
Evaluation and Application of the MIRA–qPCR Method for Rapid Detection of Norovirus Genogroup II in Shellfish

Abstract
1. Introduction
2. Materials and Methods
2.1. Norovirus GII Deoxyribonucleic Acid Standard Preparation
2.2. Design of Primers and Probes
2.3. Principle and Workflow of MIRA–qPCR Assay
2.4. MIRA–qPCR on Analytik Jena’s qTOWER3G System
2.5. Accuracy Assessment of MIRA–qPCR Method
2.6. Detection of Norovirus in Shellfish Food via MIRA–qPCR
3. Results
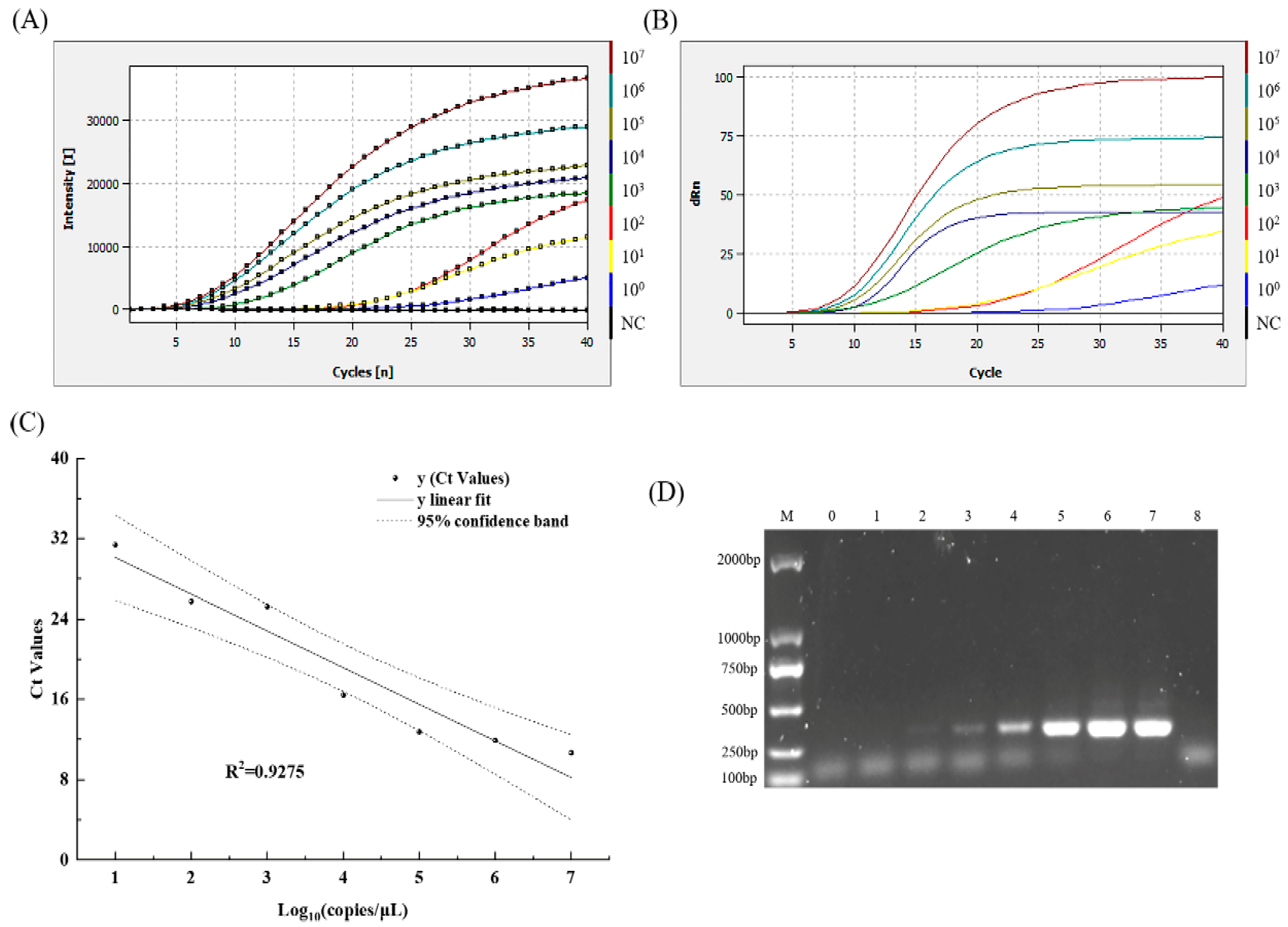
3.1. Primer Sets Screened and Systems Optimization
3.2. Accuracy Analysis of MIRA–qPCR Method
3.3. Application of MIRA–qPCR Assays of Norovirus in Shellfish Foods
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bartsch, S.M.; Lopman, B.A.; Ozawa, S.; Hall, A.J.; Lee, B.Y. Global economic burden of norovirus gastroenteritis. PLoS ONE 2016, 11, e0151219. [Google Scholar] [CrossRef] [PubMed]
- Bartsch, S.M.; O’Shea, K.J.; Lee, B.Y. The clinical and economic burden of norovirus gastroenteritis in the United States. J. Infect. Dis. 2020, 222, 1910–1919. [Google Scholar] [CrossRef] [PubMed]
- Silva, A.J.; Yang, Z.; Wolfe, J.; Hirneisen, K.A.; Ruelle, S.B.; Torres, A.; Williams-Hill, D.; Kulka, M.; Hellberg, R.S. Application of whole-genome sequencing for norovirus outbreak tracking and surveillance efforts in Orange County, CA. Food Microbiol. 2021, 98, 103796. [Google Scholar] [CrossRef] [PubMed]
- Deval, J.; Jin, Z.; Chuang, Y.-C.; Kao, C.C. Structure(s), function(s), and inhibition of the RNA-dependent RNA polymerase of noroviruses. Virus Res. 2017, 234, 21–33. [Google Scholar] [CrossRef]
- Chhabra, P.; Graaf, M.; Parra, G.I.; Chan, M.C.; Green, K.; Martella, V.; Wang, Q.; White, P.A.; Katayama, K.; Vennema, H.; et al. Corrigendum: Updated classification of norovirus genogroups and genotypes. J. Gen. Virol. 2020, 101, 893. [Google Scholar] [CrossRef]
- Yu, F.; Jiang, B.; Guo, X.; Hou, L.; Tian, Y.; Zhang, J.; Li, Q.; Jia, L.; Yang, P.; Wang, Q.; et al. Norovirus outbreaks in China, 2000-2018: A systematic review. Rev. Med. Virol. 2022, 32, e2382. [Google Scholar] [CrossRef]
- Parikh, M.P.; Vandekar, S.; Moore, C.; Thomas, L.; Britt, N.; Piya, B.; Stewart, L.S.; Batarseh, E.; Hamdan, L.; Cavallo, S.J.; et al. Temporal and Genotypic Associations of Sporadic Norovirus Gastroenteritis and Reported Norovirus Outbreaks in Middle Tennessee, 2012–2016. Clin. Infect. Dis. 2020, 71, 2398–2404. [Google Scholar] [CrossRef]
- Gao, Z.; Liu, B.; Yan, H.; Li, W.; Jia, L.; Tian, Y.; Chen, Y.; Wang, Q.; Pang, X. Norovirus outbreaks in Beijing, China, from 2014 to 2017. J. Infect. 2019, 79, 159–166. [Google Scholar] [CrossRef]
- Shukla, S.; Cho, H.; Kwon, O.J.; Chung, S.H.; Kim, M. Prevalence and evaluation strategies for viral contamination in food products: Risk to human health-a review. Crit. Rev. Food Sci. Nutr. 2018, 58, 405–419. [Google Scholar]
- Chan, M.C.W.; Shan Kwan, H.; Chan, P.K.S. Chapter 4—Structure and Genotypes of Noroviruses. In The Norovirus; Chan, P.K.S., Ed.; Academic Press: Cambridge, MA, USA, 2017; pp. 51–63. [Google Scholar]
- Campos, C.J.A.; Kershaw, S.; Morgan, O.C.; Lees, D.N. Risk factors for norovirus contamination of shellfish water catchments in England and Wales. Int. J. Food Microbiol. 2017, 241, 318–324. [Google Scholar] [CrossRef]
- David, S.T.; McIntyre, L.; MacDougal, L.; Kelly, D.; Liem, S.; Schallie, K.; McNabb, A.; Houde, A.; Mueller, P.; Ward, P.; et al. An outbreak of norovirus caused by consumption of oysters from geographically dispersed harvest sites, British Columbia, Canada, 2004. Foodborne Pathog. Dis. 2007, 4, 349–358. [Google Scholar] [CrossRef] [PubMed]
- Gyawali, P.; Fletcher, G.C.; McCoubrey, D.-J.; Hewitt, J. Norovirus in shellfish: An overview of post-harvest treatments and their challenges. Food Control 2019, 99, 171–179. [Google Scholar] [CrossRef]
- Ng, T.L.; Chan, P.P.; Phua, T.H.; Loh, J.P.; Yip, R.; Wong, C.; Liaw, C.W.; Tan, B.H.; Chiew, K.T.; Chua, S.B.; et al. Oyster-associated outbreaks of Norovirus gastroenteritis in Singapore. J. Infect. 2005, 51, 413–418. [Google Scholar] [CrossRef] [PubMed]
- Le Guyader, F.S.; Krol, J.; Ambert-Balay, K.; Ruvoen-Clouet, N.; Desaubliaux, B.; Parnaudeau, S.; Le Saux, J.-C.; Ponge, A.; Pothier, P.; Atmar, R.L.; et al. Comprehensive analysis of a norovirus-associated gastroenteritis outbreak, from the environment to the consumer. J. Clin. Microbiol. 2010, 48, 915–920. [Google Scholar] [CrossRef]
- Iritani, N.; Kaida, A.; Abe, N.; Kubo, H.; Sekiguchi, J.-I.; Yamamoto, S.P.; Goto, K.; Tanaka, T.; Noda, M. Detection and genetic characterization of human enteric viruses in oyster-associated gastroenteritis outbreaks between 2001 and 2012 in Osaka City, Japan. J. Med. Virol. 2014, 86, 2019–2025. [Google Scholar] [CrossRef]
- Loury, P.; le Guyader, F.S.; le Saux, J.C.; Ambert-Balay, K.; Parrot, P.; Hubert, B. A norovirus oyster-related outbreak in a nursing home in France, January 2012. Epidemiol. Infect. 2015, 143, 2486–2493. [Google Scholar] [CrossRef]
- Martínez-Martínez, M.; Diez-Valcarce, M.; Hernandez, M.; Rodriguez-Lazaro, D. Design and application of nucleic acid standards for quantitative detection of enteric viruses by real-time PCR. Food Environ. Virol. 2011, 3, 92–98. [Google Scholar] [CrossRef]
- Lekshmi, M.; Kumar, S.H.; Rajendran, K.V.; Nayak, B.B. Development of a reverse transcription (RT) polymerase chain reaction (PCR) method for the detection of human norovirus in bivalve molluscs. Water Sci. Technol. 2021, 83, 1103–1107. [Google Scholar] [CrossRef]
- Boehm, A.B.; Wolfe, M.K.; Wigginton, K.R.; Bidwell, A.; White, B.J.; Hughes, B.; Duong, D.; Chan-Herur, V.; Bischel, H.N.; Naughton, C.C. Human viral nucleic acids concentrations in wastewater solids from Central and Coastal California USA. Sci. Data. 2023, 10, 396. [Google Scholar] [CrossRef]
- Mangal, M.; Bansal, S.; Sharma, S.K.; Gupta, R.K. Molecular detection of foodborne pathogens: A rapid and accurate answer to food safety. Crit. Rev. Food Sci. Nutr. 2016, 56, 1568–1584. [Google Scholar] [CrossRef]
- Wang, N.; Pan, G.; Liu, P.; Rong, S.; Gao, Z.; Li, Q. Advances and future perspective on detection technology of human norovirus. Pathogens 2021, 10, 1383. [Google Scholar] [CrossRef] [PubMed]
- Yiqiang, S.; Meina, L.; Feng, Z.; Laijin, S. Research progress on biological accumulation, detection and inactivation technologies of norovirus in oysters. Foods 2023, 12, 3891. [Google Scholar] [CrossRef] [PubMed]
- Sun, M.-L.; Lai, H.-Y.; Chong, N.-Y.; Liu, D.-F.; Zhang, Z.-Y.; Pang, B.; Yao, J. Simple and feasible detection of hepatitis B virus via combination of Multienzyme Isothermal Rapid Amplification and Lateral Flow Dipstick Strip. Front. Mol. Biosci. 2021, 8, 763079. [Google Scholar] [CrossRef]
- Wang, Y.; Niu, J.; Sun, M.; Li, Z.; Wang, X.; He, Y.; Qi, J. Rapid and sensitive detection of streptococcus iniae in Trachinotus ovatus based on multienzyme isothermal rapid amplification. Int. J. Mol. Sci. 2023, 24, 7733. [Google Scholar] [CrossRef]
- Xing, Y.; Duan, Z.; Jiang, Y.; Li, M.; Lu, W.; Li, J. Development and evaluation of a real-time multienzyme isothermal rapid amplification assay for rapid detection of Streptococcus pneumoniae. Sci. Rep. 2024, 14, 17729. [Google Scholar] [CrossRef]
- Wang, X.; Zhu, J.-p.; Zhang, Q.; Xu, Z.-g.; Zhang, F.; Zhao, Z.-h.; Zheng, W.-z.; Zheng, L.-s. Detection of enterovirus 71 using reverse transcription loop-mediated isothermal amplification (RT-LAMP). J. Virol. Methods. 2012, 179, 330–334. [Google Scholar] [CrossRef]
- Chen, H.; Sun, C.; Wang, Y.; Gao, X.; You, J.; Yu, W.; Sun, N.; Yang, Y.; Li, X. Rapid Detection of SARS-CoV-2 using Duplex Reverse Transcription-Multienzyme Isothermal Rapid Amplification in a Point-of-Care Testing. Front. Cell. Infect. Microbiol. 2021, 11, 678703. [Google Scholar] [CrossRef]
- Sun, M.L.; Zhong, Y.; Li, X.N.; Yao, J.; Pan, Y.Q. Simple and feasible detection of hepatitis a virus using reverse transcription multienzyme isothermal rapid amplification and lateral flow dipsticks without standard PCR laboratory. Artif. Cells Nanomed. Biotechnol. 2023, 51, 233–240. [Google Scholar] [CrossRef]
- Ji, C.; Feng, Y.; Sun, R.; Gu, Q.; Zhang, Y.; Ma, J.; Pan, Z.; Yao, H. Development of a multienzyme isothermal rapid amplification and lateral flow dipstick combination assay for bovine coronavirus detection. Front. Vet. Sci. 2023, 9, 1059934. [Google Scholar] [CrossRef]
- Cui, H.; Zhang, C.; Tu, F.; Zhao, K.; Kong, Y.; Pu, J.; Zhang, L.; Chen, Z.; Sun, Y.; Wei, Y.; et al. Rapid detection of influenza A viruses using a real-time reverse transcription recombinase-aided amplification assay. Front. Cell. Infect. Microbiol. 2022, 12, 1071288. [Google Scholar] [CrossRef]
- ISO 15216-2:2019; Microbiology of the Food Chain—Horizontal Method for Determination of Hepatitis a Virus and Norovirus Using Real-Time RT-PCR—Part 2: Method for Detection. International Organization for Standardization: Geneva, Switzerland, 2019.
- Lai, J.; Huang, Z.; Xiao, Y.; Yu, K.; Bai, X.; Gao, H.; Dai, H.; Liu, X.; Wang, D. Development and Evaluation of Duplex MIRA-qPCR Assay for Simultaneous Detection of Staphylococcus aureus and non-aureus Staphylococci. Microorganisms 2022, 10, 1734. [Google Scholar] [CrossRef] [PubMed]
- Baert, L.; Uyttendaele, M.; Debevere, J. Evaluation of two viral extraction methods for the detection of human noroviruses in shellfish with conventional and real-time reverse transcriptase PCR. Lett. Appl. Microbiol. 2007, 44, 106–111. [Google Scholar] [CrossRef]
- Atmar, R.L.; Neill, F.H.; Romalde, J.L.; Le Guyader, F.; Woodley, C.M.; Metcalf, T.G.; Estes, M.K. Detection of Norwalk virus and hepatitis A virus in shellfish tissues with the PCR. Appl. Environ. Microbiol. 1995, 61, 3014–3018. [Google Scholar] [CrossRef] [PubMed]
- Zhan, X.; Li, Q.; Tian, P.; Wang, D. The attachment factors and attachment receptors of human noroviruses. Food Microbiol. 2024, 123, 104591. [Google Scholar] [CrossRef]
- Ye, X.; Ellender, R.D.; Wang, S.Y. A faster method to detect norovirus in oysters using probe hybridization to isolate target RNA before RT-PCR. Foodborne Pathog. Dis. 2013, 10, 362–367. [Google Scholar] [CrossRef]
- Notomi, T.; Okayama, H.; Masubuchi, H.; Yonekawa, T.; Watanabe, K.; Amino, N.; Hase, T. Loop-mediated isothermal amplification of DNA. Nucleic Acids Res. 2000, 28, E63. [Google Scholar] [CrossRef]
- Suther, C.; Stoufer, S.; Zhou, Y.; Moore, M.D. Recent developments in Isothermal Amplification Methods for the detection of foodborne viruses. Front. Microbiol. 2022, 13, 841875. [Google Scholar] [CrossRef]
- Luo, J.; Xu, Z.; Nie, K.; Ding, X.; Guan, L.; Wang, J.; Xian, Y.; Wu, X.; Ma, X. Visual detection of norovirus genogroup II by reverse transcription loop-mediated isothermal amplification with hydroxynaphthol blue dye. Food Environ. Virol. 2014, 6, 196–201. [Google Scholar] [CrossRef]
- Zaczek-Moczydlowska, M.A.; Beizaei, A.; Dillon, M.; Campbell, K. Current state-of-the-art diagnostics for Norovirus detection: Model approaches for point-of-care analysis. Trends Food Sci. Tech. 2021, 114, 684–695. [Google Scholar] [CrossRef]
- Fukuda, S.; Takao, S.; Kuwayama, M.; Shimazu, Y.; Miyazaki, K. Rapid detection of norovirus from fecal specimens by real-time reverse transcription-loop-mediated isothermal amplification assay. J. Clin. Microbiol. 2006, 44, 1376–1381. [Google Scholar] [CrossRef]
- Jia, T.; Yu, Y.; Wang, Y. A recombinase polymerase amplification-based lateral flow strip assay for rapid detection of genogroup II noroviruses in the field. Arch. Virol. 2020, 165, 2767–2776. [Google Scholar] [CrossRef] [PubMed]
- Awate, S.; Brosh, R.M., Jr. Interactive Roles of DNA Helicases and Translocases with the Single-Stranded DNA Binding Protein RPA in Nucleic Acid Metabolism. Int. J. Mol. Sci. 2017, 18, 1233. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Man, Y.; Xu, X.; Ji, J.; Wang, Y.; Yao, L.; Xie, Q.; Bi, Y. The development of a Multienzyme Isothermal Rapid Amplification assay to visually detect duck Hepatitis B Virus. Vet. Sci. 2024, 11, 191. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Wang, J.; Zhang, S.; Wang, J.; Qin, C.; Han, Y.; Xu, X. Rapid detection of norovirus genogroup II in clinical and environmental samples using recombinase polymerase amplification. Anal. Biochem. 2020, 605, 113834. [Google Scholar] [CrossRef]
- Kittigul, L.; Thamjaroen, A.; Chiawchan, S.; Chavalitshewinkoon-Petmitr, P.; Pombubpa, K.; Diraphat, P. Prevalence and Molecular Genotyping of Noroviruses in Market Oysters, Mussels, and Cockles in Bangkok, Thailand. Food Environ. Virol. 2016, 8, 133–140. [Google Scholar] [CrossRef]
- Norma, E. Perspective Chapter: Health and Safety in Oyster Aquaculture. In Aquaculture Industry; Yusuf, B., Ed.; IntechOpen: Rijeka, Croatia, 2023; Chapter 6. [Google Scholar]
- La Bella, G.; Martella, V.; Basanisi, M.G.; Nobili, G.; Terio, V.; La Salandra, G. Food-borne viruses in shellfish: Investigation on norovirus and HAV presence in Apulia (SE Italy). Food Environ. Virol. 2017, 9, 179–186. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Sequences (5′ → 3′) | Sites (X86557:4981–5460) | |
|---|---|---|---|
MIRA-qPCR qPCR PCR | Nf1 Nf2 Nf3 NR1 NR2 NR3 P1 * QNIF2 COG2R P2 PcrF PcrR | TGGCTCCCAGCTTTGTGAATGAAGATGGCG TGAGCACGTGGGAGGGCGATCGCAATCTGG TCTCAGATCTGAGCACGTGGGAGGGCGATC AGCGTTTCTAGGGGACACTGTGAACTCTCC TTTGTTGGCCCGCCACAGGTGCCGCAATAG GCCGCAATAGCGGCACCAACAACGGGCTCC GATGGGTCCGCAGCCAACCTCGTCCCAGAGG /i6FAMdT/CA/idSp//iBHQ1dT/AATGAGGATGTT CAGRTGGATGAGRTTCTCWGA TCGACGCCATCTTCATTCACA 5′ 6-FAM-AGCACGTGGGAGGGCGATCG-TAMRA-N3′ CAGATCTGAGCACGTGGGAG GGAGCGTTTCTAGGGGACAC | (5068–5097) (5041–5070) (5032–5061) (5268–5298) (5192–5221) (5172–5201) (5119–5168) (5012–5100) (ISO-2019) (5034–5053) (5281–5299) |
| Concentration (Copies/μL) | Ct Values for Replicate1 (N = 3.51) | Ct Values for Replicate2 (N = 1.23) | Ct Values for Replicate3 (N = 1.86) | Inter-Assay Reproducibility | ||
|---|---|---|---|---|---|---|
| Mean of Ct Values | SD of Ct Values | Ct CV (%) | ||||
| N × 107 | 7.87 | 8.02 | 9.73 | 8.54 | 1.03 | 12.09 |
| N × 106 | 9.00 | 8.49 | 10.35 | 9.28 | 0.96 | 10.35 |
| N × 105 | 9.39 | 9.50 | 11.02 | 9.97 | 0.91 | 9.14 |
| N × 104 | 13.14 | 10.51 | 12.95 | 12.20 | 1.47 | 12.02 |
| N × 103 | 14.41 | 13.81 | 16.16 | 14.79 | 1.22 | 8.26 |
| N × 102 | 25.12 | 24.87 | 25.86 | 25.28 | 0.51 | 2.03 |
| N × 101 | 28.86 | 29.96 | 28.43 | 29.08 | 0.79 | 2.72 |
| Detection Results | Methods | Statistical Analysis | ||
|---|---|---|---|---|
| MIRA–qPCR | qPCR | Kappa (k) | p-Value of Kappa | |
| Positive (+) | 19 | 16 | 0.900 | <0.001 |
| Negative (−) | 106 | 109 | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, Y.; Song, M.; Pan, Y.; Zhao, Y.; Liu, H. Evaluation and Application of the MIRA–qPCR Method for Rapid Detection of Norovirus Genogroup II in Shellfish. Microorganisms 2025, 13, 712. https://doi.org/10.3390/microorganisms13040712
Zhu Y, Song M, Pan Y, Zhao Y, Liu H. Evaluation and Application of the MIRA–qPCR Method for Rapid Detection of Norovirus Genogroup II in Shellfish. Microorganisms. 2025; 13(4):712. https://doi.org/10.3390/microorganisms13040712
Chicago/Turabian StyleZhu, Yanting, Mengyuan Song, Yingjie Pan, Yong Zhao, and Haiquan Liu. 2025. "Evaluation and Application of the MIRA–qPCR Method for Rapid Detection of Norovirus Genogroup II in Shellfish" Microorganisms 13, no. 4: 712. https://doi.org/10.3390/microorganisms13040712
APA StyleZhu, Y., Song, M., Pan, Y., Zhao, Y., & Liu, H. (2025). Evaluation and Application of the MIRA–qPCR Method for Rapid Detection of Norovirus Genogroup II in Shellfish. Microorganisms, 13(4), 712. https://doi.org/10.3390/microorganisms13040712