the virus and the virus-carrying strains was verified through RT-PCR, and whether the area where the virus-carrying strain was collected matched the area where the virus was detected in the sequencing results was analyzed.
4. Discussion
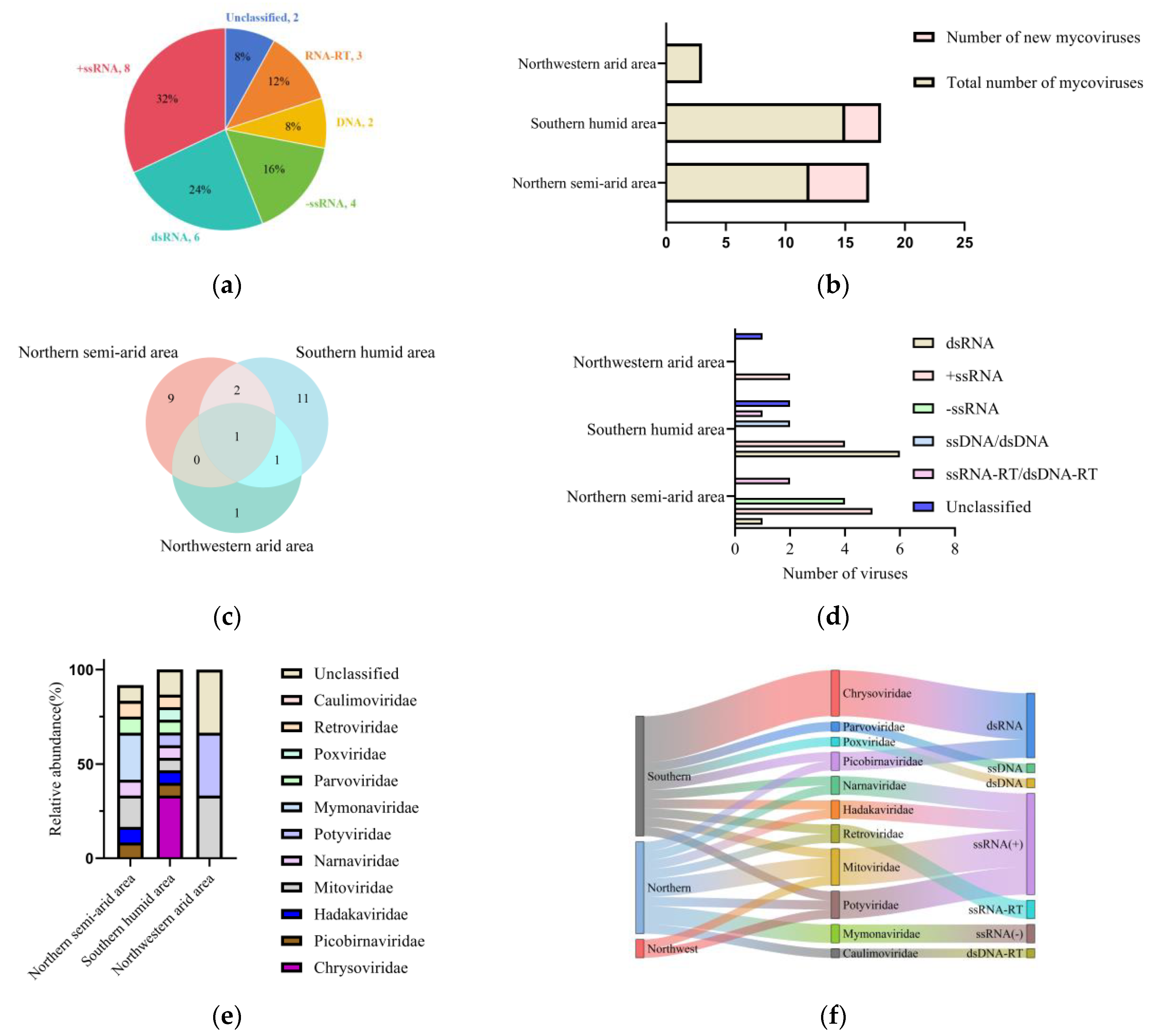
In this study, we found that the diversity of mycoviruses in
Fusarium oxysporum f. sp.
niveum differ significantly in different areas. Out of the 25 detected mycoviruses, 15 are from the southern humid area and belong to the Chrysoviridae, Parvoviridae, Narnaviridae, Picobinaviridae, Mitoviridae, Poxviridae, Potyviridae, Retroviridae and Hadakaviridae families. Twelve species are from the northern semi-arid area, belonging to the Mymonaviridae, Hadakaviridae, Mitoviridae, Narnaviridae, Caulimoviridae, Picobinaviridae, Retroviridae, and Potyviridae families. The northwestern arid area yielded a comparatively limited diversity, with only three mycoviruses identified, which were classified within the Mitoviridae and Potyviridae families. There are significant differences in the number and types of viruses among the three major areas. We speculate that this is closely related to the geographical and climatic conditions of the three areas. The southern humid area is dominated by a tropical subtropical monsoon climate, with high temperatures and humidity, and precipitation of over 800 mm. Due to the favorable growing conditions, watermelons can be planted multiple times throughout the year, providing an extended survival environment for FoN, the host of mycoviruses [
23,
24]. The favorable growing conditions and extended survival environment have made this area have the richest variety of mycoviruses. The northwest area has a temperate continental dry climate, with dry conditions, low rainfall, large diurnal temperature variations, and significant annual temperature differences. The annual precipitation is generally low, with most places having an annual precipitation of less than 200 mm [
25,
26]. Due to climate limitations, watermelons in the northwest area are generally only planted once with the growing period from May to August. Compared to the southern humid area, this provides a shorter growth environment for the host of mycoviruses FoN, the host of mycoviruses. The unfavorable growing conditions and shorter growth environment result in a lower diversity of mycoviruses in this area.
Although most mycovirus infections do not have a significant impact on their fungal hosts, a few hypovirulence-related mycoviruses can cause a decrease in the growth rate, pathogenicity, and other phenotypes of their host fungi, which has the potential to control plant fungal diseases for biocontrol purposes. In this study, we preliminarily classified the collected strains based on their growth rate, sporulation quantity, and colony morphology, and found that 18% (27/150) of the strains had abnormal biological characteristics. According to previous reports, although many mycoviruses have been identified in the genus
Fusarium, there are relatively few reported mycoviruses in
F. oxysporum. For example,
Fusarium oxysporum ourmia-like virus 1 (FoOuLV1) is a novel hypovirulence-inducing ourmia-like mycovirus. It belongs to the Botourmiaviridae family and can significantly reduce the pathogenicity of host fungi [
27]. In this study, we identified mycoviruses in FoN isolated from the three major watermelon-production areas. Among them, most mycoviruses are identified from strains with an abnormal morphology. Therefore, we speculate that mycoviral infection is likely the main reason for the abnormal colony morphology of these strains. The FoN strains carrying hypovirulence mycoviruses has biocontrol potential for controlling
Fusarium wilt of watermelon. In addition, we have also identified mycoviruses in normal strains, indicating that some viral infections do not alter the biological characteristics of fungal hosts. In this study, we found the phenomenon of co-infection in the tested strains through RT-PCR. Co-infection is a prevalent phenomenon where multiple viruses simultaneously infect the same fungus under natural conditions, and this has been observed in many fungal species [
28]. For instance, it has been reported that a strain of
Sclerotinia sclerotiorum was infected by nine different mycoviruses, which were classified into eight distinct viral families [
29]. Similarly, co-infection has also been documented in
Purpureocillium lilacinum and
Beauveria bassiana, two significant entomopathogenic fungi, as well as in various other fungal pathogens [
30,
31]. However, the effect of mycoviral co-infection on FoN needs further study.
The identification of diverse mycoviruses in FoN, particularly the eight novel viruses, opens promising avenues for biocontrol of
Fusarium wilt. At present, hypovirulence-associated mycoviruses could be deployed through two primary strategies: (1) the direct application of viral particles or infected fungal strains to soil or seedlings, or (2) horizontal transmission by engineering hypovirulent Fon isolates as “Trojan horse” vectors to spread viruses within pathogenic populations [
32]. In southern humid area, the dominance of dsRNA Chrysoviridae, which is known to dysregulate fungal virulence through RNA interference pathways [
33], supports their use as region-specific biocontrol agents. These viruses could be applied via soil drenching or hypovirulent FoN strains, with biochar amendments enhancing moisture retention to sustain viral activity. Conversely, in arid northern regions, +ssRNA viruses, like Mitoviridae, may require clay-based nanocarrier encapsulation to stabilize their viability under low humidity [
34], coupled with drip irrigation systems to optimize viral dispersal. The cross-regional
Fusarium oxysporum potyvirus (FoNPV1), exhibiting a pan-China distribution, holds promise as a broad-spectrum biocontrol agent, akin to the transcontinental success of Sclerotinia sclerotiorum hypovirulence-associated DNA virus 1 [
35]. Such geographically tailored strategies, integrating viral consortia with local agronomic practices, could maximize field efficacy while addressing climatic constraints.
The application of mycoviruses as biocontrol agents against phytopathogenic fungi faces several critical challenges. First, host specificity limits their broad-spectrum utility, as most mycoviruses exhibit narrow host ranges, often restricted to specific fungal strains or species. Second, environmental instability-particularly susceptibility to UV radiation, temperature fluctuations, and desiccation-compromises viral viability in field conditions. Third, the risk of fungal resistance evolution necessitates the long-term monitoring of pathogen-virus coevolution dynamics. Additionally, regulatory frameworks for mycovirus deployment remain underdeveloped, with unresolved biosafety concerns regarding unintended ecological impacts on non-target microorganisms. Future research should focus on multi-omics to decode hypovirulence mechanisms, such as the viral suppression of host RNA silencing. Genetic engineering (e.g., CRISPR-mediated host range expansion) and synthetic chimeric viruses could address specificity limitations. Advanced formulations, like clay-polymer nanocomposites, may enhance environmental resilience. Field trials must prioritize integrated strategies, combining mycoviruses with biocontrol microbes or resistant cultivars. Regionally tailored consortia-blending climate-adapted viral types (e.g., humidity-tolerant dsRNA and drought-resistant +ssRNA viruses)-could optimize cross-environment performance. Crucially, global collaboration is needed to establish standardized biosafety protocols and policy frameworks, ensuring the safe scaling of mycovirus biocontrol. By merging virology, agronomy, and nanotechnology, mycoviruses may evolve from experimental tools to sustainable solutions for managing crop fungal diseases in a climate-changing world.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}