
P53-Independent G1-Cell Cycle Arrest Increases SARS-CoV-2 RNA Replication
 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cells and Virus
2.2. Treatment with EIPA
2.3. Viability/Cytotoxicity Assay
2.4. Western-Blot
2.5. Flow Cytometry
2.6. Treatments
2.7. siRNA Transfection
2.8. Infection
2.9. RNA Extraction and RT-PCR Analysis
- Forward primer: 5′-ACAGGTACGTTAATAGTTAATAGCGT-3′
- Reverse primer: 5′-ATATTGCAGCAGTACGCACACA-3′
- Probe: FAM-ACACTAGCCATCCTTACTGCGCTTCG-MGB
- Forward primer: 5’-AGATTTGGACCTGCGAGCG-3’
- Reverse primer: 5’-GAGCGGCTGTCTCCACAAGT-3’
- Probe: FAM-TTCTGACCTGAAGGCTCTGCGCG-MGB
2.10. Statistical Analysis
3. Results
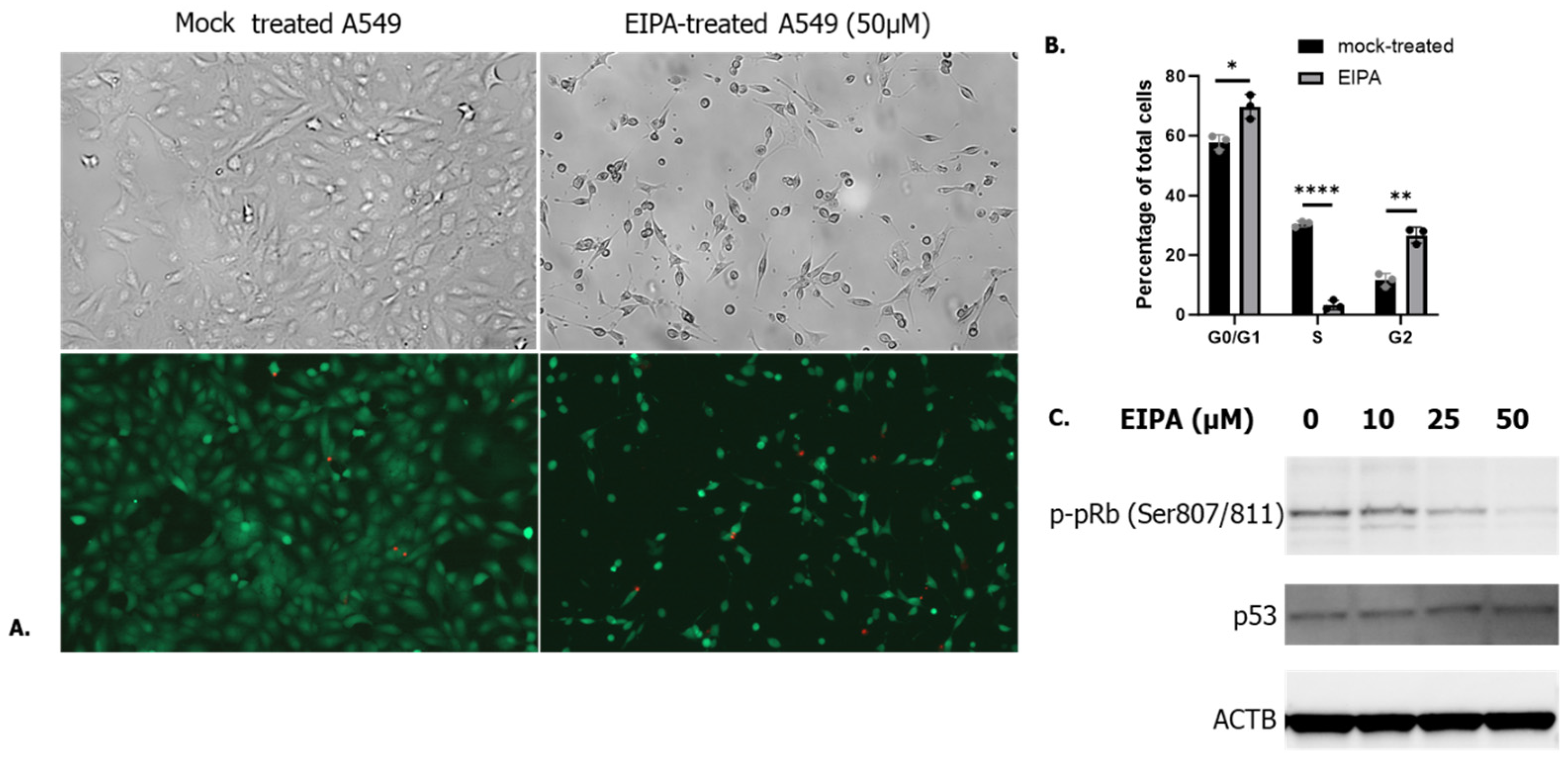
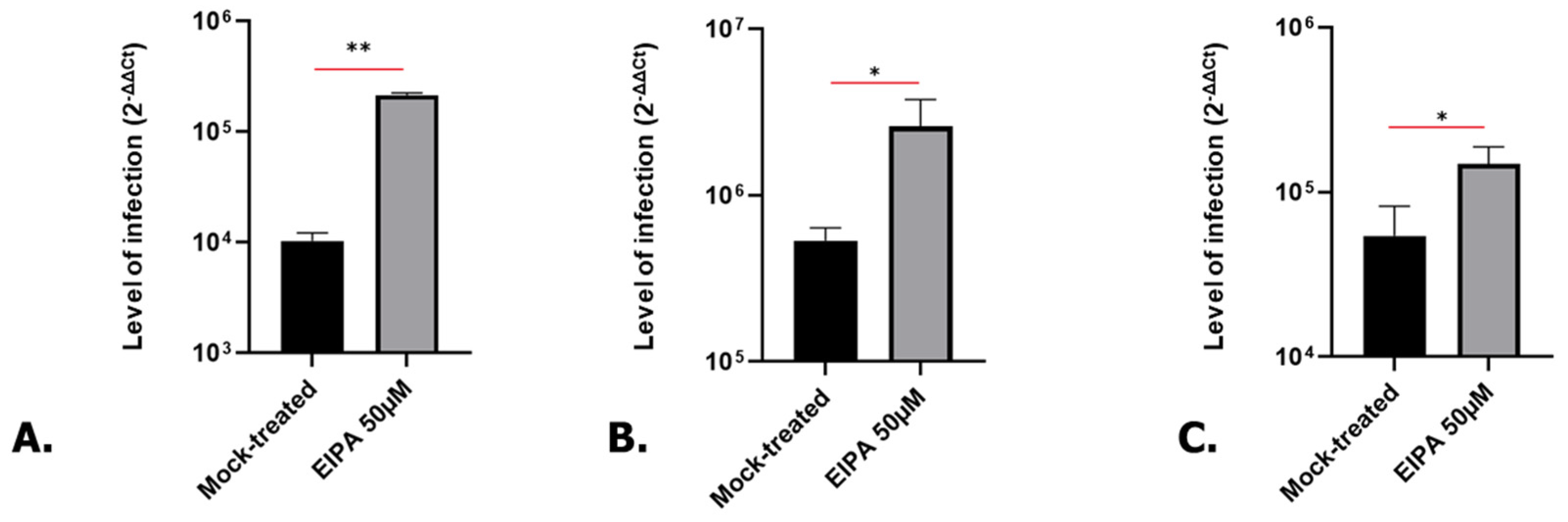
3.1. Cell Cycle Arrest in G0/G1 P53-Independent Improves SARS-CoV-2 Infection
3.2. SARS-CoV-2 Entry Step Is Not Affected by the Cell Cycle Arrest
3.3. Late G1 p53-Independent Cell Cycle Arrest Leads to an Increase in SARS-CoV-2 RNA Replication
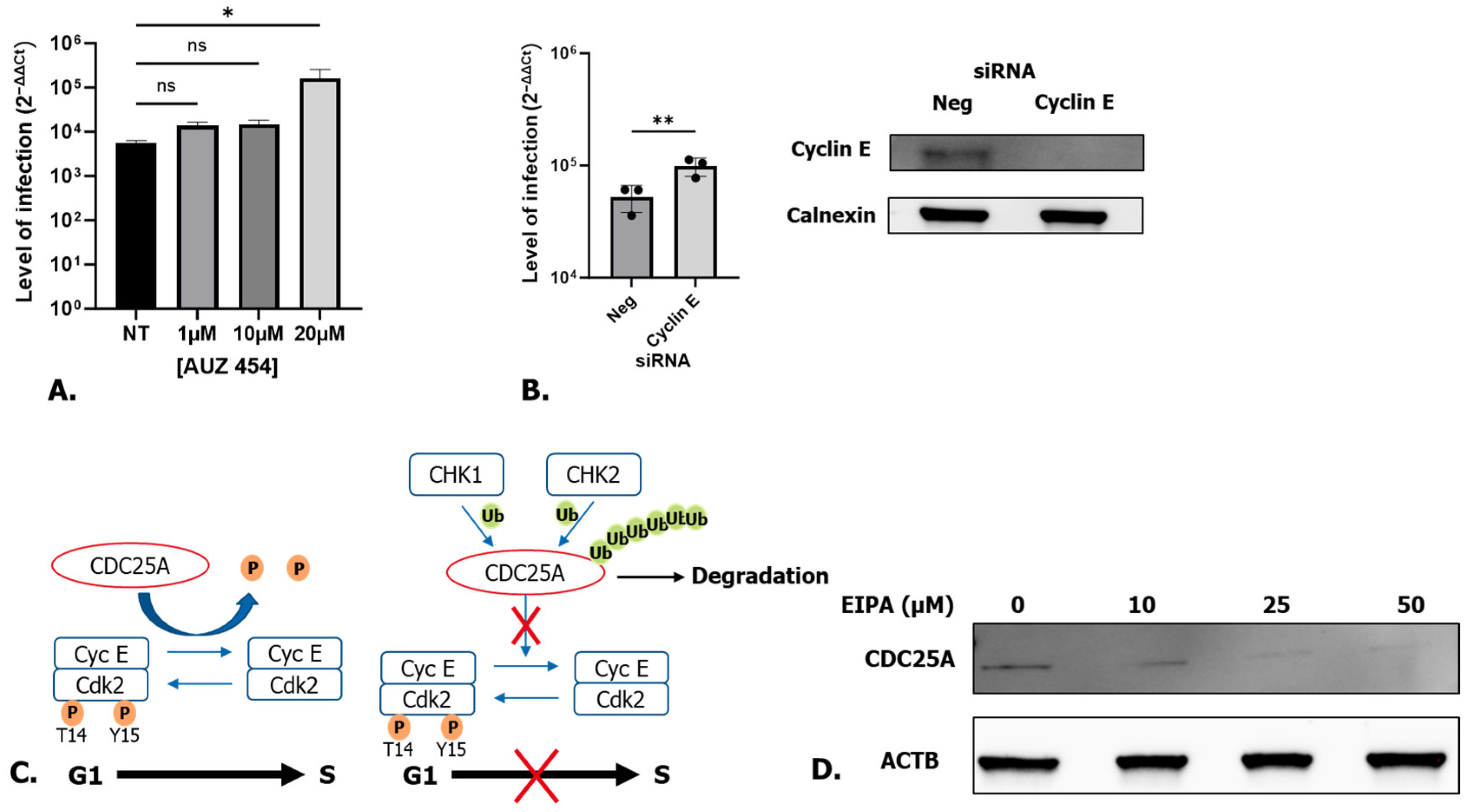
3.4. Cell Cycle Arrest at the CDK2/Cyclin E Checkpoint via CDC25A Degradation Is Responsible for Higher Levels of SARS-CoV-2 Infection
3.5. SARS-CoV-2 Infection Is Also Increased in Treated Human Kidney and Lung Organoids
4. Discussion
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Barnum, K.J.; O’Connell, M.J. Cell cycle regulation by checkpoints. Methods Mol. Biol. 2014, 1170, 29–40. [Google Scholar] [CrossRef]
- Donjerkovic, D.; Scott, D.W. Regulation of the G1 phase of the mammalian cell cycle. Cell Res. 2000, 10, 1–16. [Google Scholar] [CrossRef]
- Hume, S.; Dianov, G.L.; Ramadan, K. A unified model for the G1/S cell cycle transition. Nucleic Acids Res. 2020, 48, 12483–12501. [Google Scholar] [CrossRef]
- Bertoli, C.; Skotheim, J.M.; de Bruin, R.A. Control of cell cycle transcription during G1 and S phases. Nat. Rev. Mol. Cell Biol. 2013, 14, 518–528. [Google Scholar] [CrossRef]
- Giacinti, C.; Giordano, A. RB and cell cycle progression. Oncogene 2006, 25, 5220–5227. [Google Scholar] [CrossRef]
- Johnson, D.G.; Walker, C.L. Cyclins and cell cycle checkpoints. Annu. Rev. Pharmacol. Toxicol. 1999, 39, 295–312. [Google Scholar] [CrossRef]
- Panda, M.; Kalita, E.; Rao, A.; Prajapati, V.K. Mechanism of cell cycle regulation and cell proliferation during human viral infection. Adv. Protein Chem. Struct. Biol. 2023, 135, 497–525. [Google Scholar] [CrossRef]
- Bagga, S.; Bouchard, M.J. Cell cycle regulation during viral infection. Methods Mol. Biol. 2014, 1170, 165–227. [Google Scholar] [CrossRef]
- Nascimento, R.; Costa, H.; Parkhouse, R.M. Virus manipulation of cell cycle. Protoplasma 2012, 249, 519–528. [Google Scholar] [CrossRef]
- Chen, C.J.; Makino, S. Murine coronavirus replication induces cell cycle arrest in G0/G1 phase. J. Virol. 2004, 78, 5658–5669. [Google Scholar] [CrossRef]
- Chen, C.J.; Sugiyama, K.; Kubo, H.; Huang, C.; Makino, S. Murine coronavirus nonstructural protein p28 arrests cell cycle in G0/G1 phase. J. Virol. 2004, 78, 10410–10419. [Google Scholar] [CrossRef]
- Yuan, X.; Shan, Y.; Zhao, Z.; Chen, J.; Cong, Y. G0/G1 arrest and apoptosis induced by SARS-CoV 3b protein in transfected cells. Virol. J. 2005, 2, 66. [Google Scholar] [CrossRef]
- Yuan, X.; Wu, J.; Shan, Y.; Yao, Z.; Dong, B.; Chen, B.; Zhao, Z.; Wang, S.; Chen, J.; Cong, Y. SARS coronavirus 7a protein blocks cell cycle progression at G0/G1 phase via the cyclin D3/pRb pathway. Virology 2006, 346, 74–85. [Google Scholar] [CrossRef]
- Wang, W.; Chen, J.; Hu, D.; Pan, P.; Liang, L.; Wu, W.; Tang, Y.; Huang, X.R.; Yu, X.; Wu, J.; et al. SARS-CoV-2 N Protein Induces Acute Kidney Injury via Smad3-Dependent G1 Cell Cycle Arrest Mechanism. Adv. Sci. 2022, 9, e2103248. [Google Scholar] [CrossRef]
- Quan, L.; Sun, X.; Xu, L.; Chen, R.A.; Liu, D.X. Coronavirus RNA-dependent RNA polymerase interacts with the p50 regulatory subunit of host DNA polymerase delta and plays a synergistic role with RNA helicase in the induction of DNA damage response and cell cycle arrest in the S phase. Emerg. Microbes Infect. 2023, 12, e2176008. [Google Scholar] [CrossRef]
- Sui, L.; Li, L.; Zhao, Y.; Zhao, Y.; Hao, P.; Guo, X.; Wang, W.; Wang, G.; Li, C.; Liu, Q. Host cell cycle checkpoint as antiviral target for SARS-CoV-2 revealed by integrative transcriptome and proteome analyses. Signal Transduct. Target. Ther. 2023, 8, 21. [Google Scholar] [CrossRef]
- Grand, R.J. SARS-CoV-2 and the DNA damage response. J. Gen. Virol. 2023, 104, 001918. [Google Scholar] [CrossRef]
- Rizzo, R.; Caccuri, F.; Valacchi, G.; Zauli, G. Editorial: Cell cycle control as a new therapeutic approach for SARS-CoV-2 infection. Front. Pharmacol. 2023, 14, 1136277. [Google Scholar] [CrossRef]
- Monteil, V.; Kwon, H.; Prado, P.; Hagelkrüys, A.; Wimmer, R.A.; Stahl, M.; Leopoldi, A.; Garreta, E.; Hurtado Del Pozo, C.; Prosper, F.; et al. Inhibition of SARS-CoV-2 Infections in Engineered Human Tissues Using Clinical-Grade Soluble Human ACE2. Cell 2020, 181, 905–913.e907. [Google Scholar] [CrossRef]
- Hosogi, S.; Miyazaki, H.; Nakajima, K.; Ashihara, E.; Niisato, N.; Kusuzaki, K.; Marunaka, Y. An inhibitor of Na+/H+ exchanger (NHE), ethyl-isopropyl amiloride (EIPA), diminishes proliferation of MKN28 human gastric cancer cells by decreasing the cytosolic Cl− concentration via DIDS-sensitive pathways. Cell Physiol. Biochem. 2012, 30, 1241–1253. [Google Scholar] [CrossRef]
- Wang, B.Y.; Shen, H.T.; Lee, Y.L.; Chien, P.J.; Chang, W.W. Inhibition of Na+/H+ exchanger (NHE) 7 by 5-(N-ethyl-N-isopropyl)-Amiloride displays anti-cancer activity in non-small cell lung cancer by disrupting cancer stem cell activity and downregulating PD-L1 expression. Am. J. Cancer Res. 2023, 13, 4721–4733. [Google Scholar]
- Shang, J.; Ye, G.; Shi, K.; Wan, Y.; Luo, C.; Aihara, H.; Geng, Q.; Auerbach, A.; Li, F. Structural basis of receptor recognition by SARS-CoV-2. Nature 2020, 581, 221–224. [Google Scholar] [CrossRef]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L.; et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef]
- Wang, Q.; Zhang, Y.; Wu, L.; Niu, S.; Song, C.; Zhang, Z.; Lu, G.; Qiao, C.; Hu, Y.; Yuen, K.Y.; et al. Structural and Functional Basis of SARS-CoV-2 Entry by Using Human ACE2. Cell 2020, 181, 894–904.e899. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Cui, L.; Li, X.; Xia, C.; Li, Y.; Wang, R.; Ren, F.; Liu, H.; Chen, J. PD-0332991 combined with cisplatin inhibits nonsmall cell lung cancer and reversal of cisplatin resistance. Thorac. Cancer 2021, 12, 924–931. [Google Scholar] [CrossRef]
- Gelbert, L.M.; Cai, S.; Lin, X.; Sanchez-Martinez, C.; Del Prado, M.; Lallena, M.J.; Torres, R.; Ajamie, R.T.; Wishart, G.N.; Flack, R.S.; et al. Preclinical characterization of the CDK4/6 inhibitor LY2835219: In-vivo cell cycle-dependent/independent anti-tumor activities alone/in combination with gemcitabine. Investig. New Drugs 2014, 32, 825–837. [Google Scholar] [CrossRef]
- Hino, H.; Iriyama, N.; Kokuba, H.; Kazama, H.; Moriya, S.; Takano, N.; Hiramoto, M.; Aizawa, S.; Miyazawa, K. Abemaciclib induces atypical cell death in cancer cells characterized by formation of cytoplasmic vacuoles derived from lysosomes. Cancer Sci. 2020, 111, 2132–2145. [Google Scholar] [CrossRef]
- Hotta, K.; Tabata, M.; Kiura, K.; Kozuki, T.; Hisamoto, A.; Katayama, H.; Takigawa, N.; Fujimoto, N.; Fujiwara, K.; Ueoka, H.; et al. Gefitinib induces premature senescence in non-small cell lung cancer cells with or without EGFR gene mutation. Oncol. Rep. 2007, 17, 313–317. [Google Scholar] [CrossRef]
- Guo, M.; Liu, Z.; Si, J.; Zhang, J.; Zhao, J.; Guo, Z.; Xie, Y.; Zhang, H.; Gan, L. Cediranib Induces Apoptosis, G1 Phase Cell Cycle Arrest, and Autophagy in Non-Small-Cell Lung Cancer Cell A549 In Vitro. Biomed. Res. Int. 2021, 2021, 5582648. [Google Scholar] [CrossRef]
- Yuan, L.; Zhang, Y.; Xia, J.; Liu, B.; Zhang, Q.; Liu, J.; Luo, L.; Peng, Z.; Song, Z.; Zhu, R. Resveratrol induces cell cycle arrest via a p53-independent pathway in A549 cells. Mol. Med. Rep. 2015, 11, 2459–2464. [Google Scholar] [CrossRef]
- Xia, R.; Sheng, X.; Xu, X.; Yu, C.; Lu, H. Hesperidin induces apoptosis and G0/G1 arrest in human non-small cell lung cancer A549 cells. Int. J. Mol. Med. 2018, 41, 464–472. [Google Scholar] [CrossRef]
- Tetsu, O.; McCormick, F. Proliferation of cancer cells despite CDK2 inhibition. Cancer Cell 2003, 3, 233–245. [Google Scholar] [CrossRef]
- Garreta, E.; Prado, P.; Stanifer, M.L.; Monteil, V.; Marco, A.; Ullate-Agote, A.; Moya-Rull, D.; Vilas-Zornoza, A.; Tarantino, C.; Romero, J.P.; et al. A diabetic milieu increases ACE2 expression and cellular susceptibility to SARS-CoV-2 infections in human kidney organoids and patient cells. Cell Metab. 2022, 34, 857–873.e859. [Google Scholar] [CrossRef]
- Zhuang, Z.; Zhong, X.; Chen, Q.; Chen, H.; Liu, Z. Bioinformatics and System Biology Approach to Reveal the Interaction Network and the Therapeutic Implications for Non-Small Cell Lung Cancer Patients with COVID-19. Front. Pharmacol. 2022, 13, 857730. [Google Scholar] [CrossRef]
- Lodi, G.; Gentili, V.; Casciano, F.; Romani, A.; Zauli, G.; Secchiero, P.; Zauli, E.; Simioni, C.; Beltrami, S.; Fernandez, M.; et al. Cell cycle block by p53 activation reduces SARS-CoV-2 release in infected alveolar basal epithelial A549-hACE2 cells. Front. Pharmacol. 2022, 13, 1018761. [Google Scholar] [CrossRef]
- Milani, D.; Caruso, L.; Zauli, E.; Al Owaifeer, A.M.; Secchiero, P.; Zauli, G.; Gemmati, D.; Tisato, V. p53/NF-kB Balance in SARS-CoV-2 Infection: From OMICs, Genomics and Pharmacogenomics Insights to Tailored Therapeutic Perspectives (COVIDomics). Front. Pharmacol. 2022, 13, 871583. [Google Scholar] [CrossRef]
- Bortot, B.; Romani, A.; Ricci, G.; Biffi, S. Exploiting Extracellular Vesicles Strategies to Modulate Cell Death and Inflammation in COVID-19. Front. Pharmacol. 2022, 13, 877422. [Google Scholar] [CrossRef]
- Cooper, S. Reappraisal of serum starvation, the restriction point, G0, and G1 phase arrest points. FASEB J. 2003, 17, 333–340. [Google Scholar] [CrossRef]
- Klann, K.; Bojkova, D.; Tascher, G.; Ciesek, S.; Münch, C.; Cinatl, J. Growth Factor Receptor Signaling Inhibition Prevents SARS-CoV-2 Replication. Mol. Cell 2020, 80, 164–174.e164. [Google Scholar] [CrossRef]
- Wu, W.; Wang, X.; Yu, X.; Lan, H.Y. Smad3 Signatures in Renal Inflammation and Fibrosis. Int. J. Biol. Sci. 2022, 18, 2795–2806. [Google Scholar] [CrossRef]
- Matsuura, I.; Denissova, N.G.; Wang, G.; He, D.; Long, J.; Liu, F. Cyclin-dependent kinases regulate the antiproliferative function of Smads. Nature 2004, 430, 226–231. [Google Scholar] [CrossRef]
- Zhu, Y.; Richardson, J.A.; Parada, L.F.; Graff, J.M. Smad3 mutant mice develop metastatic colorectal cancer. Cell 1998, 94, 703–714. [Google Scholar] [CrossRef]
- Han, S.U.; Kim, H.T.; Seong, D.H.; Kim, Y.S.; Park, Y.S.; Bang, Y.J.; Yang, H.K.; Kim, S.J. Loss of the Smad3 expression increases susceptibility to tumorigenicity in human gastric cancer. Oncogene 2004, 23, 1333–1341. [Google Scholar] [CrossRef]
- Ray, D.; Terao, Y.; Nimbalkar, D.; Chu, L.H.; Donzelli, M.; Tsutsui, T.; Zou, X.; Ghosh, A.K.; Varga, J.; Draetta, G.F.; et al. Transforming growth factor beta facilitates beta-TrCP-mediated degradation of Cdc25A in a Smad3-dependent manner. Mol. Cell Biol. 2005, 25, 3338–3347. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Husser, C.; Kwon, H.; Andersson, K.; Appelberg, S.; Montserrat, N.; Mirazimi, A.; Monteil, V.M. P53-Independent G1-Cell Cycle Arrest Increases SARS-CoV-2 RNA Replication. Microorganisms 2024, 12, 443. https://doi.org/10.3390/microorganisms12030443
Husser C, Kwon H, Andersson K, Appelberg S, Montserrat N, Mirazimi A, Monteil VM. P53-Independent G1-Cell Cycle Arrest Increases SARS-CoV-2 RNA Replication. Microorganisms. 2024; 12(3):443. https://doi.org/10.3390/microorganisms12030443
Chicago/Turabian StyleHusser, Clara, Hyesoo Kwon, Klara Andersson, Sofia Appelberg, Nuria Montserrat, Ali Mirazimi, and Vanessa M. Monteil. 2024. "P53-Independent G1-Cell Cycle Arrest Increases SARS-CoV-2 RNA Replication" Microorganisms 12, no. 3: 443. https://doi.org/10.3390/microorganisms12030443
APA StyleHusser, C., Kwon, H., Andersson, K., Appelberg, S., Montserrat, N., Mirazimi, A., & Monteil, V. M. (2024). P53-Independent G1-Cell Cycle Arrest Increases SARS-CoV-2 RNA Replication. Microorganisms, 12(3), 443. https://doi.org/10.3390/microorganisms12030443