
Metagenomic and Metabolomic Analyses Reveal the Role of a Bacteriocin-Producing Strain of Enterococcus faecalis DH9003 in Regulating Gut Microbiota in Mice


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials and Strain Culture Conditions
2.2. E. faecalis DH9003 Preparation
2.3. Fecal Sample Collection
2.4. Detection of Antimicrobial Activity
2.5. DNA Isolation
2.6. Metagenomic Sequencing Analysis
2.7. Sequencing Quality Control
2.8. Metabolomic Analysis
2.9. Statistics Analysis
3. Results
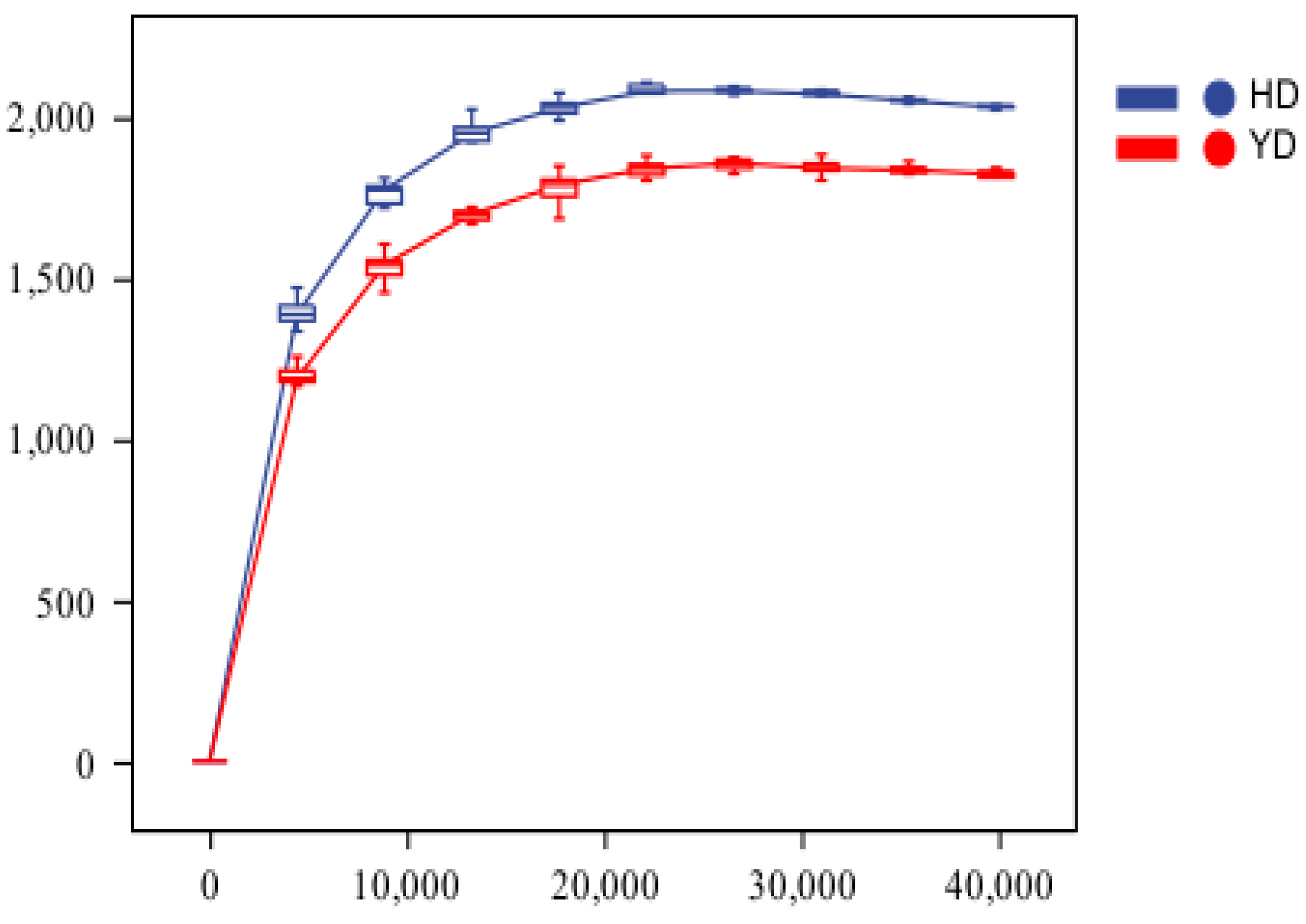
3.1. Metagenomic Sequencing Data
3.2. Alpha Diversity Analysis
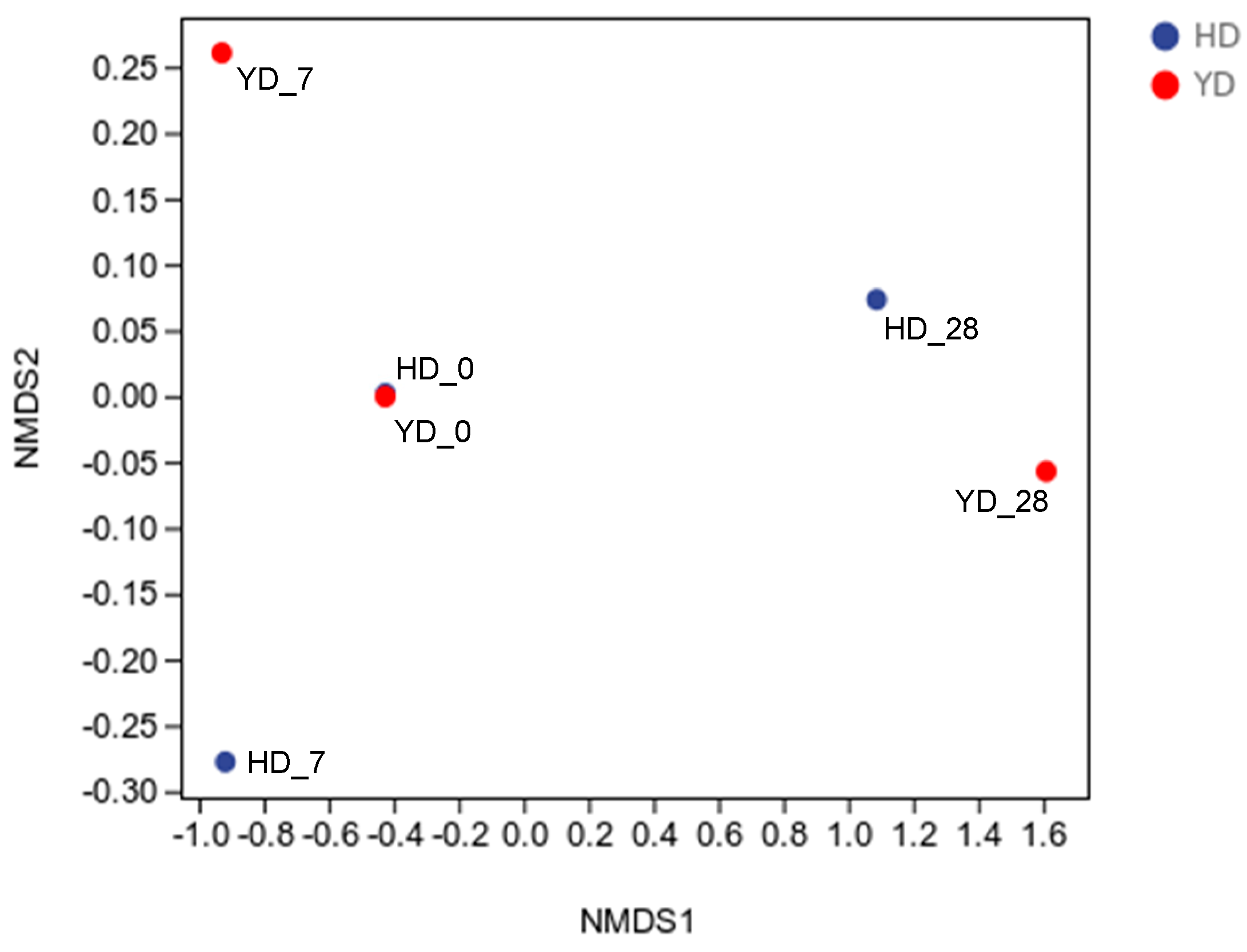
3.3. Beta Diversity Analysis
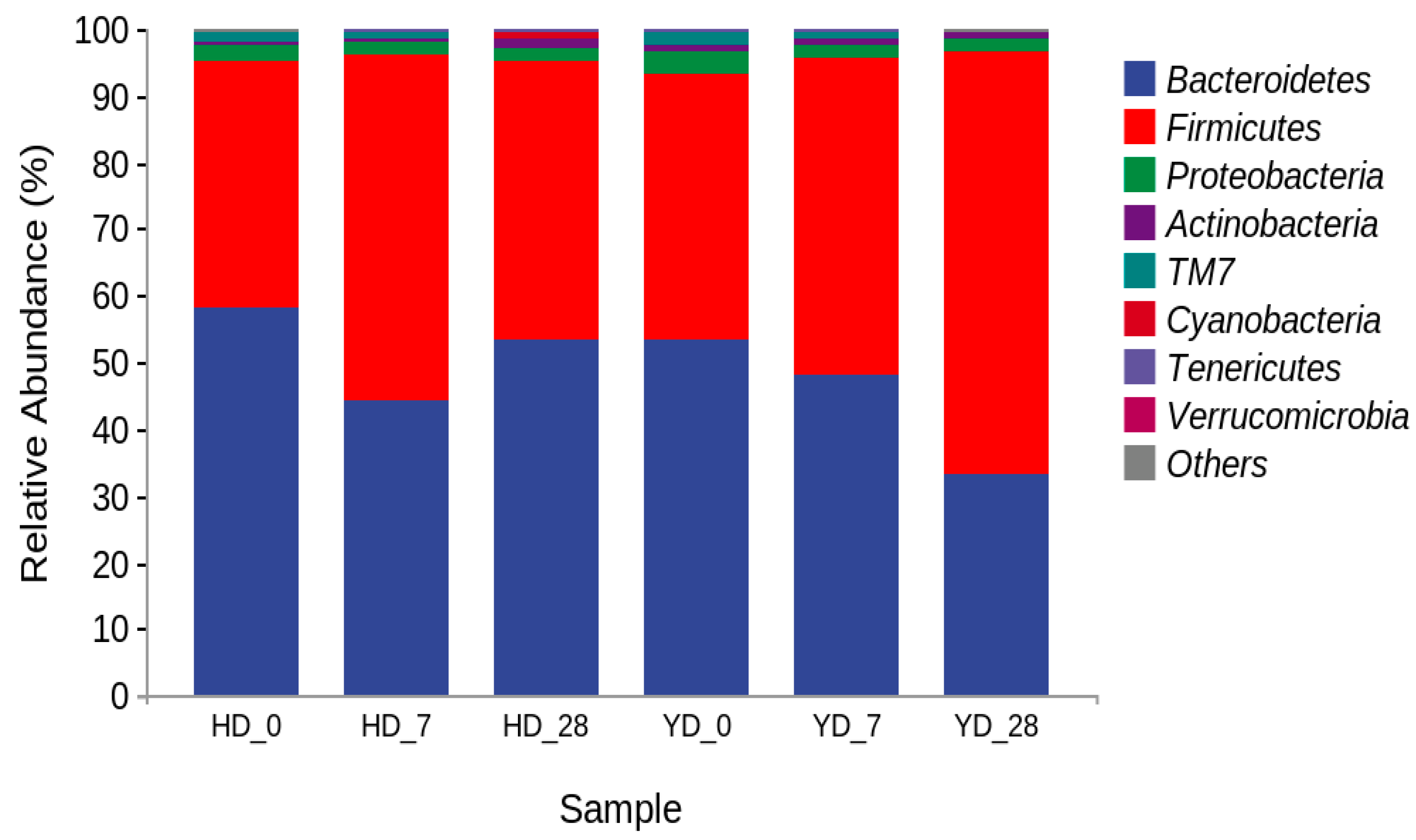
3.4. Microorganism Composition Analysis
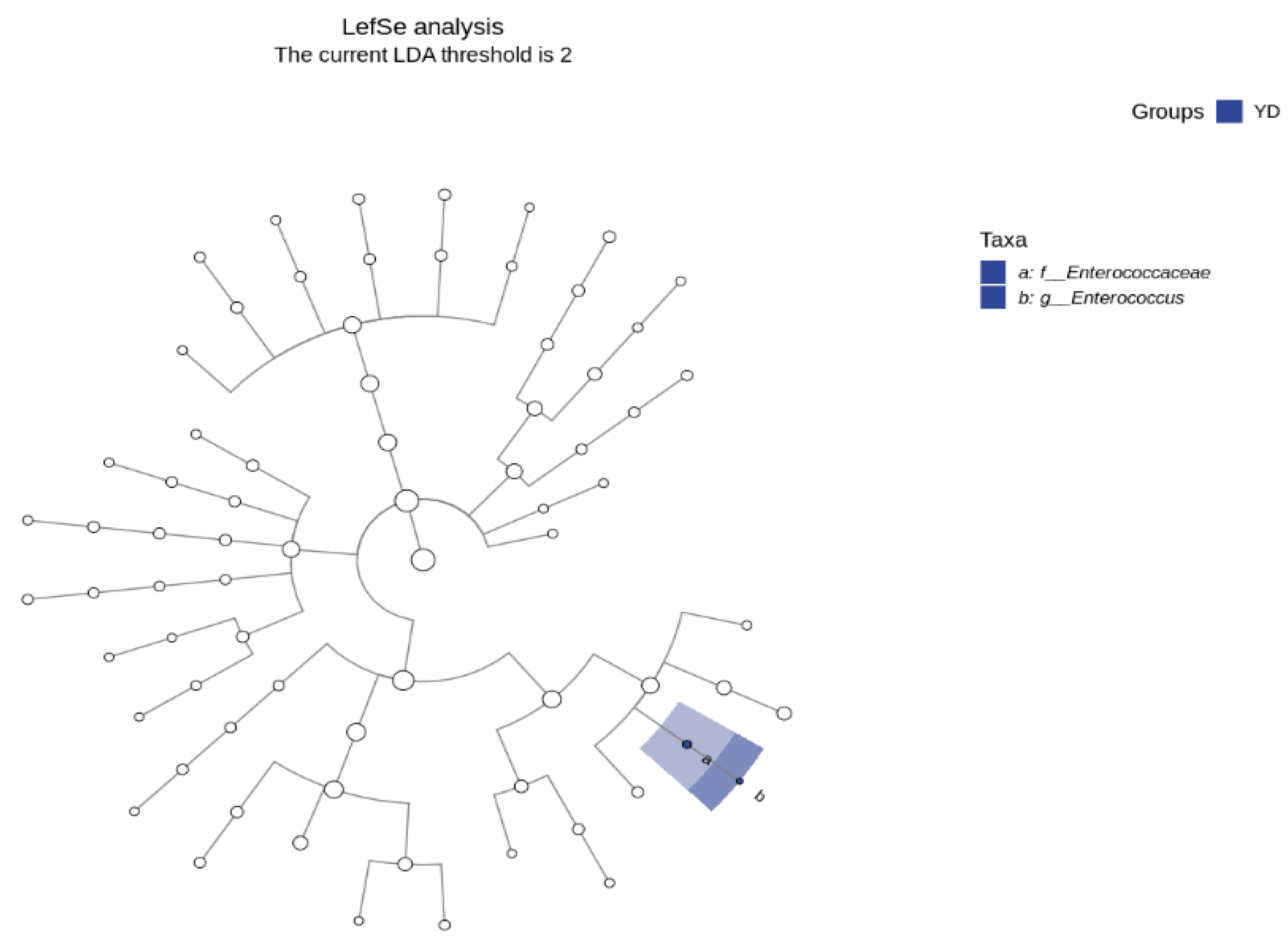
3.5. LDA Effect Size (Lefse) Analysis and Marker Species Determination
3.6. E. faecalis DH9003 Isolation and Its Antimicrobial Activity
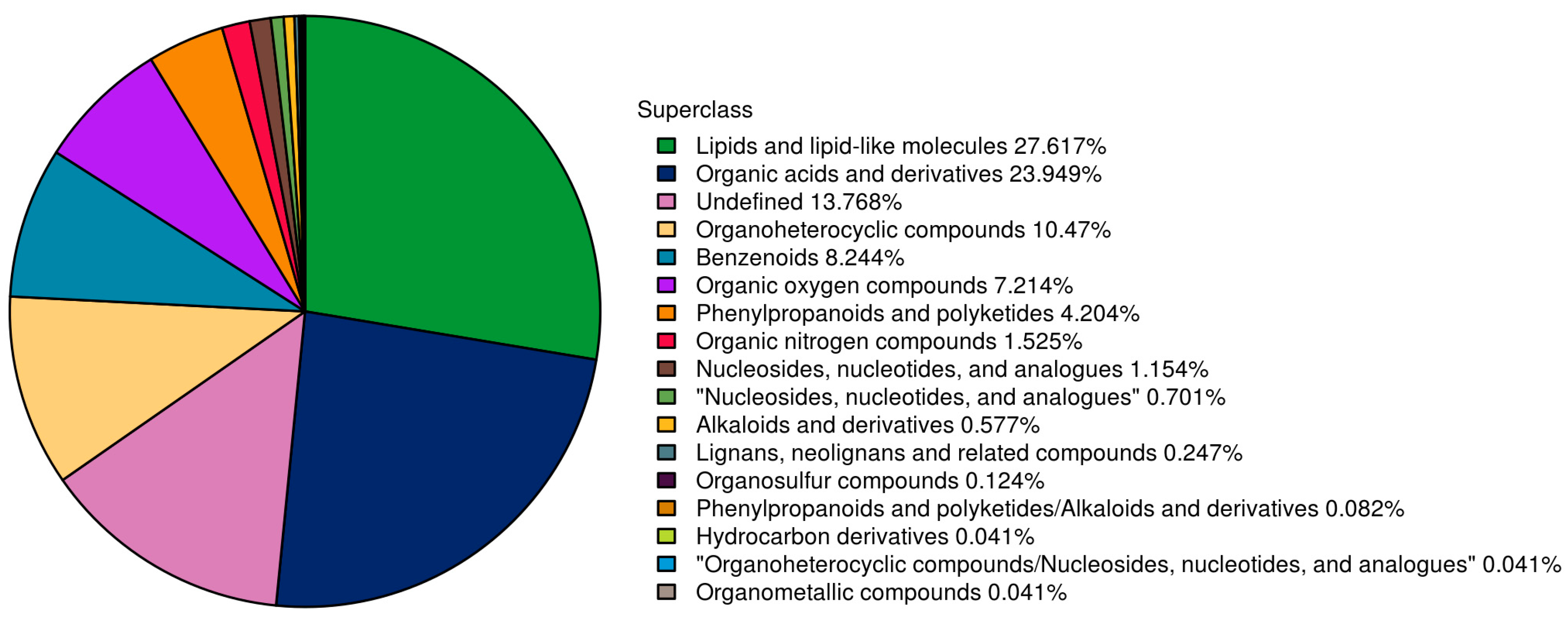
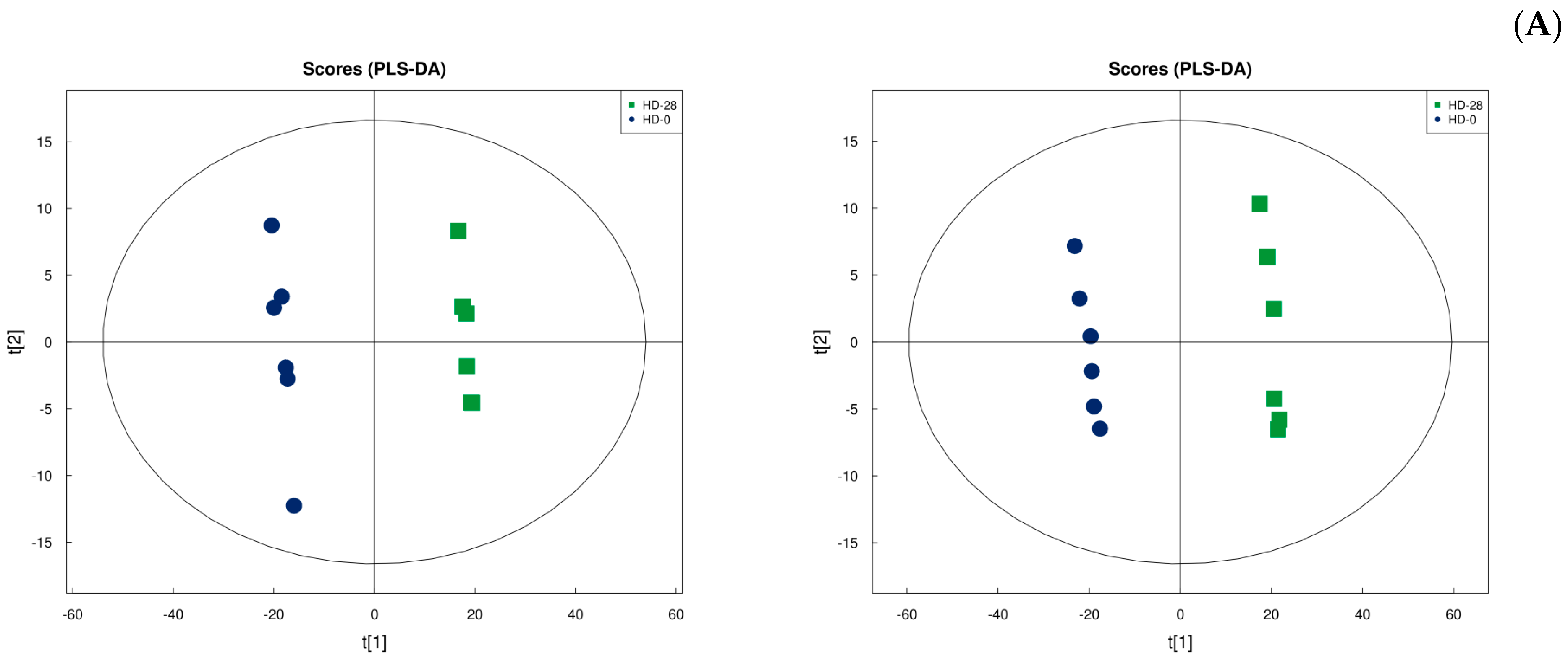
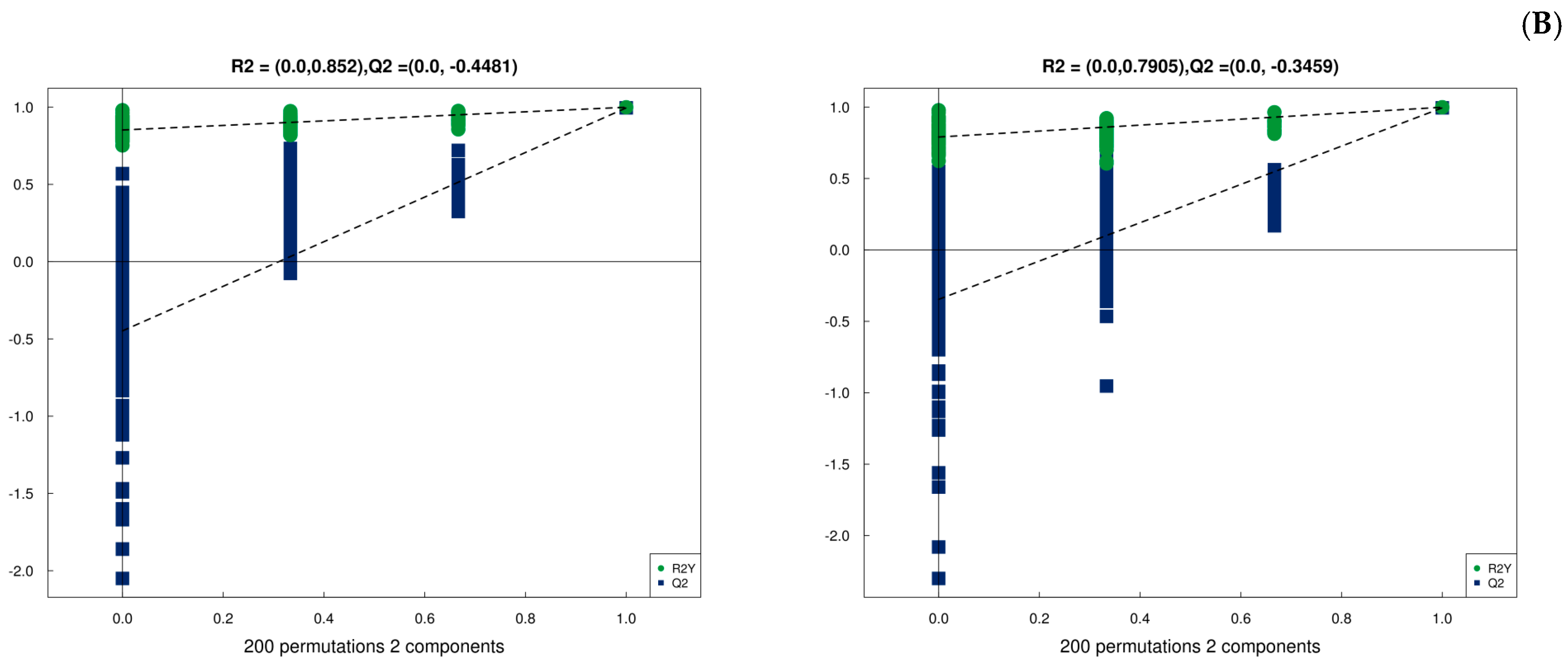
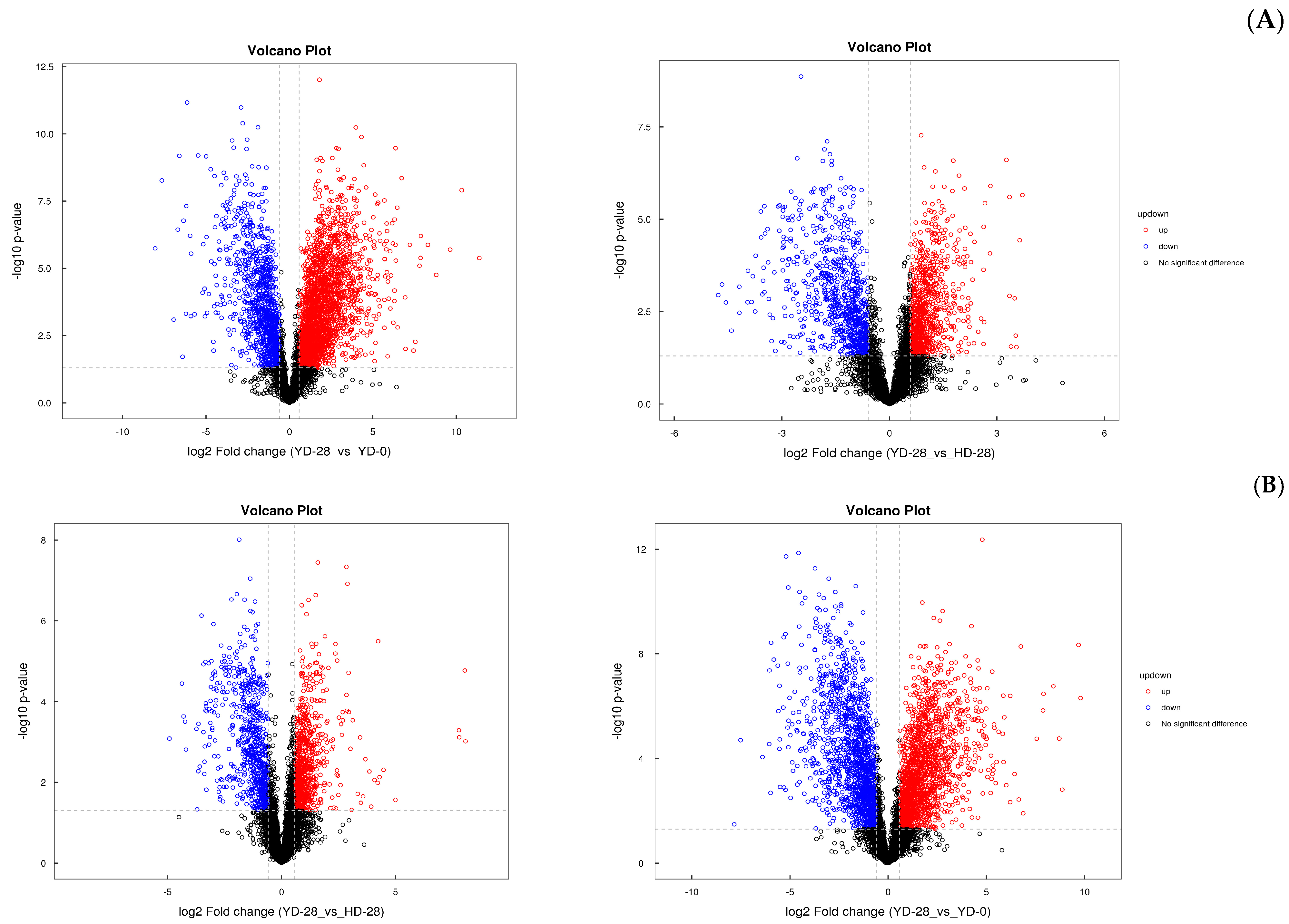
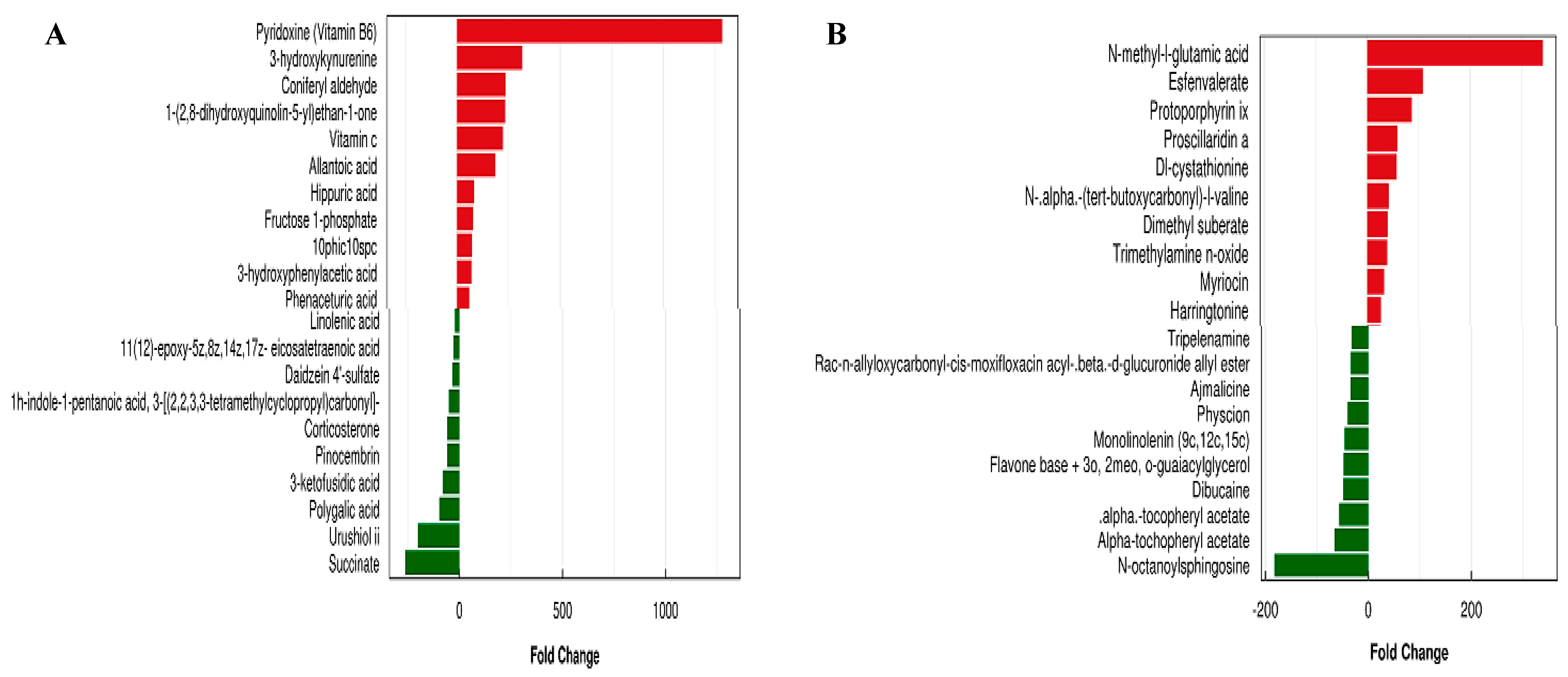
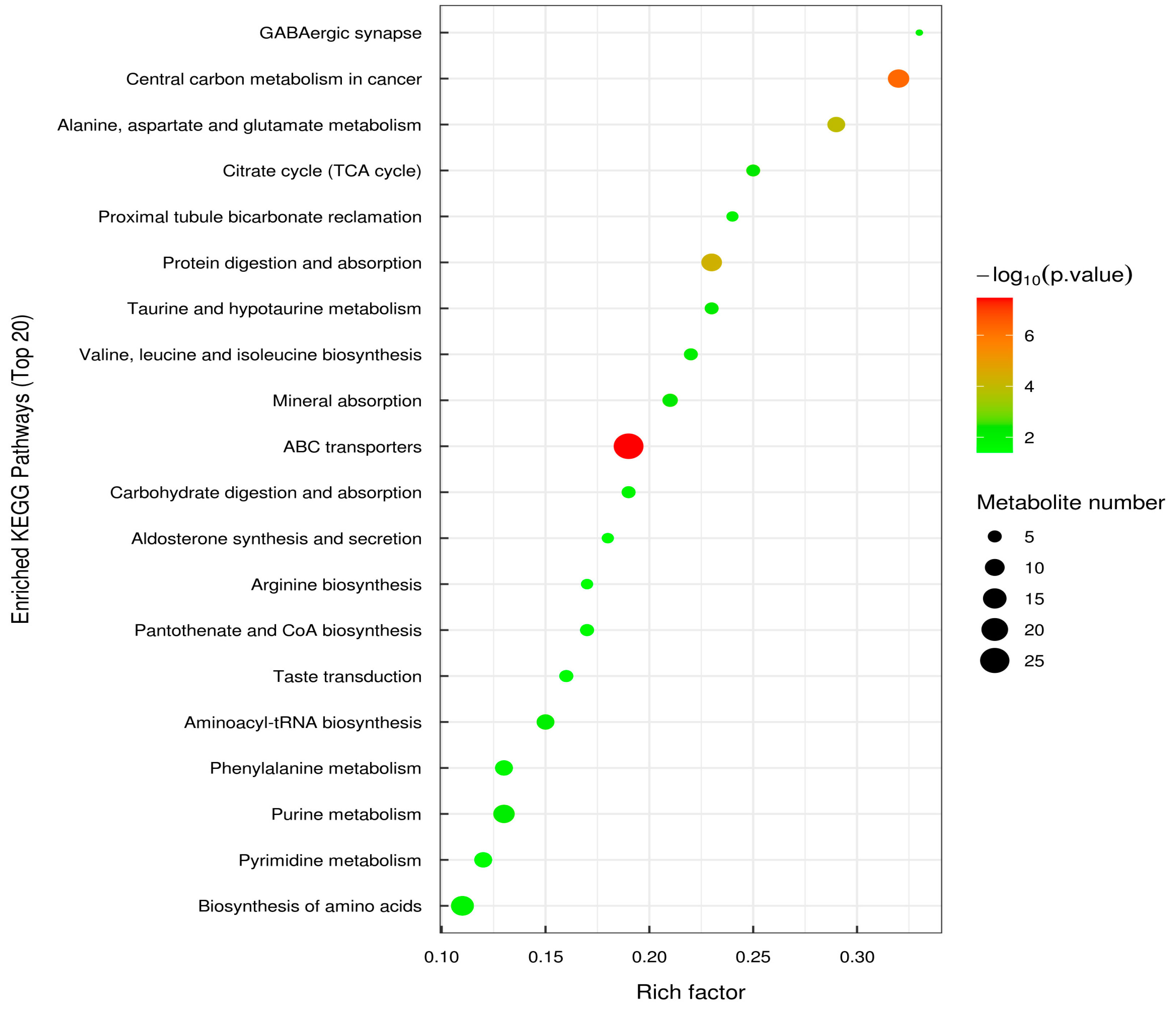
3.7. Metabolic Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mokoena, M.P. Lactic Acid Bacteria and Their Bacteriocins: Classification, Biosynthesis and Applications against Uropathogens: A Mini-Review. Molecules 2017, 22, 1255. [Google Scholar] [CrossRef]
- Raman, J.; Kim, J.-S.; Choi, K.R.; Eun, H.; Yang, D.; Ko, Y.-J.; Kim, S.-J. Application of Lactic Acid Bacteria (LAB) in Sustainable Agriculture: Advantages and Limitations. Int. J. Mol. Sci. 2022, 23, 7784. [Google Scholar] [CrossRef]
- Chi, H.; Holo, H. Synergistic Antimicrobial Activity Between the Broad Spectrum Bacteriocin Garvicin KS and Nisin, Farnesol and Polymyxin B Against Gram-Positive and Gram-Negative Bacteria. Curr. Microbiol. 2018, 75, 272–277. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, A.; Jobby, R. Bacteriocins from lactic acid bacteria and their potential clinical applications. Appl. Biochem. Biotechnol. 2022, 194, 4377–4399. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Moore, R.J.; Stanley, D.; Chousalkar, K.K. The Gut Microbiota of Laying Hens and Its Manipulation with Prebiotics and Probiotics To Enhance Gut Health and Food Safety. Appl. Environ. Microbiol. 2020, 86, e00600-20. [CrossRef]
- Anjana; Tiwari, S.K. Bacteriocin-Producing Probiotic Lactic Acid Bacteria in Controlling Dysbiosis of the Gut Microbiota. Front. Cell Infect. Microbiol. 2022, 12, 851140. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.; Ed-Dra, A.; Song, Y.; Elbediwi, M.; Nambiar, R.B.; Zhou, X.; Yue, M. Lacticaseibacillus rhamnosus alleviates intestinal inflammation and promotes microbiota-mediated protection against Salmonella fatal infections. Front. Immunol. 2022, 13, 973224. [Google Scholar] [CrossRef] [PubMed]
- Anastasilakis, C.D.; Ioannidis, O.; Gkiomisi, A.I.; Botsios, D. Artificial nutrition and intestinal mucosal barrier functionality. Digestion 2013, 88, 193–208. [Google Scholar] [CrossRef]
- Sanders, M.E.; Merenstein, D.J.; Reid, G.; Gibson, G.R.; Rastall, R.A. Probiotics and prebiotics in intestinal health and disease: From biology to the clinic. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 605–616. [Google Scholar] [CrossRef]
- Parvez, S.; Malik, K.A.; Kang, S.; Kim, H.Y. Probiotics and their fermented food products are beneficial for health. J. Appl. Microbiol. 2006, 100, 1171–1185. [Google Scholar] [CrossRef]
- Markelova, N.N.; Semenova, E.F.; Sineva, O.N.; Sadykova, V.S. The Role of Cyclomodulins and Some Microbial Metabolites in Bacterial Microecology and Macroorganism Carcinogenesis. Int. J. Mol. Sci. 2022, 23, 11706. [Google Scholar] [CrossRef] [PubMed]
- Viesser, J.A.; de Melo Pereira, G.V.; de Carvalho Neto, D.P.; Rogez, H.; Góes-Neto, A.; Azevedo, V.; Brenig, B.; Aburjaile, F.; Soccol, C.R. Co-culturing fructophilic lactic acid bacteria and yeast enhanced sugar metabolism and aroma formation during cocoa beans fermentation. Int. J. Food Microbiol. 2021, 339, 109015. [Google Scholar] [CrossRef] [PubMed]
- Loncke, C.; Ortigues-Marty, I.; Vernet, J.; Lapierre, H.; Sauvant, D.; Nozière, P. Empirical prediction of net portal appearance of volatile fatty acids, glucose, and their secondary metabolites (beta-hydroxybutyrate, lactate) from dietary characteristics in ruminants: A meta-analysis approach. J. Anim. Sci. 2009, 87, 253–268. [Google Scholar] [CrossRef] [PubMed]
- Ducarmon, Q.R.; Zwittink, R.D.; Hornung, B.V.H.; van Schaik, W.; Young, V.B.; Kuijper, E.J. Gut Microbiota and Colonization Resistance against Bacterial Enteric Infection. Microbiol. Mol. Biol. Rev. 2019, 83, e00007-19. [Google Scholar] [CrossRef]
- Wu, H.; Xie, S.; Miao, J.; Li, Y.; Wang, Z.; Wang, M.; Yu, Q. Lactobacillus reuteri maintains intestinal epithelial regeneration and repairs damaged intestinal mucosa. Gut Microbes 2020, 11, 997–1014. [Google Scholar] [CrossRef] [PubMed]
- Yi, H.; Wang, L.; Xiong, Y.; Wen, X.; Wang, Z.; Yang, X.; Gao, K.; Jiang, Z. Effects of Lactobacillus reuteri LR1 on the growth performance, intestinal morphology, and intestinal barrier function in weaned pigs. J. Anim. Sci. 2018, 96, 2342–2351. [Google Scholar] [CrossRef] [PubMed]
- Rokana, N.; Singh, R.; Mallappa, R.H.; Batish, V.K.; Grover, S. Modulation of intestinal barrier function to ameliorate Salmonella infection in mice by oral administration of fermented milks produced with Lactobacillus plantarum MTCC 5690—A probiotic strain of Indian gut origin. J. Med. Microbiol. 2016, 65, 1482–1493. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.; Wang, L.C.; Di, S.; Stein, D.C.; Song, W. Gonococcal invasion into epithelial cells depends on both cell polarity and ezrin. PLoS Pathog. 2021, 17, e1009592. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Li, Q.; Cheng, L.; Buch, H.; Zhang, F. Akkermansia muciniphila is a promising probiotic. Microb. Biotechnol. 2019, 12, 1109–1125. [Google Scholar] [CrossRef]
- Kim, S.Y. Production of Fermented Kale Juices with Lactobacillus Strains and Nutritional Composition. Prev. Nutr. Food Sci. 2017, 22, 231–236. [Google Scholar] [CrossRef]
- Guo, X.; Okpara, E.S.; Hu, W.; Yan, C.; Wang, Y.; Liang, Q.; Chiang, J.Y.L.; Han, S. Interactive Relationships between Intestinal Flora and Bile Acids. Int. J. Mol. Sci. 2022, 23, 8343. [Google Scholar] [CrossRef]
- Trevelline, B.K.; Kohl, K.D. The gut microbiome influences host diet selection behavior. Proc. Natl. Acad. Sci. USA 2022, 119, e2117537119. [Google Scholar] [CrossRef]
- Brown, E.M.; Clardy, J.; Xavier, R.J. Gut microbiome lipid metabolism and its impact on host physiology. Cell Host Microbe 2023, 31, 173–186. [Google Scholar] [CrossRef] [PubMed]
- Krautkramer, K.A.; Fan, J.; Bäckhed, F. Gut microbial metabolites as multi-kingdom intermediates. Nat. Rev. Microbiol. 2021, 19, 77–94. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P. Influence of Foods and Nutrition on the Gut Microbiome and Implications for Intestinal Health. Int. J. Mol. Sci. 2022, 23, 9588. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.; Lyu, L.; Wang, Y.; Zhang, Y.; Guo, X.; Chen, Q.; Liu, C. Safety assessment and probiotic characteristics of Enterococcus lactis JDM1. Microb. Pathog. 2022, 163, 105380. [Google Scholar] [CrossRef]
- Tao, Y.; Huang, F.; Zhang, Z.; Tao, X.; Wu, Q.; Qiu, L.; Wei, H. Probiotic Enterococcus faecalis Symbioflor 1 ameliorates pathobiont-induced miscarriage through bacterial antagonism and Th1-Th2 modulation in pregnant mice. Appl. Microbiol. Biotechnol. 2020, 104, 5493–5504. [Google Scholar] [CrossRef]
- Yang, Z.; Ni, L.; Tian, W.; Chi, H. Screening and Identification of Goat-Milk-Derived Lactic Acid Bacteria with Bacteriocin-like Activity and Probiotic Potentials. Microorganisms 2023, 11, 849. [Google Scholar] [CrossRef] [PubMed]
- Kong, C.; Liang, L.; Liu, G.; Du, L.; Yang, Y.; Liu, J.; Shi, D.; Li, X.; Ma, Y. Integrated metagenomic and metabolomic analysis reveals distinct gut-microbiome-derived phenotypes in early-onset colorectal cancer. Gut 2023, 72, 1129–1142. [Google Scholar] [CrossRef] [PubMed]
- Hall, M.; Beiko, R.G. 16S rRNA Gene Analysis with QIIME2. Methods Mol. Biol. 2018, 1849, 113–129. [Google Scholar] [CrossRef]
- Mattoli, L.; Gianni, M.; Burico, M. Mass spectrometry-based metabolomic analysis as a tool for quality control of natural complex products. Mass Spectrom. Rev. 2023, 42, 1358–1396. [Google Scholar] [CrossRef] [PubMed]
- Bauermeister, A.; Mannochio-Russo, H.; Costa-Lotufo, L.V.; Jarmusch, A.K.; Dorrestein, P.C. Mass spectrometry-based metabolomics in microbiome investigations. Nat. Rev. Microbiol. 2022, 20, 143–160. [Google Scholar] [CrossRef] [PubMed]
- Hills, R.D.; Pontefract, B.A.; Mishcon, H.R.; Black, C.A.; Sutton, S.C.; Theberge, C.R. Gut Microbiome: Profound Implications for Diet and Disease. Nutrients 2019, 11, 1613. [Google Scholar] [CrossRef]
- Yang, J.; Liu, S.; Zhao, Q.; Li, X.; Jiang, K. Gut microbiota-related metabolite alpha-linolenic acid mitigates intestinal inflammation induced by oral infection with Toxoplasma gondii. Microbiome 2023, 11, 273. [Google Scholar] [CrossRef]
- Qu, H.; Long, Y.; Wang, X.; Wang, K.; Chen, L.; Yang, Y.; Chen, L. Diversity and Abundance of Bacterial and Fungal Communities Inhabiting Camellia sinensis Leaf, Rhizospheric Soil, and Gut of Agriophara rhombata. Microorganisms 2023, 11, 2188. [Google Scholar] [CrossRef]
- Jing, Z.; Zheng, W.; Jianwen, S.; Hong, S.; Xiaojian, Y.; Qiang, W.; Yunfeng, Y.; Xinyue, W.; Shuwen, H.; Feimin, Z. Gut microbes on the risk of advanced adenomas. BMC Microbiol. 2024, 24, 264. [Google Scholar] [CrossRef]
- Pan, T.; Guo, H.Y.; Zhang, H.; Liu, A.P.; Wang, X.X.; Ren, F.Z. Oral administration of Lactobacillus paracasei alleviates clinical symptoms of colitis induced by dextran sulphate sodium salt in BALB/c mice. Benef. Microbes 2014, 5, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Erawijantari, P.P.; Mizutani, S.; Shiroma, H.; Shiba, S.; Nakajima, T.; Sakamoto, T.; Saito, Y.; Fukuda, S.; Yachida, S.; Yamada, T. Influence of gastrectomy for gastric cancer treatment on faecal microbiome and metabolome profiles. Gut 2020, 69, 1404–1415. [Google Scholar] [CrossRef]
- Yang, L.; Xiang, Z.; Zou, J.; Zhang, Y.; Ni, Y.; Yang, J. Comprehensive Analysis of the Relationships Between the Gut Microbiota and Fecal Metabolome in Individuals With Primary Sjogren’s Syndrome by 16S rRNA Sequencing and LC-MS-Based Metabolomics. Front. Immunol. 2022, 13, 874021. [Google Scholar] [CrossRef] [PubMed]
- Umu, Ö.C.O.; Rudi, K.; Diep, D.B. Modulation of the gut microbiota by prebiotic fibres and bacteriocins. Microb. Ecol. Health Dis. 2017, 28, 1348886. [Google Scholar] [CrossRef]
- Rolfe, R.D. The role of probiotic cultures in the control of gastrointestinal health. J. Nutr. 2000, 130, 396S–402S. [Google Scholar] [CrossRef] [PubMed]
- Myhill, L.J.; Stolzenbach, S.; Mejer, H.; Krych, L.; Jakobsen, S.R.; Kot, W.; Skovgaard, K.; Canibe, N.; Nejsum, P.; Nielsen, D.S.; et al. Parasite-Probiotic Interactions in the Gut: Bacillus sp. and Enterococcus faecium Regulate Type-2 Inflammatory Responses and Modify the Gut Microbiota of Pigs During Helminth Infection. Front. Immunol. 2022, 12, 793260. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Liang, Z.T.; Yi, T.; Ma, Y.; Zhao, Z.Z.; Guo, B.L.; Zhang, J.Y.; Chen, H.B. Comparison of chemical profiles between the root and aerial parts from three Bupleurum species based on a UHPLC-QTOF-MS metabolomics approach. BMC Complement. Altern. Med. 2017, 17, 305. [Google Scholar] [CrossRef] [PubMed]
- Rinschen, M.M.; Ivanisevic, J.; Giera, M.; Siuzdak, G. Identification of bioactive metabolites using activity metabolomics. Nat. Rev. Mol. Cell Biol. 2019, 20, 353–367. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, J.; Zhang, C.; Deng, C.; Wen, A.; Zhou, H.; You, J.; Li, G. Dietary vitamin B6 supplementation alleviates heat stress-induced intestinal barrier impairment by regulating the gut microbiota and metabolites in broilers. Poult. Sci. 2024, 103, 104202. [Google Scholar] [CrossRef] [PubMed]
- Sharma, V.; Rodionov, D.A.; Leyn, S.A.; Tran, D.; Iablokov, S.N.; Ding, H.; Peterson, D.A.; Osterman, A.L.; Peterson, S.N. B-Vitamin Sharing Promotes Stability of Gut Microbial Communities. Front. Microbiol. 2019, 10, 1485. [Google Scholar] [CrossRef]
- Mandić, M.; Mitić, K.; Nedeljković, P.; Perić, M.; Božić, B.; Lunić, T.; Bačić, A.; Rajilić-Stojanović, M.; Peković, S.; Božić Nedeljković, B. Vitamin B Complex and Experimental Autoimmune Encephalomyelitis–Attenuation of the Clinical Signs and Gut Microbiota Dysbiosis. Nutrients 2022, 14, 1273. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fu, Z.; Zhang, H.; Yang, Z.; Liu, Y.; Wang, P.; Zhang, J.; Chi, H. Metagenomic and Metabolomic Analyses Reveal the Role of a Bacteriocin-Producing Strain of Enterococcus faecalis DH9003 in Regulating Gut Microbiota in Mice. Microorganisms 2025, 13, 372. https://doi.org/10.3390/microorganisms13020372
Fu Z, Zhang H, Yang Z, Liu Y, Wang P, Zhang J, Chi H. Metagenomic and Metabolomic Analyses Reveal the Role of a Bacteriocin-Producing Strain of Enterococcus faecalis DH9003 in Regulating Gut Microbiota in Mice. Microorganisms. 2025; 13(2):372. https://doi.org/10.3390/microorganisms13020372
Chicago/Turabian StyleFu, Zhiyu, Haitao Zhang, Zhenzhu Yang, Yujun Liu, Peng Wang, Junjie Zhang, and Hai Chi. 2025. "Metagenomic and Metabolomic Analyses Reveal the Role of a Bacteriocin-Producing Strain of Enterococcus faecalis DH9003 in Regulating Gut Microbiota in Mice" Microorganisms 13, no. 2: 372. https://doi.org/10.3390/microorganisms13020372
APA StyleFu, Z., Zhang, H., Yang, Z., Liu, Y., Wang, P., Zhang, J., & Chi, H. (2025). Metagenomic and Metabolomic Analyses Reveal the Role of a Bacteriocin-Producing Strain of Enterococcus faecalis DH9003 in Regulating Gut Microbiota in Mice. Microorganisms, 13(2), 372. https://doi.org/10.3390/microorganisms13020372