
Global Archaeal Diversity Revealed Through Massive Data Integration: Uncovering Just Tip of Iceberg

 ,
,
Abstract
1. Introduction
2. Materials and Methods
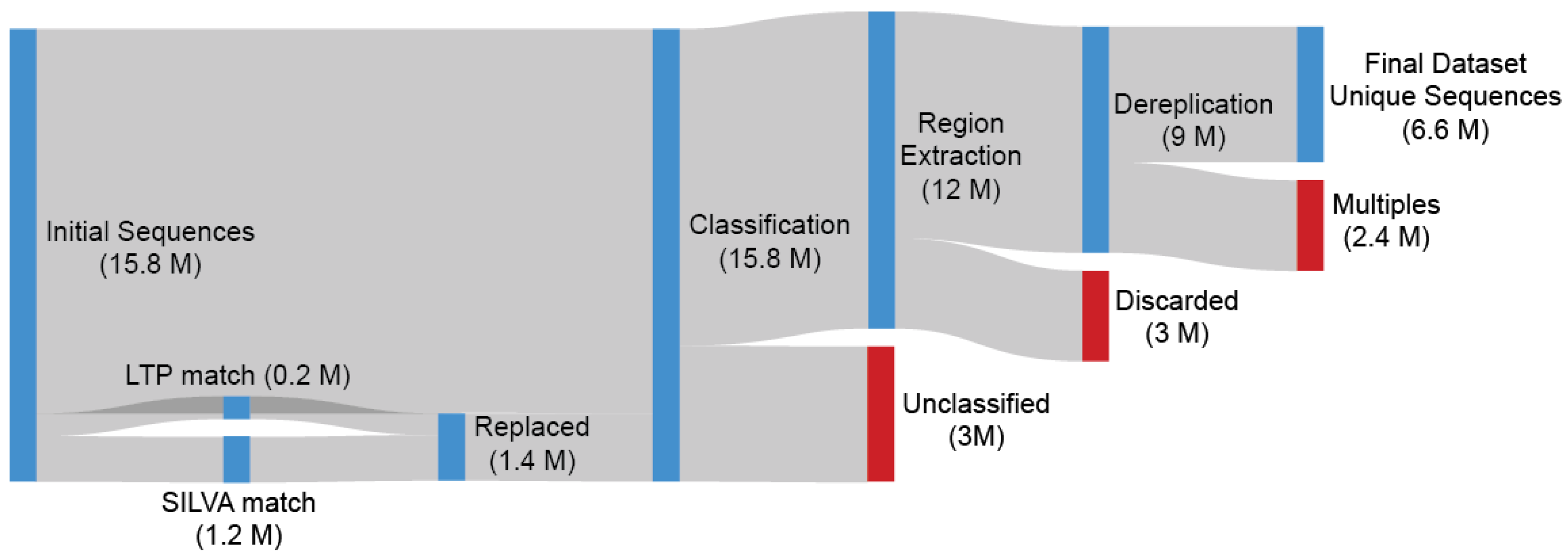
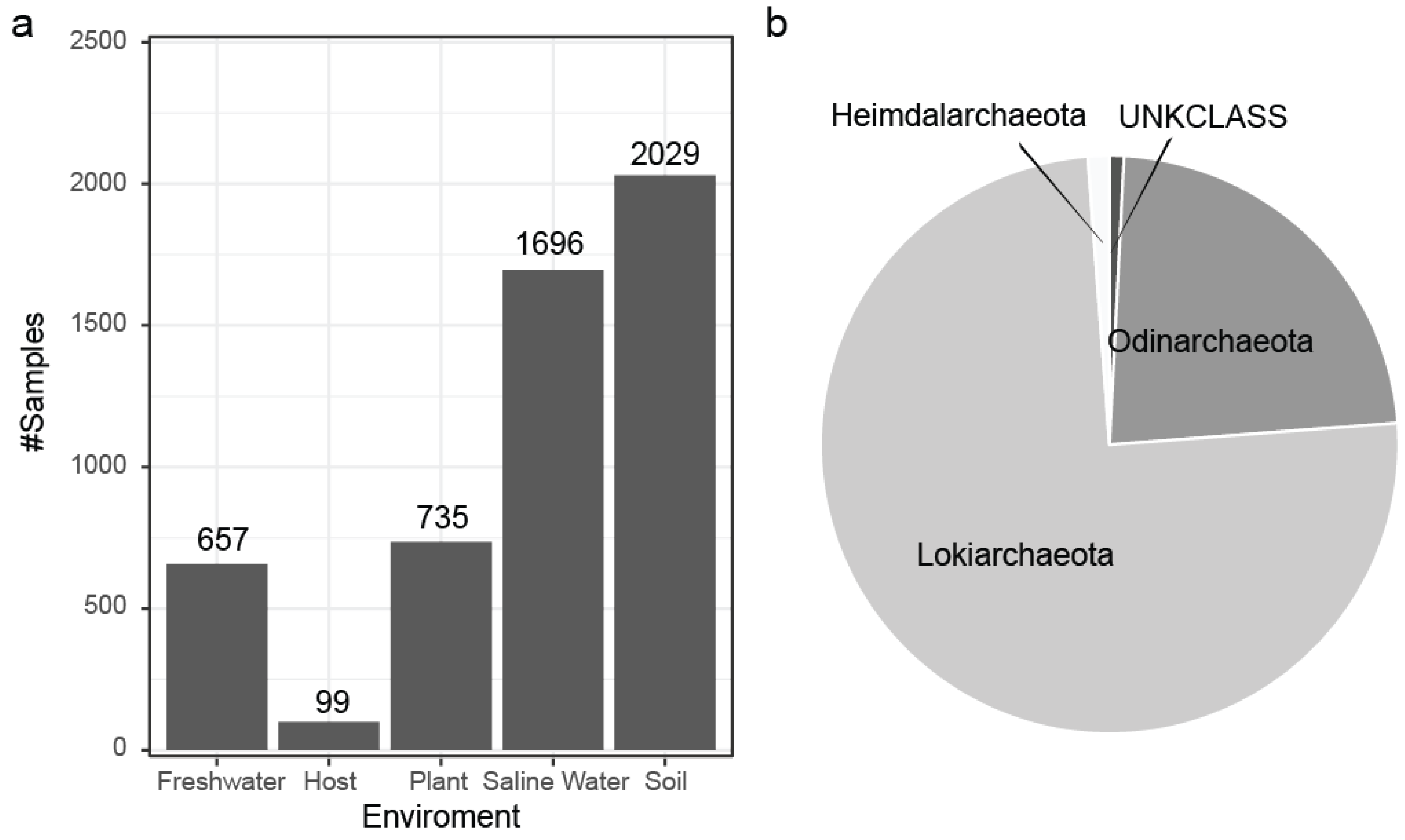
2.1. Dataset Creation
2.2. Clustering Limits Identification
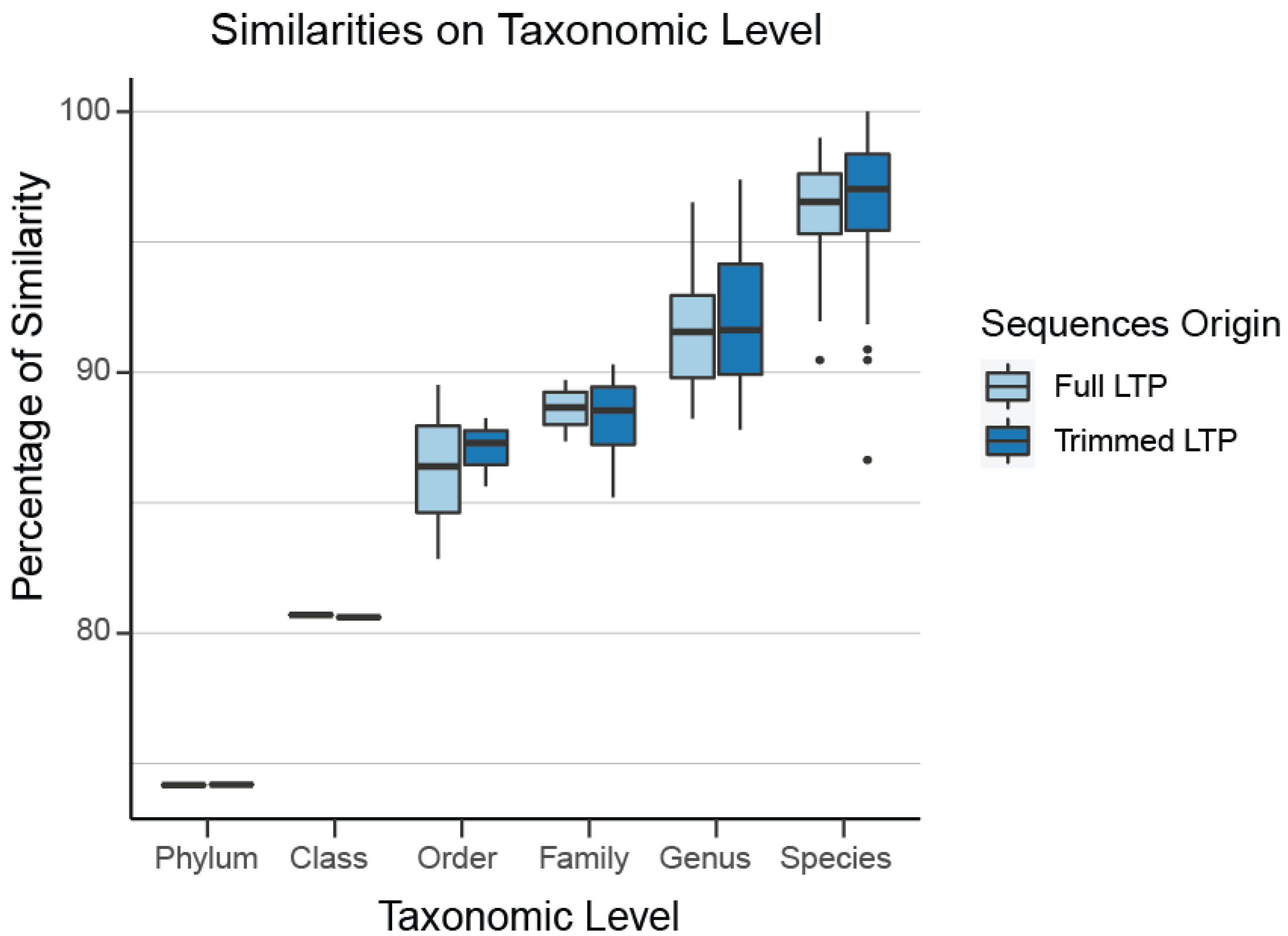
2.3. Verification
3. Results
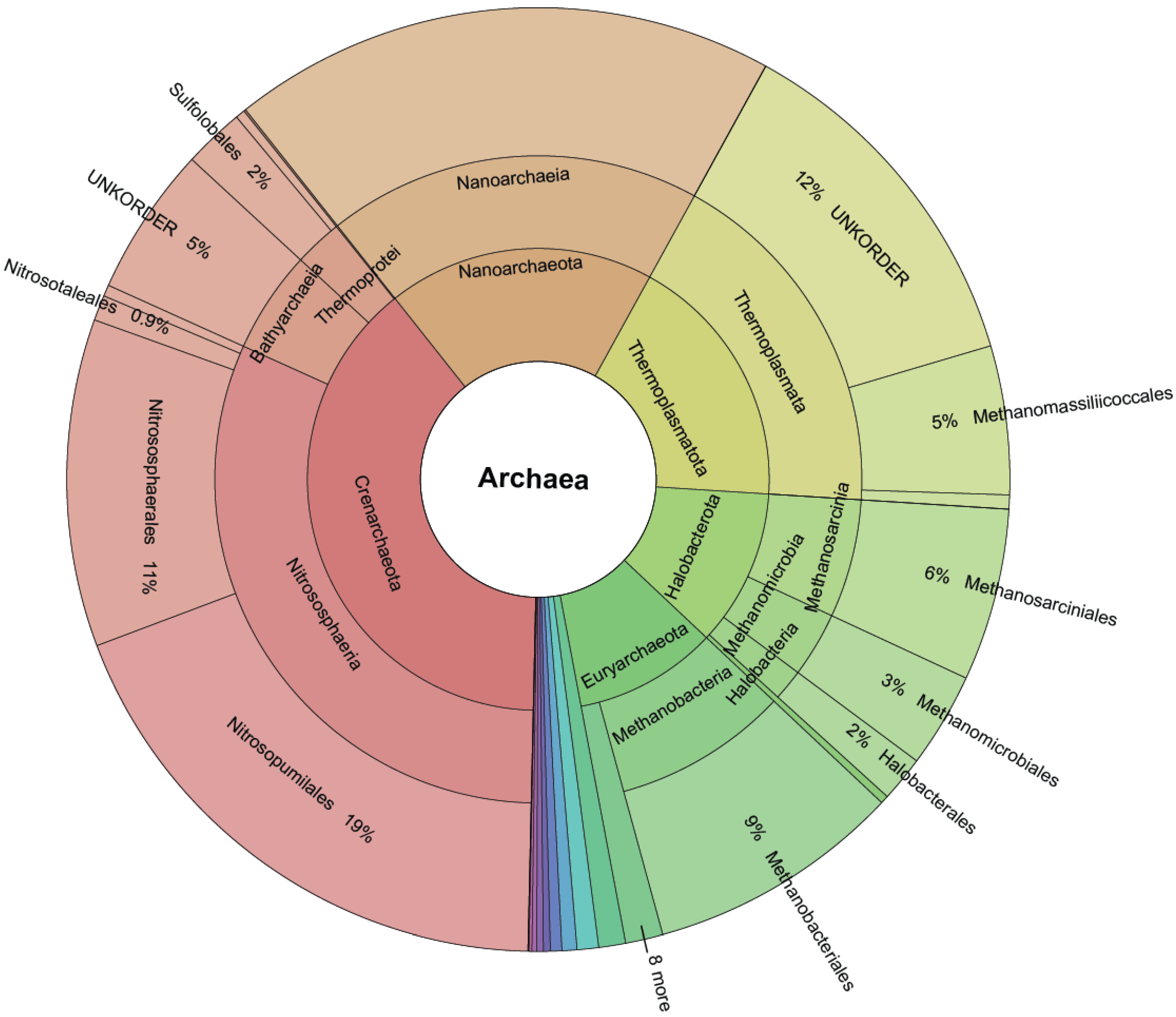
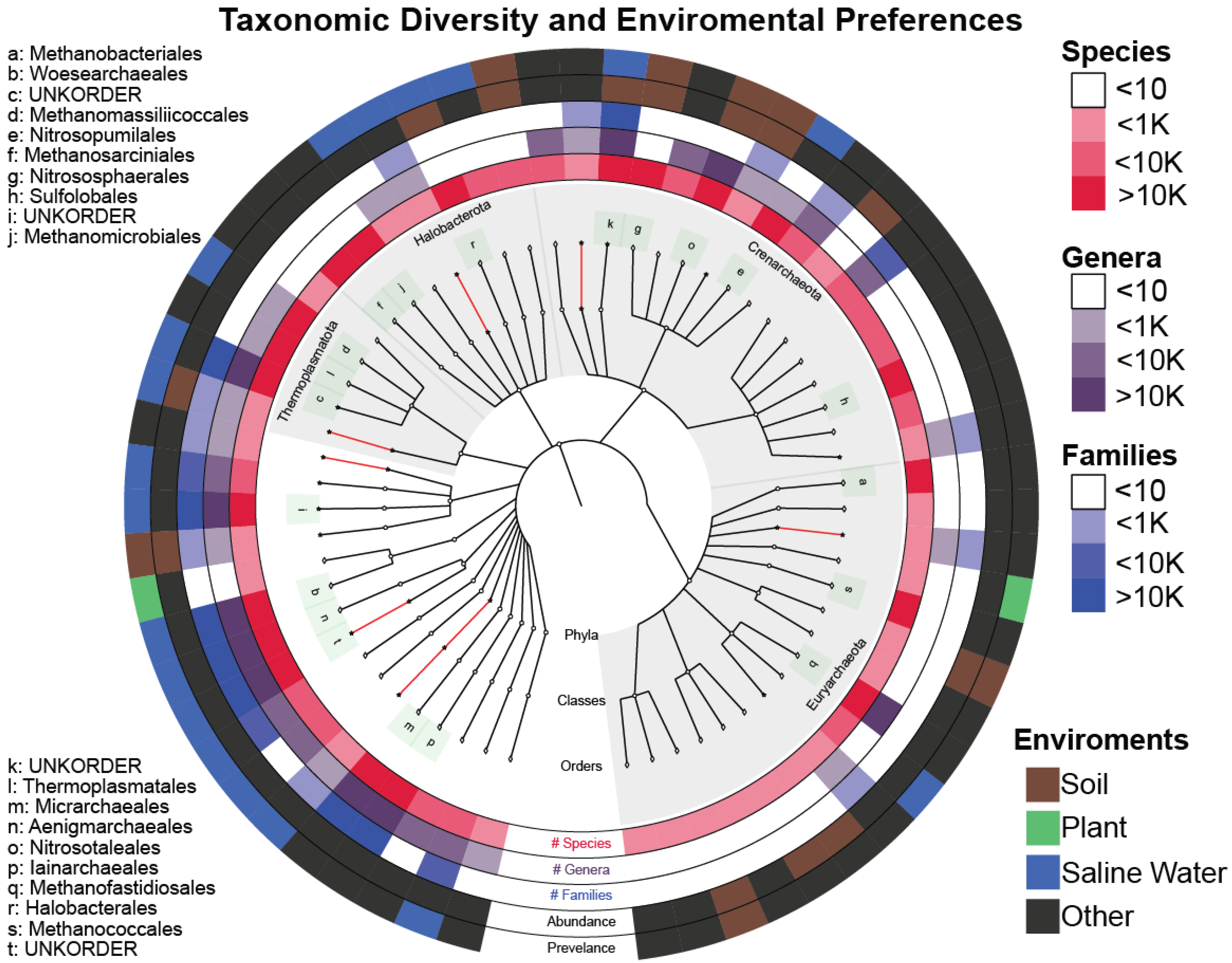
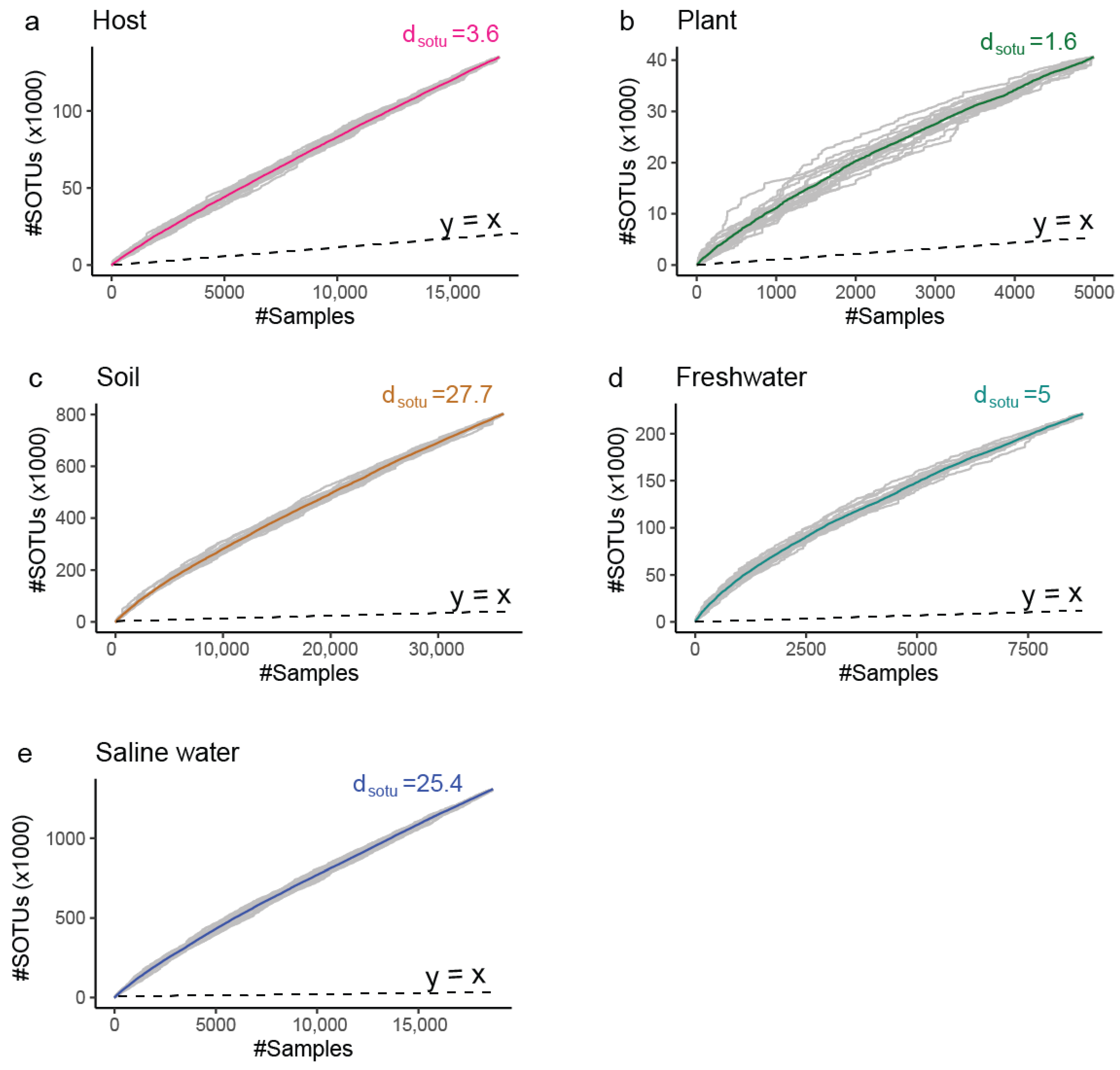
3.1. Archaea Knowledge Expansion
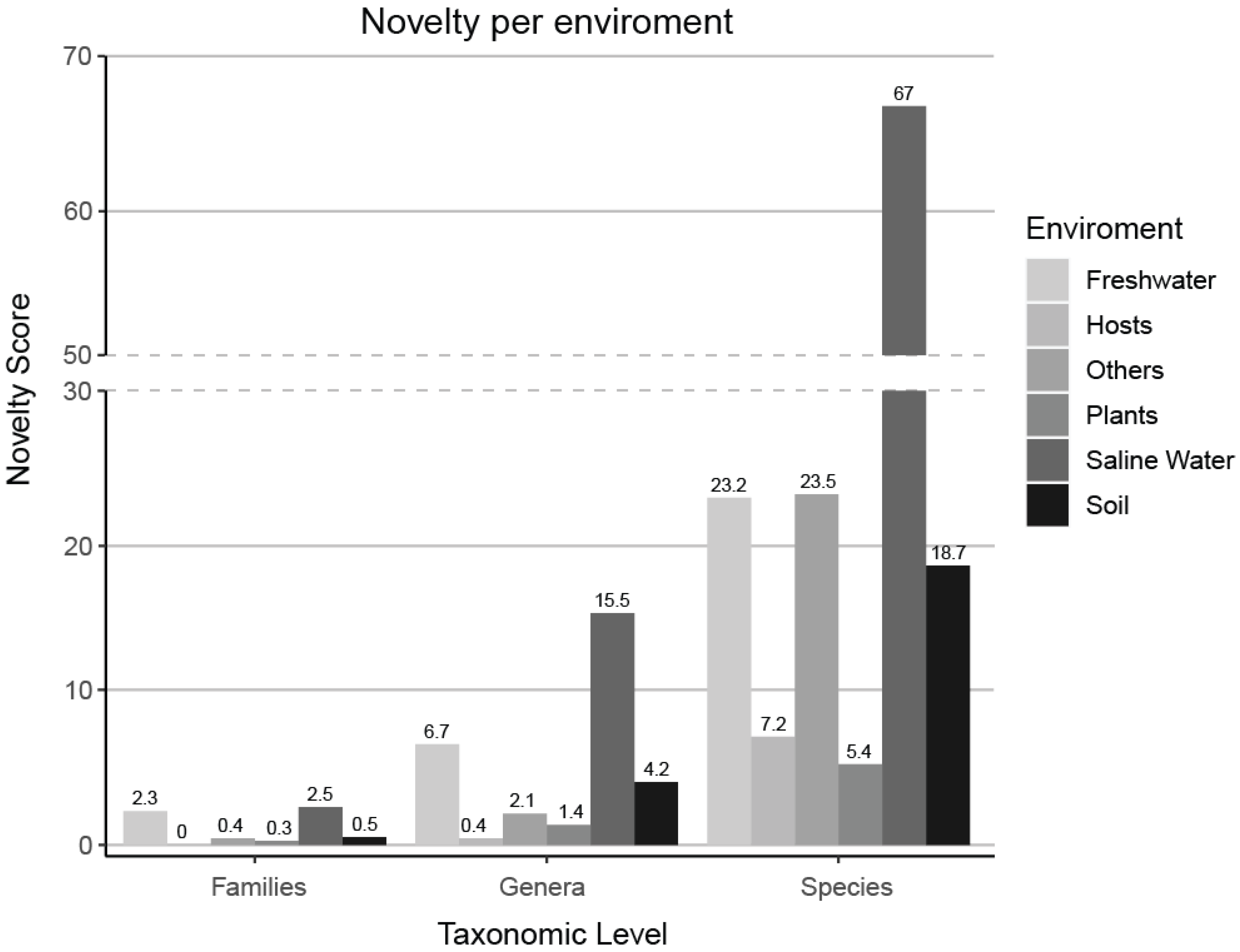
3.2. Novelty
3.3. Verification
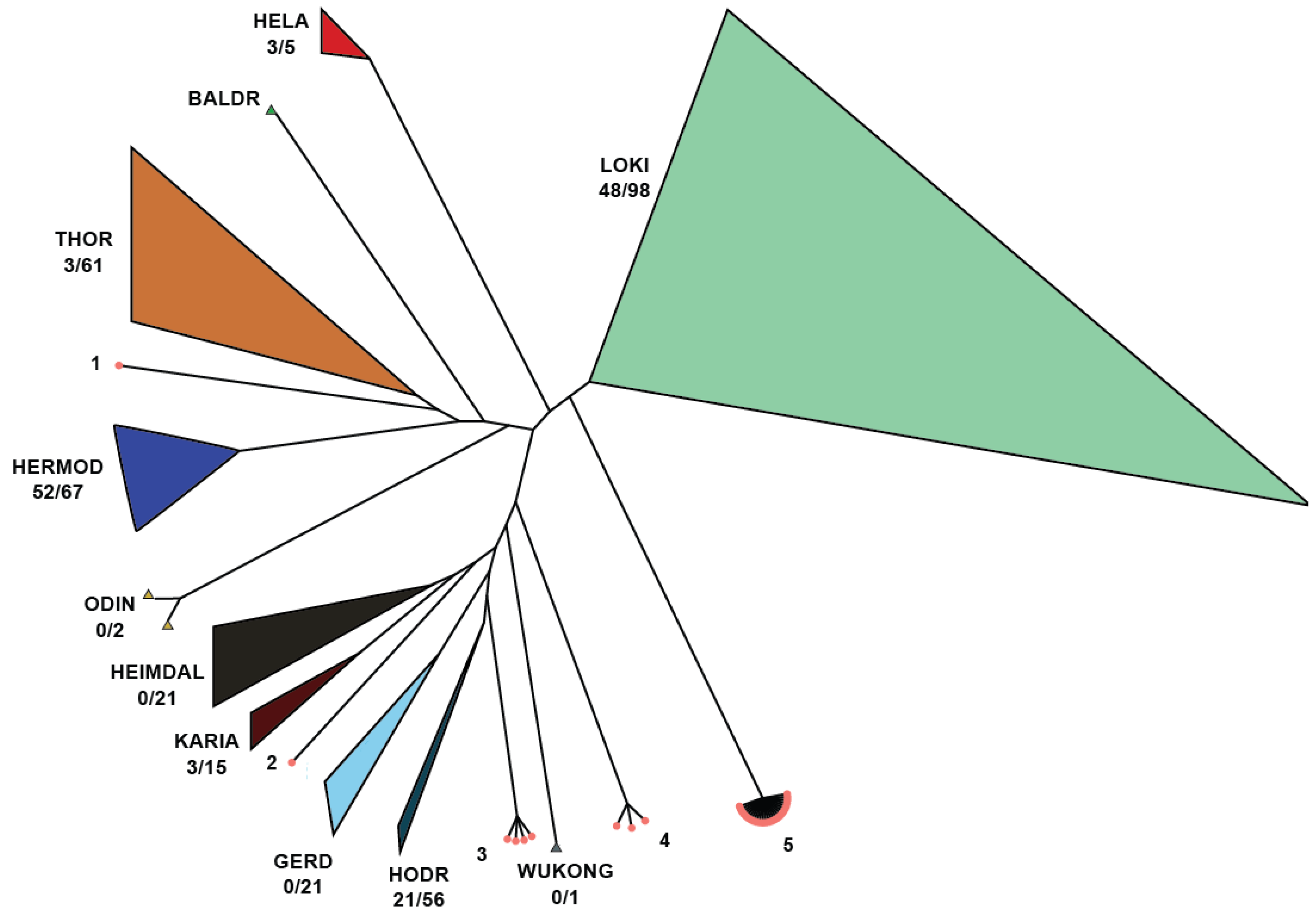
3.4. Asgardarchaeota Case Study
4. Discussion
4.1. Lower Boundary of Archaea Diversity
4.2. Sleipnirarchaeota
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Woese, C.R.; Kandler, O.; Wheelis, M.L. Towards a natural system of organisms: Proposal for the domains Archaea, Bacteria, and Eucarya. Proc. Natl. Acad. Sci. USA 1990, 87, 4576–4579. [Google Scholar] [CrossRef]
- Doolittle, W.F.; Logsdon, J.M., Jr. Archaeal genomics: Do archaea have a mixed heritage? Curr. Biol. 1998, 8, R209–R211. [Google Scholar] [CrossRef] [PubMed]
- DeLong, E.F. Archaea in coastal marine environments. Proc. Natl. Acad. Sci. USA 1992, 89, 5685–5689. [Google Scholar] [CrossRef]
- MacGregor, B.J.; Moser, D.P.; Alm, E.W.; Nealson, K.H.; Stahl, D.A. Crenarchaeota in lake Michigan sediment. Appl. Environ. Microbiol. 1997, 63, 1178–1181. [Google Scholar] [CrossRef]
- Rappé, M.S.; Giovannoni, S.J. The uncultured microbial majority. Annu. Rev. Microbiol. 2003, 57, 369–394. [Google Scholar] [CrossRef] [PubMed]
- Hugenholtz, P.; Goebel, B.M.; Pace, N.R. Impact of culture-independent studies on the emerging phylogenetic view of bacterial diversity. J. Bacteriol. 1998, 180, 4765–4774. [Google Scholar] [CrossRef]
- Pace, N.R. A molecular view of microbial diversity and the biosphere. Science 1997, 276, 734–740. [Google Scholar] [CrossRef] [PubMed]
- Baker, B.J.; Dick, G.J. Omic approaches in microbial ecology: Charting the unknown. Microbe 2013, 8, 353–359. [Google Scholar] [CrossRef]
- Barns, S.M.; Fundyga, R.E.; Jeffries, M.W.; Pace, N.R. Remarkable archaeal diversity detected in a Yellowstone National Park hot spring environment. Proc. Natl. Acad. Sci. USA 1994, 91, 1609–1613. [Google Scholar] [CrossRef]
- Fuhrman, J.A.; McCallum, K.; Davis, A.A. Novel major archaebacterial group from marine plankton. Nature 1992, 356, 148–149. [Google Scholar] [CrossRef]
- Barns, S.M.; Delwiche, C.F.; Palmer, J.D.; Pace, N.R. Perspectives on archaeal diversity, thermophily and monophyly from environmental rRNA sequences. Proc. Natl. Acad. Sci. USA 1996, 93, 9188–9193. [Google Scholar] [CrossRef] [PubMed]
- Rinke, C.; Schwientek, P.; Sczyrba, A.; Ivanova, N.N.; Anderson, I.J.; Cheng, J.F.; Darling, A.; Malfatti, S.; Swan, B.K.; Gies, E.A.; et al. Insights into the phylogeny and coding potential of microbial dark matter. Nature 2013, 499, 431–437. [Google Scholar] [CrossRef] [PubMed]
- Adam, P.S.; Borrel, G.; Brochier-Armanet, C.; Gribaldo, S. The growing tree of Archaea: New perspectives on their diversity, evolution and ecology. ISME J. 2017, 11, 2407–2425. [Google Scholar] [CrossRef] [PubMed]
- Stepanauskas, R.; Sieracki, M.E. Matching phylogeny and metabolism in the uncultured marine bacteria, one cell at a time. Proc. Natl. Acad. Sci. USA 2007, 104, 9052–9057. [Google Scholar] [CrossRef]
- Dick, G.J.; Andersson, A.F.; Baker, B.J.; Simmons, S.L.; Thomas, B.C.; Yelton, A.P.; Banfield, J.F. Community-wide analysis of microbial genome sequence signatures. Genome Biol. 2009, 10, 1–16. [Google Scholar] [CrossRef]
- Venter, J.C.; Remington, K.; Heidelberg, J.F.; Halpern, A.L.; Rusch, D.; Eisen, J.A.; Wu, D.; Paulsen, I.; Nelson, K.E.; Nelson, W.; et al. Environmental genome shotgun sequencing of the Sargasso Sea. Science 2004, 304, 66–74. [Google Scholar] [CrossRef]
- Tyson, G.W.; Chapman, J.; Hugenholtz, P.; Allen, E.E.; Ram, R.J.; Richardson, P.M.; Solovyev, V.V.; Rubin, E.M.; Rokhsar, D.S.; Banfield, J.F. Community structure and metabolism through reconstruction of microbial genomes from the environment. Nature 2004, 428, 37–43. [Google Scholar] [CrossRef]
- Peng, Y.; Leung, H.C.; Yiu, S.M.; Chin, F.Y. IDBA-UD: A de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics 2012, 28, 1420–1428. [Google Scholar] [CrossRef] [PubMed]
- Schloss, P.D.; Girard, R.A.; Martin, T.; Edwards, J.; Thrash, J.C. Status of the archaeal and bacterial census: An update. mBio 2016, 7, e00201-16. [Google Scholar] [CrossRef]
- Amann, R.; Rosselló-Móra, R. After all, only millions? mBio 2016, 7, e00999-16. [Google Scholar] [CrossRef]
- Dombrowski, N.; Teske, A.P.; Baker, B.J. Expansive microbial metabolic versatility and biodiversity in dynamic Guaymas Basin hydrothermal sediments. Nat. Commun. 2018, 9, 4999. [Google Scholar] [CrossRef] [PubMed]
- Hug, L.A.; Baker, B.J.; Anantharaman, K.; Brown, C.T.; Probst, A.J.; Castelle, C.J.; Butterfield, C.N.; Hernsdorf, A.W.; Amano, Y.; Ise, K.; et al. A new view of the tree of life. Nat. Microbiol. 2016, 1, 16048. [Google Scholar] [CrossRef]
- Lagkouvardos, I.; Joseph, D.; Kapfhammer, M.; Giritli, S.; Horn, M.; Haller, D.; Clavel, T. IMNGS: A comprehensive open resource of processed 16S rRNA microbial profiles for ecology and diversity studies. Sci. Rep. 2016, 6, 33721. [Google Scholar] [CrossRef] [PubMed]
- Leinonen, R.; Sugawara, H.; Shumway, M.; Collaboration, I.N.S.D. The sequence read archive. Nucleic Acids Res. 2010, 39, D19–D21. [Google Scholar] [CrossRef] [PubMed]
- Kopylova, E.; Noé, L.; Touzet, H. SortMeRNA: Fast and accurate filtering of ribosomal RNAs in metatranscriptomic data. Bioinformatics 2012, 28, 3211–3217. [Google Scholar] [CrossRef]
- Edgar, R.C. UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 2013, 10, 996–998. [Google Scholar] [CrossRef]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef]
- Lan, Y.; Wang, Q.; Cole, J.R.; Rosen, G.L. Using the RDP classifier to predict taxonomic novelty and reduce the search space for finding novel organisms. PLoS ONE 2012, 7, e32491. [Google Scholar] [CrossRef]
- Yarza, P.; Richter, M.; Peplies, J.; Euzeby, J.; Amann, R.; Schleifer, K.H.; Ludwig, W.; Glöckner, F.O.; Rosselló-Móra, R. The All-Species Living Tree project: A 16S rRNA-based phylogenetic tree of all sequenced type strains. Syst. Appl. Microbiol. 2008, 31, 241–250. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2012, 41, D590–D596. [Google Scholar] [CrossRef]
- Kioukis, A.; Pourjam, M.; Neuhaus, K.; Lagkouvardos, I. Taxonomy Informed Clustering, an Optimized Method for Purer and More Informative Clusters in Diversity Analysis and Microbiome Profiling. Front. Bioinform. 2022, 2, 864597. [Google Scholar] [CrossRef] [PubMed]
- Chen, I.M.A.; Chu, K.; Palaniappan, K.; Ratner, A.; Huang, J.; Huntemann, M.; Hajek, P.; Ritter, S.; Varghese, N.; Seshadri, R.; et al. The IMG/M data management and analysis system v. 6.0: New tools and advanced capabilities. Nucleic Acids Res. 2021, 49, D751–D763. [Google Scholar] [CrossRef] [PubMed]
- Clum, A.; Huntemann, M.; Bushnell, B.; Foster, B.; Foster, B.; Roux, S.; Hajek, P.P.; Varghese, N.; Mukherjee, S.; Reddy, T.B.K.; et al. DOE JGI Metagenome Workflow. mSystems 2021, 6, e00804-20. [Google Scholar] [CrossRef]
- Parks, D.H.; Chuvochina, M.; Rinke, C.; Mussig, A.J.; Chaumeil, P.A.; Hugenholtz, P. GTDB: An ongoing census of bacterial and archaeal diversity through a phylogenetically consistent, rank normalized and complete genome-based taxonomy. Nucleic Acids Res. 2022, 50, D785–D794. [Google Scholar] [CrossRef] [PubMed]
- Chaumeil, P.A.; Mussig, A.J.; Hugenholtz, P.; Parks, D.H. GTDB-Tk: A toolkit to classify genomes with the Genome Taxonomy Database. Bioinformatics 2020, 36, 1925–1927. [Google Scholar] [CrossRef]
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahé, F. VSEARCH: A versatile open source tool for metagenomics. PeerJ 2016, 4, e2584. [Google Scholar] [CrossRef]
- Baker, B.J.; De Anda, V.; Seitz, K.W.; Dombrowski, N.; Santoro, A.E.; Lloyd, K.G. Diversity, ecology and evolution of Archaea. Nat. Microbiol. 2020, 5, 887–900. [Google Scholar] [CrossRef]
- Zhou, Z.; Liu, Y.; Lloyd, K.G.; Pan, J.; Yang, Y.; Gu, J.D.; Li, M. Genomic and transcriptomic insights into the ecology and metabolism of benthic archaeal cosmopolitan, Thermoprofundales (MBG-D archaea). ISME J. 2019, 13, 885–901. [Google Scholar] [CrossRef]
- Lloyd, K.G.; Schreiber, L.; Petersen, D.G.; Kjeldsen, K.U.; Lever, M.A.; Steen, A.D.; Stepanauskas, R.; Richter, M.; Kleindienst, S.; Lenk, S.; et al. Predominant archaea in marine sediments degrade detrital proteins. Nature 2013, 496, 215–218. [Google Scholar] [CrossRef]
- Tully, B.J. Metabolic diversity within the globally abundant Marine Group II Euryarchaea offers insight into ecological patterns. Nat. Commun. 2019, 10, 271. [Google Scholar] [CrossRef]
- Rinke, C.; Rubino, F.; Messer, L.F.; Youssef, N.; Parks, D.H.; Chuvochina, M.; Brown, M.; Jeffries, T.; Tyson, G.W.; Seymour, J.R.; et al. A phylogenomic and ecological analysis of the globally abundant Marine Group II archaea (Ca. Poseidoniales ord. nov.). ISME J. 2019, 13, 663–675. [Google Scholar] [CrossRef]
- Spang, A.; Stairs, C.W.; Dombrowski, N.; Eme, L.; Lombard, J.; Caceres, E.F.; Greening, C.; Baker, B.J.; Ettema, T.J. Proposal of the reverse flow model for the origin of the eukaryotic cell based on comparative analyses of Asgard archaeal metabolism. Nat. Microbiol. 2019, 4, 1138–1148. [Google Scholar] [CrossRef]
- Imachi, H.; Nobu, M.K.; Nakahara, N.; Morono, Y.; Ogawara, M.; Takaki, Y.; Takano, Y.; Uematsu, K.; Ikuta, T.; Ito, M.; et al. Isolation of an archaeon at the prokaryote–eukaryote interface. Nature 2020, 577, 519–525. [Google Scholar] [CrossRef] [PubMed]
- Zaremba-Niedzwiedzka, K.; Caceres, E.F.; Saw, J.H.; Bäckström, D.; Juzokaite, L.; Vancaester, E.; Seitz, K.W.; Anantharaman, K.; Starnawski, P.; Kjeldsen, K.U.; et al. Asgard archaea illuminate the origin of eukaryotic cellular complexity. Nature 2017, 541, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Akıl, C.; Robinson, R.C. Genomes of Asgard archaea encode profilins that regulate actin. Nature 2018, 562, 439–443. [Google Scholar] [CrossRef] [PubMed]
- Seitz, K.W.; Dombrowski, N.; Eme, L.; Spang, A.; Lombard, J.; Sieber, J.R.; Teske, A.P.; Ettema, T.J.; Baker, B.J. Asgard archaea capable of anaerobic hydrocarbon cycling. Nat. Commun. 2019, 10, 1822. [Google Scholar] [CrossRef]
- Seitz, K.W.; Lazar, C.S.; Hinrichs, K.U.; Teske, A.P.; Baker, B.J. Genomic reconstruction of a novel, deeply branched sediment archaeal phylum with pathways for acetogenesis and sulfur reduction. ISME J. 2016, 10, 1696–1705. [Google Scholar] [CrossRef]
- Parte, A.C.; Carbasse, J.S.; Meier-Kolthoff, J.P.; Reimer, L.C.; Göker, M. List of Prokaryotic names with Standing in Nomenclature (LPSN) moves to the DSMZ. Int. J. Syst. Evol. Microbiol. 2020, 70, 5607. [Google Scholar] [CrossRef]
- Liu, Y.; Makarova, K.S.; Huang, W.C.; Wolf, Y.I.; Nikolskaya, A.N.; Zhang, X.; Cai, M.; Zhang, C.J.; Xu, W.; Luo, Z.; et al. Expanded diversity of Asgard archaea and their relationships with eukaryotes. Nature 2021, 593, 553–557. [Google Scholar] [CrossRef]
- Xie, R.; Wang, Y.; Huang, D.; Hou, J.; Li, L.; Hu, H.; Zhao, X.; Wang, F. Expanding Asgard members in the domain of Archaea sheds new light on the origin of eukaryotes. Sci. China Life Sci. 2021, 65, 818–829. [Google Scholar] [CrossRef]
- Barbera, P.; Kozlov, A.M.; Czech, L.; Morel, B.; Darriba, D.; Flouri, T.; Stamatakis, A. EPA-ng: Massively parallel evolutionary placement of genetic sequences. Syst. Biol. 2019, 68, 365–369. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, M.D.; Dumontier, M.; Aalbersberg, I.J.; Appleton, G.; Axton, M.; Baak, A.; Blomberg, N.; Boiten, J.W.; da Silva Santos, L.B.; Bourne, P.E.; et al. The FAIR Guiding Principles for scientific data management and stewardship. Sci. Data 2016, 3, 1–9. [Google Scholar] [CrossRef]
- Mourgela, R.N.; Kioukis, A.; Pourjam, M.; Lagkouvardos, I. Large-scale integration of amplicon data reveals massive diversity within saprospirales, mostly originating from saline environments. Microorganisms 2023, 11, 1767. [Google Scholar] [CrossRef]
- Lagkouvardos, I.; Weinmaier, T.; Lauro, F.M.; Cavicchioli, R.; Rattei, T.; Horn, M. Integrating metagenomic and amplicon databases to resolve the phylogenetic and ecological diversity of the Chlamydiae. ISME J. 2014, 8, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Spang, A.; Saw, J.H.; Jørgensen, S.L.; Zaremba-Niedzwiedzka, K.; Martijn, J.; Lind, A.E.; Van Eijk, R.; Schleper, C.; Guy, L.; Ettema, T.J. Complex archaea that bridge the gap between prokaryotes and eukaryotes. Nature 2015, 521, 173–179. [Google Scholar] [CrossRef]
- Ettema, T.J.; Lindås, A.C.; Bernander, R. An actin-based cytoskeleton in archaea. Mol. Microbiol. 2011, 80, 1052–1061. [Google Scholar] [CrossRef]
- Koonin, E.V.; Yutin, N. The dispersed archaeal eukaryome and the complex archaeal ancestor of eukaryotes. Cold Spring Harb. Perspect. Biol. 2014, 6, a016188. [Google Scholar] [CrossRef]
- Dalziel, M.; Crispin, M.; Scanlan, C.N.; Zitzmann, N.; Dwek, R.A. Emerging principles for the therapeutic exploitation of glycosylation. Science 2014, 343, 1235681. [Google Scholar] [CrossRef] [PubMed]
- Eme, L.; Spang, A.; Lombard, J.; Stairs, C.W.; Ettema, T.J. Archaea and the origin of eukaryotes. Nat. Rev. Microbiol. 2017, 15, 711–723. [Google Scholar] [CrossRef]
- Williams, T.A.; Cox, C.J.; Foster, P.G.; Szöllősi, G.J.; Embley, T.M. Phylogenomics provides robust support for a two-domains tree of life. Nat. Ecol. Evol. 2020, 4, 138–147. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Database | #Families | #Genera | #Species |
|---|---|---|---|
| LTP | 35 | 129 | 490 |
| SILVA | 82 | 161 | - |
| GTDB | 2621 | 2769 | 3044 |
| GAD (singletons in) | 98,172 | 561,788 | 2,807,013 |
| GAD (singletons out) | 30,887 | 87,200 | 419,934 |
| SOTUs | 99 | 97 | 93 | 89 | |
|---|---|---|---|---|---|
| Singletons | 2,398,351 | 17,986 (0.7%) | 254,283 (10%) | 1,485,595 (61%) | 2,246,610 (93%) |
| Doubletons | 151,600 | 8157 (5%) | 52,428 (34%) | 112,662 (74%) | 144,255 (95%) |
| Tripletons | 61,824 | 4905 (7%) | 22,172 (35%) | 41,752 (67%) | 58,978 (95%) |
| Moretons | 195,238 | 36,579 (18%) | 61,631 (31%) | 151,397 (77%) | 189,018 (96%) |
| Total | 2,807,013 | 67,627 (2%) | 390,514 (13%) | 1,791,406 (63%) | 2,638,461 (93%) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kioukis, A.; Camargo, A.P.; Pavlidis, P.; Iliopoulos, I.; Kyrpides, N.C.; Lagkouvardos, I. Global Archaeal Diversity Revealed Through Massive Data Integration: Uncovering Just Tip of Iceberg. Microorganisms 2025, 13, 598. https://doi.org/10.3390/microorganisms13030598
Kioukis A, Camargo AP, Pavlidis P, Iliopoulos I, Kyrpides NC, Lagkouvardos I. Global Archaeal Diversity Revealed Through Massive Data Integration: Uncovering Just Tip of Iceberg. Microorganisms. 2025; 13(3):598. https://doi.org/10.3390/microorganisms13030598
Chicago/Turabian StyleKioukis, Antonios, Antonio Pedro Camargo, Pavlos Pavlidis, Ioannis Iliopoulos, Nikos C Kyrpides, and Ilias Lagkouvardos. 2025. "Global Archaeal Diversity Revealed Through Massive Data Integration: Uncovering Just Tip of Iceberg" Microorganisms 13, no. 3: 598. https://doi.org/10.3390/microorganisms13030598
APA StyleKioukis, A., Camargo, A. P., Pavlidis, P., Iliopoulos, I., Kyrpides, N. C., & Lagkouvardos, I. (2025). Global Archaeal Diversity Revealed Through Massive Data Integration: Uncovering Just Tip of Iceberg. Microorganisms, 13(3), 598. https://doi.org/10.3390/microorganisms13030598