
Harnessing the Power of Our Immune System: The Antimicrobial and Antibiofilm Properties of Nitric Oxide

Abstract
:1. Introduction
2. The Innate Nitric Oxide System
2.1. The Immune Response to Pathogens
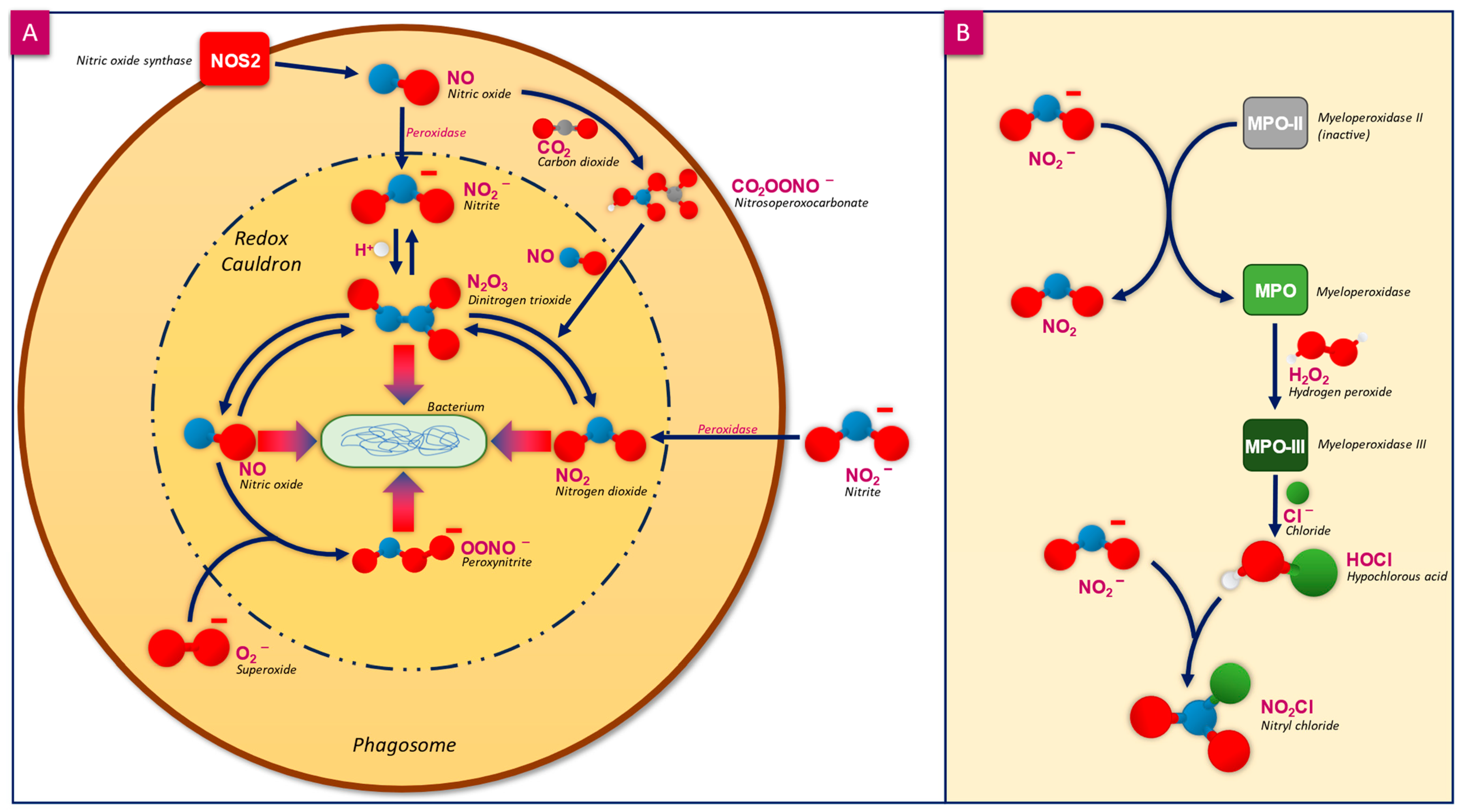
2.2. Mammalian Tolerance to NO and Other RNS
3. Antimicrobial Action
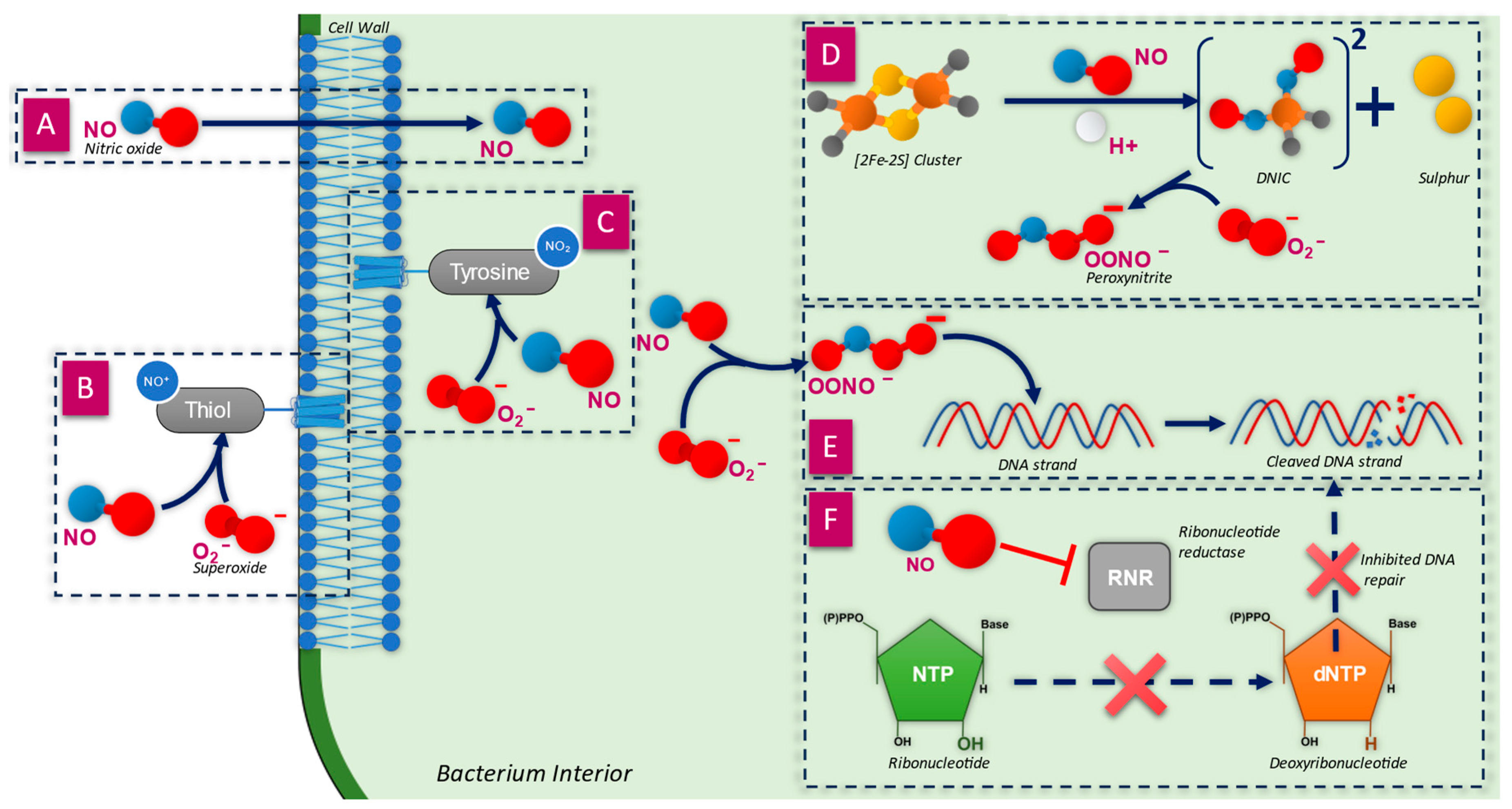
3.1. Penetration of the Cell Wall
3.2. Inactivation of Membrane Proteins
3.3. Breakdown of the Cell Wall
3.4. Inactivation of Iron–Sulphur-Containing Proteins
3.5. Direct Damage to DNA Strands
3.6. Inhibition of DNA Synthesis and Repair
4. Antibiofilm Action
4.1. Biofilm Dispersal—c-di-GMP Pathway
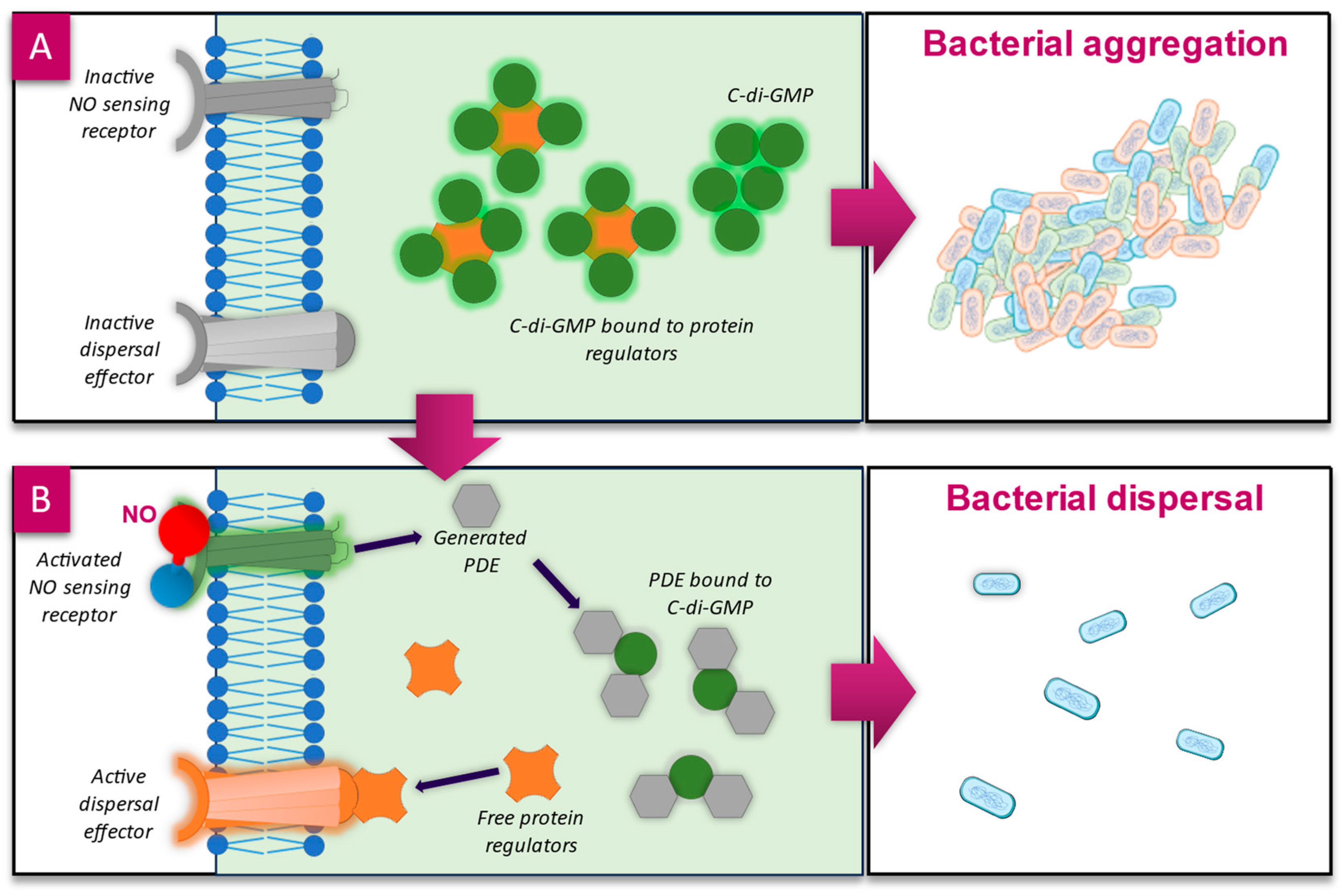
4.2. Influencing Quorum Sensing Pathways
4.3. Interactions with Extracellular Polymeric Substances
5. Conclusions and Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Bowler, P.G. Antibiotic resistance and biofilm tolerance: A combined threat in the treatment of chronic infections. J. Wound Care 2018, 27, 273–277. [Google Scholar] [CrossRef] [PubMed]
- Choi, V.; Rohn, J.L.; Stoodley, P.; Carugo, D.; Stride, E. Drug delivery strategies for antibiofilm therapy. Nat. Rev. Microbiol. 2023, 21, 555–572. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Mohler, J.; Mahajan, S.D.; Schwartz, S.A.; Bruggemann, L.; Aalinkeel, R. Microbial Biofilm: A Review on Formation, Infection, Antibiotic Resistance, Control Measures, and Innovative Treatment. Microorganisms 2023, 11, 1614. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.; Wang, H.; Pan, X.; Zhang, C.; Zhang, K.; Chen, Z.; Dong, W.; Xie, A.; Qi, X. Dendritic Hydrogels with Robust Inherent Antibacterial Properties for Promoting Bacteria-Infected Wound Healing. ACS Appl. Mater. Interfaces 2022, 14, 11144–11155. [Google Scholar] [CrossRef] [PubMed]
- Ding, L.; Wang, J.; Cai, S.; Smyth, H.; Cui, Z. Pulmonary biofilm-based chronic infections and inhaled treatment strategies. Int. J. Pharm. 2021, 604, 120768. [Google Scholar] [CrossRef]
- Weigelt, M.A.; McNamara, S.A.; Sanchez, D.; Hirt, P.A.; Kirsner, R.S. Evidence-Based Review of Antibiofilm Agents for Wound Care. Adv. Wound Care 2021, 10, 13–23. [Google Scholar] [CrossRef]
- Bowler, P.G.; Parsons, D. Combatting wound biofilm and recalcitrance with a novel anti-biofilm Hydrofiber® wound dressing. Wound Med. 2016, 14, 6–11. [Google Scholar] [CrossRef]
- Murphy, C.; Atkin, L.; Dissemond, J.; Hurlow, J.; Tan, Y.K.; Apelqvist, J.; James, G.; Salles, N.; Wu, J.; Tachi, M.; et al. Defying hard-to-heal wounds with an early antibiofilm intervention strategy: ‘wound hygiene’. J. Wound Care 2019, 28, 818–822. [Google Scholar] [CrossRef]
- Torkington-Stokes, R.; Moran, K.; Martinez, D.S.; Granara, D.C.; Metcalf, D.G. Improving outcomes for patients with hard-to-heal wounds following adoption of the Wound Hygiene Protocol: Real-world evidence. J. Wound Care 2024, 33, 304–310. [Google Scholar] [CrossRef]
- Barnea, Y. A review of the applications of the hydrofiber dressing with silver (Aquacel Ag®) in wound care. Ther. Clin. Risk Manag. 2010, 6, 21–27. [Google Scholar] [CrossRef]
- Heras, K.L.; Igartua, M.; Santos-Vizcaino, E.; Hernandez, R.M. Chronic wounds: Current status, available strategies and emerging therapeutic solutions. J. Control. Release 2020, 328, 532–550. [Google Scholar] [CrossRef] [PubMed]
- Freedman, B.R.; Hwang, C.; Talbot, S.; Hibler, B.; Matoori, S.; Mooney, D.J. Breakthrough treatments for accelerated wound healing. Sci. Adv. 2023, 9, eade7007. [Google Scholar] [CrossRef] [PubMed]
- Wounds International. International Consensus. Appropriate Use of Silver Dressings in Wounds. An Expert Working Group Consensus. London 2012. Available online: www.woundsinternational.com/consensus-documents/international-consensus-appropriate-use-silver-dressings-wounds-english-en/ (accessed on 9 December 2024).
- Heras, K.L.; Garcia-Orue, I.; Rancan, F.; Igartua, M.; Santos-Vizcaino, E.; Hernandez, R.M. Modulating the immune system towards a functional chronic wound healing: A biomaterials and Nanomedicine perspective. Adv. Drug Deliv. Rev. 2024, 210, 115342. [Google Scholar] [CrossRef] [PubMed]
- Burnet, M.; Metcalf, D.G.; Milo, S.; Gamerith, C.; Heinzle, A.; Sigl, E.; Eitel, K.; Haalboom, M.; Bowler, P.G. A Host-Directed Approach to the Detection of Infection in Hard-to-Heal Wounds. Diagnostics 2022, 12, 2408. [Google Scholar] [CrossRef]
- Zasloff, M. Antimicrobial peptides of multicellular organisms. Nature 2002, 415, 389–395. [Google Scholar] [CrossRef]
- Joachim, D. Wound cleansing: Benefits of hypochlorous acid. J. Wound Care 2020, 29, S4–S8. [Google Scholar] [CrossRef]
- Culotta, E.; Koshland, D.E. NO News Is Good News. Science 1992, 258, 1862–1865. [Google Scholar] [CrossRef]
- Del Sorbo, L.; Michaelsen, V.S.; Ali, A.; Wang, A.; Ribeiro, R.V.P.; Cypel, M. High Doses of Inhaled Nitric Oxide as an Innovative Antimicrobial Strategy for Lung Infections. Biomedicines 2022, 10, 1525. [Google Scholar] [CrossRef]
- Edmonds, M.E.; Bodansky, H.J.; Boulton, A.J.; Chadwick, P.J.; Dang, C.N.; D’Costa, R.; Johnston, A.; Kennon, B.; Leese, G.; Rajbhandari, S.M.; et al. Multicenter, randomized controlled, observer-blinded study of a nitric oxide generating treatment in foot ulcers of patients with diabetes—ProNOx1 study. Wound Repair Regen. 2018, 26, 228–237. [Google Scholar] [CrossRef]
- Davis, S.C.; Gil, J.; Solis, M. Nitric Oxide as an Efficient Antimicrobial Treatment for Second-Degree Burn Wounds. Mil. Med. 2024, usae402. [Google Scholar] [CrossRef]
- Chernoff, G. The Utilization of a Nitric Oxide Generating Serum in the Treatment of Active Acne and Acne Scarred Patients. Int. J. Pharma. Anal. Acta 2020, 3, 10–14. [Google Scholar]
- Nitric Oxide Footbath for Treatment of Diabetic Foot Ulcers. Available online: https://clinicaltrials.gov/study/NCT04755647 (accessed on 3 December 2024).
- Sullivan, R. A Registry-based Study Assessing the Efficacy of a Nitric Oxide Delivering Foam for Wound Healing. In Proceedings of the Poster CR-048, Las Vegas, NV, USA, 23 May 2024. [Google Scholar]
- Miller, C.M.; James, G.; Bell, D.; Schultz, G. Antimicrobial effects of an acidified nitrite foam on drip flow reactor biofilm. J. Wound Manag. 2024, 25, 16–21. [Google Scholar] [CrossRef]
- Bogdan, C. Nitric oxide and the immune response NO production in the immune system. Nat. Immunol. 2001, 2, 907–916. [Google Scholar] [CrossRef] [PubMed]
- Fang, F.C. Mechanisms of nitric oxide-related antimicrobial activity. J. Clin. Investig. 1997, 99, 2818–2825. [Google Scholar] [CrossRef] [PubMed]
- Möller, M.N.; Denicola, A. Diffusion of nitric oxide and oxygen in lipoproteins and membranes studied by pyrene fluorescence quenching. Free. Radic. Biol. Med. 2018, 128, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Coleman, J.W. Nitric oxide in immunity and inflammation. Int. Immunopharmacol. 2001, 1, 1397–1406. [Google Scholar] [CrossRef]
- Thomas, D.D. Breathing new life into nitric oxide signaling: A brief overview of the interplay between oxygen and nitric oxide. Redox Biol. 2015, 5, 225–233. [Google Scholar] [CrossRef]
- MacMicking, J.; Xie, Q.-W.; Nathan, C. Nitric oxide and macrophage function. Annu. Rev. Immunol. 1997, 15, 323–350. [Google Scholar] [CrossRef]
- Chang, C.-I.; Liao, J.C.; Kuo, L. Arginase modulates nitric oxide production in activated macrophages. Am. J. Physiol. Circ. Physiol. 1998, 274, H342–H348. [Google Scholar] [CrossRef]
- Eiserich, J.P.; Hristova, M.; Cross, C.E.; Jones, A.D.; Freeman, B.A.; Halliwell, B.; van der Vliet, A. Formation of nitric oxide-derived inflammatory oxidants by myeloperoxidase in neutrophils. Nature 1998, 391, 393–397. [Google Scholar] [CrossRef]
- Al-Shehri, S.S. Reactive oxygen and nitrogen species and innate immune response. Biochimie 2020, 181, 52–64. [Google Scholar] [CrossRef] [PubMed]
- A Wink, D.; Hines, H.B.; Cheng, R.Y.S.; Switzer, C.H.; Flores-Santana, W.; Vitek, M.P.; A Ridnour, L.; A Colton, C. Nitric oxide and redox mechanisms in the immune response. J. Leukoc. Biol. 2011, 89, 873–891. [Google Scholar] [CrossRef] [PubMed]
- Hampton, M.B.; Dickerhof, N. Inside the phagosome: A bacterial perspective. Immunol. Rev. 2023, 314, 197–209. [Google Scholar] [CrossRef]
- Uribe-Querol, E.; Rosales, C. Phagocytosis. Encycl. Infect. Immun. 2022, 1, 99–109. [Google Scholar] [CrossRef]
- Prolo, C.; Álvarez, M.N.; Radi, R. Peroxynitrite, a potent macrophage-derived oxidizing cytotoxin to combat invading pathogens. BioFactors 2014, 40, 215–225. [Google Scholar] [CrossRef]
- Eiserich, J.P.; Cross, C.E.; Jones, A.D.; Halliwell, B.; van der Vliet, A. Formation of nitrating and chlorinating species by reaction of nitrite with hypochlorous acid. A novel mechanism for nitric oxide-mediated protein modification. J. Biol. Chem. 1996, 271, 19199–19208. [Google Scholar] [CrossRef]
- Wink, D.A.; Cook, J.A.; Pacelli, R.; Liebmann, J.; Krishna, M.C.; Mitchell, J.B. Nitric oxide (NO) protects against cellular damage by reactive oxygen species. Toxicol. Lett. 1995, 82–83, 221–226. [Google Scholar] [CrossRef]
- Kröncke, K.-D.; Fehsel, K.; Kolb-Bachofen, V. Nitric Oxide: Cytotoxicity versus Cytoprotection—How, Why, When, and Where? Nitric Oxide 1997, 1, 107–120. [Google Scholar] [CrossRef]
- Chaturvedi, R.; Asim, M.; Lewis, N.D.; Algood, H.M.S.; Cover, T.L.; Kim, P.Y.; Wilson, K.T. l-Arginine Availability Regulates Inducible Nitric Oxide Synthase-Dependent Host Defense against Helicobacter pylori. Infect. Immun. 2007, 75, 4305–4315. [Google Scholar] [CrossRef]
- Gantner, B.N.; LaFond, K.M.; Bonini, M.G. Nitric oxide in cellular adaptation and disease. Redox Biol. 2020, 34, 101550. [Google Scholar] [CrossRef]
- Murphy, M.P. Nitric oxide and cell death. Biochim. Biophys. Acta (BBA)—Bioenerg. 1999, 1411, 401–414. [Google Scholar] [CrossRef]
- Beckman, J.S.; Koppenol, W.H. Nitric oxide, superoxide, and peroxynitrite: The good, the bad, and ugly. Am. J. Physiol. Cell Physiol. 1996, 271, C1424–C1437. [Google Scholar] [CrossRef] [PubMed]
- Urzedo, A.L.; Gonçalves, M.C.; Nascimento, M.H.M.; Lombello, C.B.; Nakazato, G.; Seabra, A.B. Cytotoxicity and Antibacterial Activity of Alginate Hydrogel Containing Nitric Oxide Donor and Silver Nanoparticles for Topical Applications. ACS Biomater. Sci. Eng. 2020, 6, 2117–2134. [Google Scholar] [CrossRef] [PubMed]
- Miranda, K.M.; Ridnour, L.A.; McGinity, C.L.; Bhattacharyya, D.; Wink, D.A. Nitric Oxide and Cancer: When to Give and When to Take Away? Inorg. Chem. 2021, 60, 15941–15947. [Google Scholar] [CrossRef] [PubMed]
- Rong, F.; Tang, Y.; Wang, T.; Feng, T.; Song, J.; Li, P.; Huang, W. Nitric oxide-releasing polymeric materials for antimicrobial applications: A review. Antioxidants 2019, 8, 556. [Google Scholar] [CrossRef]
- Fitzpatrick, J.; Kim, E. Synthetic Modeling Chemistry of Iron–Sulfur Clusters in Nitric Oxide Signaling. Acc. Chem. Res. 2015, 48, 2453–2461. [Google Scholar] [CrossRef]
- Burney, S.; Caulfield, J.L.; Niles, J.C.; Wishnok, J.S.; Tannenbaum, S.R. The chemistry of DNA damage from nitric oxide and peroxynitrite. Mutat. Res. Mol. Mech. Mutagen. 1999, 424, 37–49. [Google Scholar] [CrossRef]
- Lepoivre, M.; Fieschi, F.; Coves, J.; Thelander, L.; Fontecave, M. Inactivation of ribonucleotide reductase by nitric oxide. Biochem. Biophys. Res. Commun. 1991, 179, 442–448. [Google Scholar] [CrossRef]
- Hall, J.R.; Rouillard, K.R.; Suchyta, D.J.; Brown, M.D.; Ahonen, M.J.R.; Schoenfisch, M.H. Mode of Nitric Oxide Delivery Affects Antibacterial Action. ACS Biomater. Sci. Eng. 2020, 6, 433–441. [Google Scholar] [CrossRef]
- Ahonen, M.J.R.; Suchyta, D.J.; Zhu, H.; Schoenfisch, M.H. Nitric Oxide-Releasing Alginates. Biomacromolecules 2018, 19, 1189–1197. [Google Scholar] [CrossRef]
- Michelon, D.; Abraham, S.; Ebel, B.; De Coninck, J.; Husson, F.; Feron, G.; Gervais, P.; Cachon, R. Contribution of exofacial thiol groups in the reducing activity of Lactococcus lactis. FEBS J. 2010, 277, 2282–2290. [Google Scholar] [CrossRef]
- Soufi, B.; Macek, B. Global analysis of bacterial membrane proteins and their modifications. Int. J. Med. Microbiol. 2015, 305, 203–208. [Google Scholar] [CrossRef]
- Schulz, G.E. The structure of bacterial outer membrane proteins. Biochim. Biophys. Acta (BBA)—Biomembr. 2002, 1565, 308–317. [Google Scholar] [CrossRef]
- Li, H.; Xu, H. Mechanisms of bacterial resistance to environmental silver and antimicrobial strategies for silver: A review. Environ. Res. 2024, 248, 118313. [Google Scholar] [CrossRef]
- Sun, L.; Bertelshofer, F.; Greiner, G.; Böckmann, R.A. Characteristics of Sucrose Transport through the Sucrose-Specific Porin ScrY Studied by Molecular Dynamics Simulations. Front. Bioeng. Biotechnol. 2016, 4, 9. [Google Scholar] [CrossRef]
- Morris, S.L.; Walsh, R.C.; Hansen, J.N. Identification and characterization of some bacterial membrane sulfhydryl groups which are targets of bacteriostatic and antibiotic action. J. Biol. Chem. 1984, 259, 13590–13594. [Google Scholar] [CrossRef]
- Larraga, V.; Muñoz, E. Molecular Organization in Bacterial Cell Membranes. Eur. J. Biochem. 1975, 54, 207–218. [Google Scholar] [CrossRef]
- Andrés, C.M.C.; de la Lastra, J.M.P.; Juan, C.A.; Plou, F.J.; Pérez-Lebeña, E. Impact of Reactive Species on Amino Acids—Biological Relevance in Proteins and Induced Pathologies. Int. J. Mol. Sci. 2022, 23, 14049. [Google Scholar] [CrossRef]
- E Whitmore, S.; Lamont, R.J. Tyrosine phosphorylation and bacterial virulence. Int. J. Oral Sci. 2012, 4, 1–6. [Google Scholar] [CrossRef]
- Deupree, S.M.; Schoenfisch, M.H. Morphological analysis of the antimicrobial action of nitric oxide on Gram-negative pathogens using atomic force microscopy. Acta Biomater. 2009, 5, 1405–1415. [Google Scholar] [CrossRef]
- Freeman, B.A.; White, C.R.; Gutierrez, H.; Paler-Martínez, A.; Tarpey, M.M.; Rubbo, H. Oxygen Radical-Nitric Oxide Reactions in Vascular Diseases. Adv. Pharmacol. 1995, 34, 45–69. [Google Scholar] [CrossRef] [PubMed]
- Rouillard, K.R.; Novak, O.P.; Pistiolis, A.M.; Yang, L.; Ahonen, M.J.R.; McDonald, R.A.; Schoenfisch, M.H. Exogenous Nitric Oxide Improves Antibiotic Susceptibility in Resistant Bacteria. ACS Infect. Dis. 2021, 7, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Lehnert, N.; Kim, E.; Dong, H.T.; Harland, J.B.; Hunt, A.P.; Manickas, E.C.; Oakley, K.M.; Pham, J.; Reed, G.C.; Alfaro, V.S. The Biologically Relevant Coordination Chemistry of Iron and Nitric Oxide: Electronic Structure and Reactivity. Chem. Rev. 2021, 121, 14682–14905. [Google Scholar] [CrossRef] [PubMed]
- Ayala-Castro, C.; Saini, A.; Outten, F.W. Fe-S Cluster Assembly Pathways in Bacteria. Microbiol. Mol. Biol. Rev. 2008, 72, 110–125. [Google Scholar] [CrossRef]
- Wächtershäuser, G. Groundworks for an evolutionary biochemistry: The iron-sulphur world. Prog. Biophys. Mol. Biol. 1992, 58, 85–201. [Google Scholar] [CrossRef]
- Imlay, J.A. Iron-sulphur clusters and the problem with oxygen. Mol. Microbiol. 2006, 59, 1073–1082. [Google Scholar] [CrossRef]
- Moulis, J.-M.; Davasse, V.; Golinelli, M.-P.; Meyer, J.; QUintkal, I. The coordination sphere of iron-sulfur clusters. JBIC 1996, 1, 2–14. [Google Scholar] [CrossRef]
- Beinert, H.; Kiley, P.J. Fe-S proteins in sensing and regulatory functions. Curr. Opin. Chem. Biol. 1999, 3, 152–157. [Google Scholar] [CrossRef]
- Kiley, P.J.; Beinert, H. The role of Fe–S proteins in sensing and regulation in bacteria. Curr. Opin. Microbiol. 2003, 6, 181–185. [Google Scholar] [CrossRef]
- Jordan, P.A.; Tang, Y.; Bradbury, A.J.; Thomson, A.J.; Guest, J.R. Biochemical and spectroscopic characterization of Escherichia coli aconitases (AcnA and AcnB). Biochem. J. 1999, 344, 739–746. [Google Scholar] [CrossRef]
- Tang, Y.; Guest, J.R. Direct evidence for mRNA binding and post-transcriptional regulation by Escherichia coli aconitases. Microbiology 1999, 145, 3069–3079. [Google Scholar] [CrossRef] [PubMed]
- Foster, M.W.; Cowan, J.A. Chemistry of Nitric Oxide with Protein-Bound Iron Sulfur Centers. Insights on Physiological Reactivity. J. Am. Chem. Soc. 1999, 121, 4093–4100. [Google Scholar] [CrossRef]
- Ren, B.; Zhang, N.; Yang, J.; Ding, H. Nitric oxide-induced bacteriostasis and modification of iron-sulphur proteins in Escherichia coli. Mol. Microbiol. 2008, 70, 953–964. [Google Scholar] [CrossRef] [PubMed]
- Pacelli, R.; A Wink, D.; A Cook, J.; Krishna, M.C.; DeGraff, W.; Friedman, N.; Tsokos, M.; Samuni, A.; Mitchell, J.B. Nitric oxide potentiates hydrogen peroxide-induced killing of Escherichia coli. J. Exp. Med. 1995, 182, 1469–1479. [Google Scholar] [CrossRef] [PubMed]
- Radi, R. Protein Tyrosine Nitration: Biochemical Mechanisms and Structural Basis of Functional Effects. Acc. Chem. Res. 2013, 46, 550–559. [Google Scholar] [CrossRef]
- Christen, S.; Gee, P.; Ames, B.N. Mutagenicity of nitric oxide in base pair-specific Salmonella tester strains: TA7000 series. Methods Enzym. 1996, 269, 267–278. [Google Scholar] [CrossRef]
- Szabó, C.; Ohshima, H. DNA Damage Induced by Peroxynitrite: Subsequent Biological Effects. Nitric Oxide 1997, 1, 373–385. [Google Scholar] [CrossRef]
- Lindahl, T.; Andersson, A. Rate of chain breakage at apurinic sites in double-stranded deoxyribonucleic acid. Biochemistry 1972, 11, 3618–3623. [Google Scholar] [CrossRef]
- Snyder, R.R.; Kocsis, J.J.; Sipes, I.G.; Kalf, G.F.; Jollow, D.J.; Greim, H.; Monks, T.J.; Witmer, C.M. Biological Reactive Intermediates V: Basic Mechanistic Research in Toxicology and Human Risk Assessment; Springer: Berlin/Heidelberg, Germany, 1996. [Google Scholar]
- Torrents, E. Ribonucleotide reductases: Essential enzymes for bacterial life. Front. Cell. Infect. Microbiol. 2014, 4, 52. [Google Scholar] [CrossRef]
- Roy, B.; Lepoivre, M.; Henry, Y.; Fontecave, M. Inhibition of Ribonucleotide Reductase by Nitric Oxide Derived from Thionitrites: Reversible Modifications of Both Subunits. Biochemistry 1995, 34, 5411–5418. [Google Scholar] [CrossRef]
- Peng, Y.; Pei, H. DNA alkylation lesion repair: Outcomes and implications in cancer chemotherapy. J. Zhejiang Univ. Sci. B 2021, 22, 47–62. [Google Scholar] [CrossRef]
- Mattossovich, R.; Merlo, R.; Miggiano, R.; Valenti, A.; Perugino, G. O6-alkylguanine-DNA Alkyltransferases in Microbes Living on the Edge: From Stability to Applicability. Int. J. Mol. Sci. 2020, 21, 2878. [Google Scholar] [CrossRef]
- Laval, F.; Wink, D.A. Inhibition by nitric oxide of the repair protein, O6-DNA-methyltransferase. Carcinogenesis 1994, 15, 443–447. [Google Scholar] [CrossRef]
- Liu, L.; Xu-Welliver, M.; Kanugula, S.; E Pegg, A. Inactivation and degradation of O6-alkylguanine-DNA alkyltransferase after reaction with nitric oxide. Cancer Res. 2002, 62, 3037–3043. [Google Scholar]
- Penesyan, A.; Paulsen, I.T.; Kjelleberg, S.; Gillings, M.R. Three faces of biofilms: A microbial lifestyle, a nascent multicellular organism, and an incubator for diversity. npj Biofilms Microbiomes 2021, 7, 80. [Google Scholar] [CrossRef]
- Costerton, J. Cystic fibrosis pathogenesis and the role of biofilms in persistent infection. Trends Microbiol. 2001, 9, 50–52. [Google Scholar] [CrossRef]
- Malone, M.; Bjarnsholt, T.; McBain, A.J.; James, G.A.; Stoodley, P.; Leaper, D.; Tachi, M.; Schultz, G.; Swanson, T.; Wolcott, R.D. The prevalence of biofilms in chronic wounds: A systematic review and meta-analysis of published data. J. Wound Care 2017, 26, 20–25. [Google Scholar] [CrossRef]
- Hall-Stoodley, L.; Costerton, J.W.; Stoodley, P. Bacterial biofilms: From the Natural environment to infectious diseases. Nat. Rev. Microbiol. 2004, 2, 95–108. [Google Scholar] [CrossRef]
- Park, S.; Sauer, K. Controlling Biofilm Development Through Cyclic di-GMP Signaling. In Advances in Experimental Medicine and Biology; Springer: Cham, Switzerland, 2022; Volume 1386. [Google Scholar] [CrossRef]
- Valentini, M.; Filloux, A. Biofilms and Cyclic di-GMP (c-di-GMP) signaling: Lessons from Pseudomonas aeruginosa and other bacteria. J. Biol. Chem. 2016, 291, 12547–12555. [Google Scholar] [CrossRef]
- Arora, D.P.; Hossain, S.; Xu, Y.; Boon, E.M. Nitric Oxide Regulation of Bacterial Biofilms. Biochemistry 2015, 54, 3717–3728. [Google Scholar] [CrossRef]
- McDougald, D.; Rice, S.A.; Barraud, N.; Steinberg, P.D.; Kjelleberg, S. Should we stay or should we go: Mechanisms and ecological consequences for biofilm dispersal. Nat. Rev. Microbiol. 2011, 10, 39–50. [Google Scholar] [CrossRef] [PubMed]
- Rinaldo, S.; Giardina, G.; Mantoni, F.; Paone, A.; Cutruzzolà, F. Beyond nitrogen metabolism: Nitric oxide, cyclic-di-GMP and bacterial biofilms. FEMS Microbiol. Lett. 2018, 365, fny029. [Google Scholar] [CrossRef] [PubMed]
- Sadrearhami, Z.; Nguyen, T.-K.; Namivandi-Zangeneh, R.; Jung, K.; Wong, E.H.H.; Boyer, C. Recent advances in nitric oxide delivery for antimicrobial applications using polymer-based systems. J. Mater. Chem. B 2018, 6, 2945–2959. [Google Scholar] [CrossRef] [PubMed]
- Barraud, N.; Kelso, M.; Rice, S.; Kjelleberg, S. Nitric Oxide: A Key Mediator of Biofilm Dispersal with Applications in Infectious Diseases. Curr. Pharm. Des. 2014, 21, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Barraud, N.; Storey, M.V.; Moore, Z.P.; Webb, J.S.; Rice, S.A.; Kjelleberg, S. Nitric oxide-mediated dispersal in single- and multi-species biofilms of clinically and industrially relevant microorganisms. Microb. Biotechnol. 2009, 2, 370–378. [Google Scholar] [CrossRef]
- Soren, O.; Rineh, A.; Silva, D.G.; Cai, Y.; Howlin, R.P.; Allan, R.N.; Feelisch, M.; Davies, J.C.; Connett, G.J.; Faust, S.N.; et al. Cephalosporin nitric oxide-donor prodrug DEA-C3D disperses biofilms formed by clinical cystic fibrosis isolates of Pseudomonas aeruginosa. J. Antimicrob. Chemother. 2020, 75, 117–125. [Google Scholar] [CrossRef]
- Barraud, N.; Kardak, B.G.; Yepuri, N.R.; Howlin, R.P.; Webb, J.S.; Faust, S.N.; Kjelleberg, S.; Rice, S.A.; Kelso, M.J. Cephalosporin-3′-diazeniumdiolates: Targeted NO-Donor Prodrugs for Dispersing Bacterial Biofilms. Angew. Chem. Int. Ed. 2012, 51, 9057–9060. [Google Scholar] [CrossRef]
- Barraud, N.; Hassett, D.J.; Hwang, S.-H.; Rice, S.A.; Kjelleberg, S.; Webb, J.S. Involvement of nitric oxide in biofilm dispersal of Pseudomonas aeruginosa. J. Bacteriol. 2006, 188, 7344–7353. [Google Scholar] [CrossRef]
- Howlin, R.P.; Cathie, K.; Hall-Stoodley, L.; Cornelius, V.; Duignan, C.; Allan, R.N.; Fernandez, B.O.; Barraud, N.; Bruce, K.D.; Jefferies, J.; et al. Low-Dose Nitric Oxide as Targeted Anti-biofilm Adjunctive Therapy to Treat Chronic Pseudomonas aeruginosa Infection in Cystic Fibrosis. Mol. Ther. 2017, 25, 2104–2116. [Google Scholar] [CrossRef]
- Heckler, I.; Boon, E.M. Insights Into Nitric Oxide Modulated Quorum Sensing Pathways. Front. Microbiol. 2019, 10, 2174. [Google Scholar] [CrossRef]
- Miller, M.B.; Bassler, B.L. Quorum Sensing in Bacteria. Annu. Rev. Microbiol. 2001, 55, 165–199. [Google Scholar] [CrossRef] [PubMed]
- Cutruzzolà, F.; Frankenberg-Dinkel, N. Origin and impact of nitric oxide in Pseudomonas aeruginosa biofilms. J. Bacteriol. 2016, 198, 55–65. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, A.W.; Schoenfisch, M.H. Nitric oxide release: Part II. Therapeutic applications. Chem. Soc. Rev. 2012, 41, 3742–3752. [Google Scholar] [CrossRef]
- Partridge, J.D.; Bodenmiller, D.M.; Humphrys, M.S.; Spiro, S. NsrR targets in the Escherichia coli genome: New insights into DNA sequence requirements for binding and a role for NsrR in the regulation of motility. Mol. Microbiol. 2009, 73, 680–694. [Google Scholar] [CrossRef] [PubMed]
- Sulemankhil, I.; Ganopolsky, J.G.; Dieni, C.A.; Dan, A.F.; Jones, M.L.; Prakash, S. Prevention and treatment of virulent bacterial biofilms with an enzymatic nitric oxide-releasing dressing. Antimicrob. Agents Chemother. 2012, 56, 6095–6103. [Google Scholar] [CrossRef] [PubMed]
- Jardeleza, C.; Foreman, A.; Baker, L.; Paramasivan, S.; Field, J.; Tan, L.W.; Wormald, P. The effects of nitric oxide on Staphylococcus aureus biofilm growth and its implications in chronic rhinosinusitis. Int. Forum Allergy Rhinol. 2011, 1, 438–444. [Google Scholar] [CrossRef] [PubMed]
- Hetrick, E.M.; Shin, J.H.; Paul, H.S.; Schoenfisch, M.H. Anti-biofilm efficacy of nitric oxide-releasing silica nanoparticles. Biomaterials 2009, 30, 2782–2789. [Google Scholar] [CrossRef]
- Lu, Y.; Slomberg, D.L.; Schoenfisch, M.H. Nitric oxide-releasing chitosan oligosaccharides as antibacterial agents. Biomaterials 2014, 35, 1716–1724. [Google Scholar] [CrossRef]
- Lu, Y.; Slomberg, D.L.; Shah, A.; Schoenfisch, M.H. Nitric Oxide-Releasing Amphiphilic Poly(amidoamine) (PAMAM) Dendrimers as Antibacterial Agents. Biomacromolecules 2013, 14, 3589–3598. [Google Scholar] [CrossRef]
- de la Fuente-Núñez, C.; Reffuveille, F.; Fairfull-Smith, K.E.; Hancock, R.E.W. Effect of Nitroxides on Swarming Motility and Biofilm Formation, Multicellular Behaviors in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2013, 57, 4877–4881. [Google Scholar] [CrossRef]
- Kutty, S.K.; Barraud, N.; Pham, A.; Iskander, G.; Rice, S.A.; Black, D.S.; Kumar, N. Design, Synthesis, and Evaluation of Fimbrolide–Nitric Oxide Donor Hybrids as Antimicrobial Agents. J. Med. Chem. 2013, 56, 9517–9529. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, I.W. Biofilm exopolysaccharides: A strong and sticky framework. Microbiology 2001, 147, 3–9. [Google Scholar] [CrossRef]
- Donlan, R.M. Biofilms: Microbial Life on Surfaces. Emerg. Infect. Dis. 2002, 8, 881–890. [Google Scholar] [CrossRef] [PubMed]
- Vu, B.; Chen, M.; Crawford, R.J.; Ivanova, E.P. Bacterial extracellular polysaccharides involved in biofilm formation. Molecules 2009, 14, 2535–2554. [Google Scholar] [CrossRef] [PubMed]
- Uruén, C.; Chopo-Escuin, G.; Tommassen, J.; Mainar-Jaime, R.C.; Arenas, J. Biofilms as promoters of bacterial antibiotic resistance and tolerance. Antibiotics 2021, 10, 3. [Google Scholar] [CrossRef]
- Divakar, S.; Lama, M.; Khan, A.U. Antibiotics versus biofilm: An emerging battleground in microbial communities. Antimicrob. Resist. Infect. Control 2019, 8, 76. [Google Scholar]
- Anantharaman, S.; Guercio, D.; Mendoza, A.G.; Withorn, J.M.; Boon, E.M. Negative regulation of biofilm formation by nitric oxide sensing proteins. Biochem. Soc. Trans. 2023, 51, 1447–1458. [Google Scholar] [CrossRef]
- Andrabi, S.M.; Andrabi, S.M.; Sharma, N.S.; Sharma, N.S.; Karan, A.; Karan, A.; Shahriar, S.M.S.; Shahriar, S.M.S.; Cordon, B.; Cordon, B.; et al. Nitric Oxide: Physiological Functions, Delivery, and Biomedical Applications. Adv. Sci. 2023, 10, e2303259. [Google Scholar] [CrossRef]
- Tricoire, L.; Vitalis, T. Neuronal nitric oxide synthase expressing neurons: A journey from birth to neuronal circuits. Front. Neural Circuits 2012, 6, 30605. [Google Scholar] [CrossRef]
- National Center for Biotechnology Information. PubChem Compound Summary for CID 145068. Nitric Oxide. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Nitric-Oxide (accessed on 10 October 2024).
- National Center for Biotechnology Information. PubChem Compound Summary for CID 977. Oxygen. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Oxygen (accessed on 10 October 2024).
- National Center for Biotechnology Information. PubChem Compound Summary for CID 807. Iodine. Available online: https://pubchem.ncbi.nlm.nih.gov/compound/Iodine (accessed on 10 October 2024).
- Gilbert, P.; Moore, L. Cationic antiseptics: Diversity of action under a common epithet. J. Appl. Microbiol. 2005, 99, 703–715. [Google Scholar] [CrossRef]
- Miranda, J.E.A.; Sotomayor, C.E.; Albesa, I.; Paraje, M.G. Oxidative and nitrosative stress in Staphylococcus aureus biofilm. FEMS Microbiol. Lett. 2011, 315, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Duan, J.; Kasper, D.L. Oxidative depolymerization of polysaccharides by reactive oxygen/nitrogen species. Glycobiology 2011, 21, 401–409. [Google Scholar] [CrossRef] [PubMed]
- Jinno, A.; Park, P.W. Role of Glycosaminoglycans in Infectious Disease. In Methods in Molecular Biology; Humana Press: New York, NY, USA, 2015; Volume 1229, pp. 567–585. [Google Scholar] [CrossRef]
- Lindberg, B.; Lindqvist, B.; Lönngren, J.; Powell, D.A. Structural studies of the capsular polysaccharide from streptococcus pneumoniae type 1. Carbohydr. Res. 1980, 78, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Velez, C.D.; Lewis, C.J.; Kasper, D.L.; Cobb, B.A. Type I Streptococcus pneumoniae carbohydrate utilizes a nitric oxide and MHC II-dependent pathway for antigen presentation. Immunology 2009, 127, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Chew, S.C.; Kundukad, B.; Seviour, T.; van der Maarel, J.R.C.; Yang, L.; Rice, S.A.; Doyle, P.; Kjelleberg, S. Dynamic Remodeling of Microbial Biofilms by Functionally Distinct Exopolysaccharides. mBio 2014, 5, 1–11. [Google Scholar] [CrossRef]
- Fong, J.N.C.; Yildiz, F.H. Biofilm matrix proteins. Microb. Biofilms 2015, 3, 201–222. [Google Scholar] [CrossRef]
- Reighard, K.P.; Hill, D.B.; Dixon, G.A.; Worley, B.V.; Schoenfisch, M.H. Disruption and eradication of P. aeruginosa biofilms using nitric oxide-releasing chitosan oligosaccharides. Biofouling 2015, 31, 775–787. [Google Scholar] [CrossRef]
- Wang, S.; Liu, X.; Liu, H.; Zhang, L.; Guo, Y.; Yu, S.; Wozniak, D.J.; Ma, L.Z. The exopolysaccharide Psl–eDNA interaction enables the formation of a biofilm skeleton in Pseudomonas aeruginosa. Environ. Microbiol. Rep. 2015, 7, 330–340. [Google Scholar] [CrossRef]
- Butler, A.R.; Ridd, J.H. Formation of nitric oxide from nitrous acid in ischemic tissue and skin. Nitric Oxide 2004, 10, 20–24. [Google Scholar] [CrossRef]
- Chislett, M.; Guo, J.; Bond, P.L.; Yuan, Z. Structural changes in model compounds of sludge extracellular polymeric substances caused by exposure to free nitrous acid. Water Res. 2021, 188, 116553. [Google Scholar] [CrossRef]
- Wu, M.; Lu, Z.; Wu, K.; Nam, C.; Zhang, L.; Guo, J. Recent advances in the development of nitric oxide-releasing biomaterials and their application potentials in chronic wound healing. J. Mater. Chem. B 2021, 9, 7063–7075. [Google Scholar] [CrossRef] [PubMed]
- Waite, R.D.; Stewart, J.E.; Stephen, A.S.; Allaker, R.P. Activity of a nitric oxide-generating wound treatment system against wound pathogen biofilms. Int. J. Antimicrob. Agents 2018, 52, 338–343. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Product/Prototype | NO Technology | Status | Reference |
|---|---|---|---|
| EDX 110 | Acidified nitrite in two-part superabsorbent wound dressing | Prototype wound dressing reported in the ProNOx1 clinical study in DFUs | [20] |
| N1O1 Nitric Oxide Activating Serum with Antioxidants | Acidified nitrite in a two-part aqueous formulation | Available over-the-counter (OTC) | [22] |
| NoWonder™ Foot Cleanser | Nitric oxide in two-part aqueous solution footbath | Described as available in the US. Clinical proof of concept completed in a 40-patient DFU study | [23] |
| NOX 1416 | Acidified nitrite in a surfactant-containing aqueous foam formulation delivered in a two-part pump dispenser | Prototype wound care product subject to ongoing clinical registry [24] | [25] |
| Species | NO Source | NO Donor Concentration | Approximate NO Concentration | Effect on Biofilm | Reference |
|---|---|---|---|---|---|
| Gram-Negative | |||||
| Pseudomonas aeruginosa | Sodium nitroprusside (SNP) | 25 nM–2.5 mM | 0.025–2500 nM * | Reduction | [103] |
| Pseudomonas aeruginosa | SNP | >25 mM | >25,000 nM * | Reduction | [103] |
| Serratia marcescens | SNP | 25–500 nM | 0.025–0.5 nM * | Reduction | [103] |
| Escherichia coli | SNP | 500 nM | 0.5 nM * | Reduction | [100] |
| Escherichia coli | N-diazeniumdiolate (NONOate) | 100 μM | ~100–300 nM | Reduction | [109] |
| Acinetobacter baumannii | Gaseous NO | 200 ppm | ~7 mM | Reduction | [110] |
| Gram-Positive | |||||
| Staphylococcus epidermidis | SNP | 10 μM | 10 nM * | Reduction | [100] |
| Staphylococcus aureus | Gaseous NO | 200 ppm | ~7 mM | Reduction | [110] |
| Staphylococcus aureus | NONOate | 1–1000 μM | >0.125 mM | Reduction | [111] |
| Staphylococcus aureus | NONOate | 1–1000 μM | ~900–2000 nM | Reduction | [111] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roberts, J.M.; Milo, S.; Metcalf, D.G. Harnessing the Power of Our Immune System: The Antimicrobial and Antibiofilm Properties of Nitric Oxide. Microorganisms 2024, 12, 2543. https://doi.org/10.3390/microorganisms12122543
Roberts JM, Milo S, Metcalf DG. Harnessing the Power of Our Immune System: The Antimicrobial and Antibiofilm Properties of Nitric Oxide. Microorganisms. 2024; 12(12):2543. https://doi.org/10.3390/microorganisms12122543
Chicago/Turabian StyleRoberts, Jonathan Matthew, Scarlet Milo, and Daniel Gary Metcalf. 2024. "Harnessing the Power of Our Immune System: The Antimicrobial and Antibiofilm Properties of Nitric Oxide" Microorganisms 12, no. 12: 2543. https://doi.org/10.3390/microorganisms12122543
APA StyleRoberts, J. M., Milo, S., & Metcalf, D. G. (2024). Harnessing the Power of Our Immune System: The Antimicrobial and Antibiofilm Properties of Nitric Oxide. Microorganisms, 12(12), 2543. https://doi.org/10.3390/microorganisms12122543