
Geographic Distribution Pattern Determines Soil Microbial Community Assembly Process in Acanthopanax senticosus Rhizosphere Soil


Abstract
1. Introduction
2. Materials and Methods
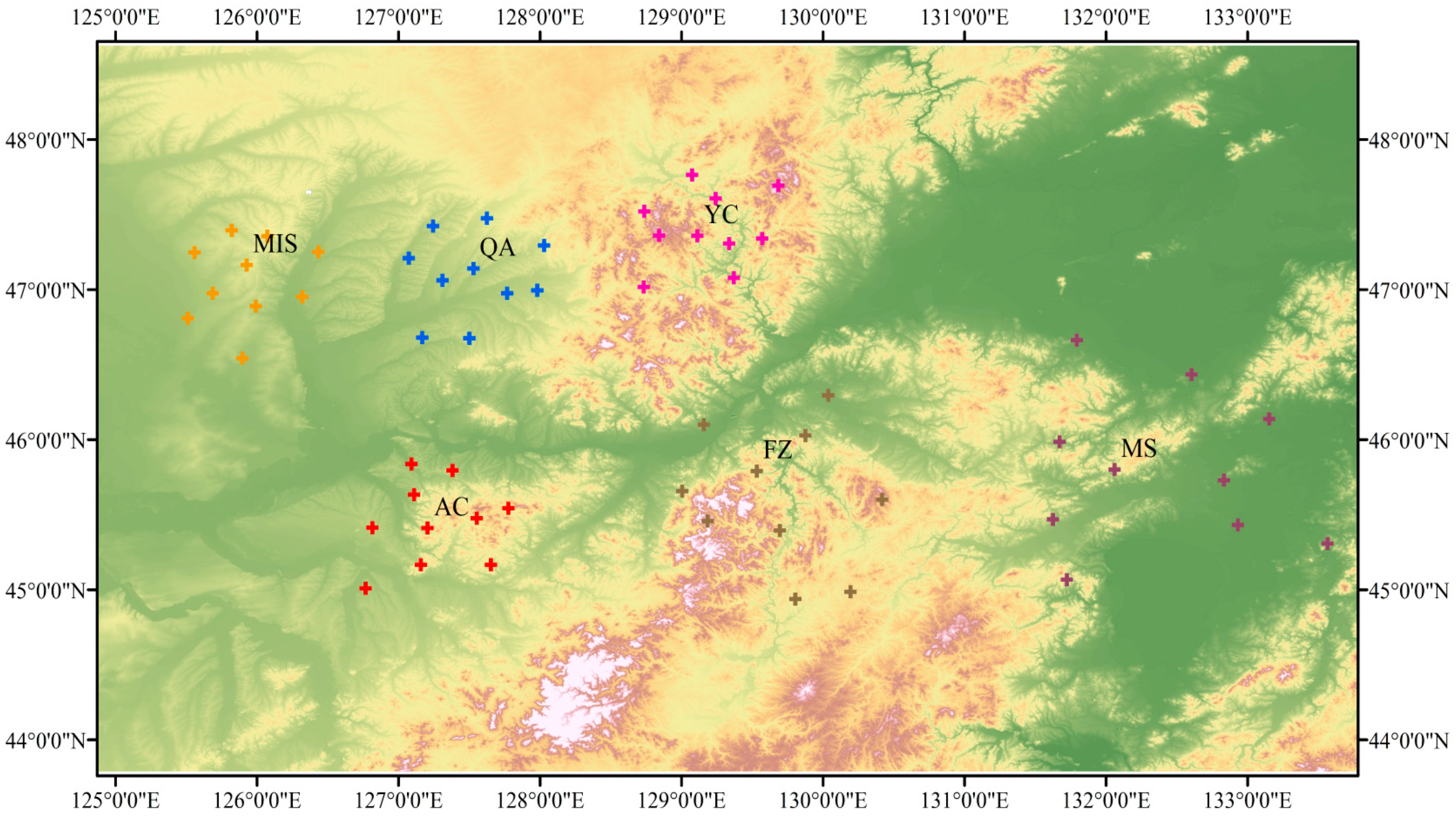
2.1. Study Sites
2.2. Determination of Soil Chemical Properties
2.3. Determination of Plant Biomass and Secondary Metabolites
2.4. DNA Extraction and High-Throughput Sequencing
2.5. Bioinformatic and Statistical Analysis
3. Results
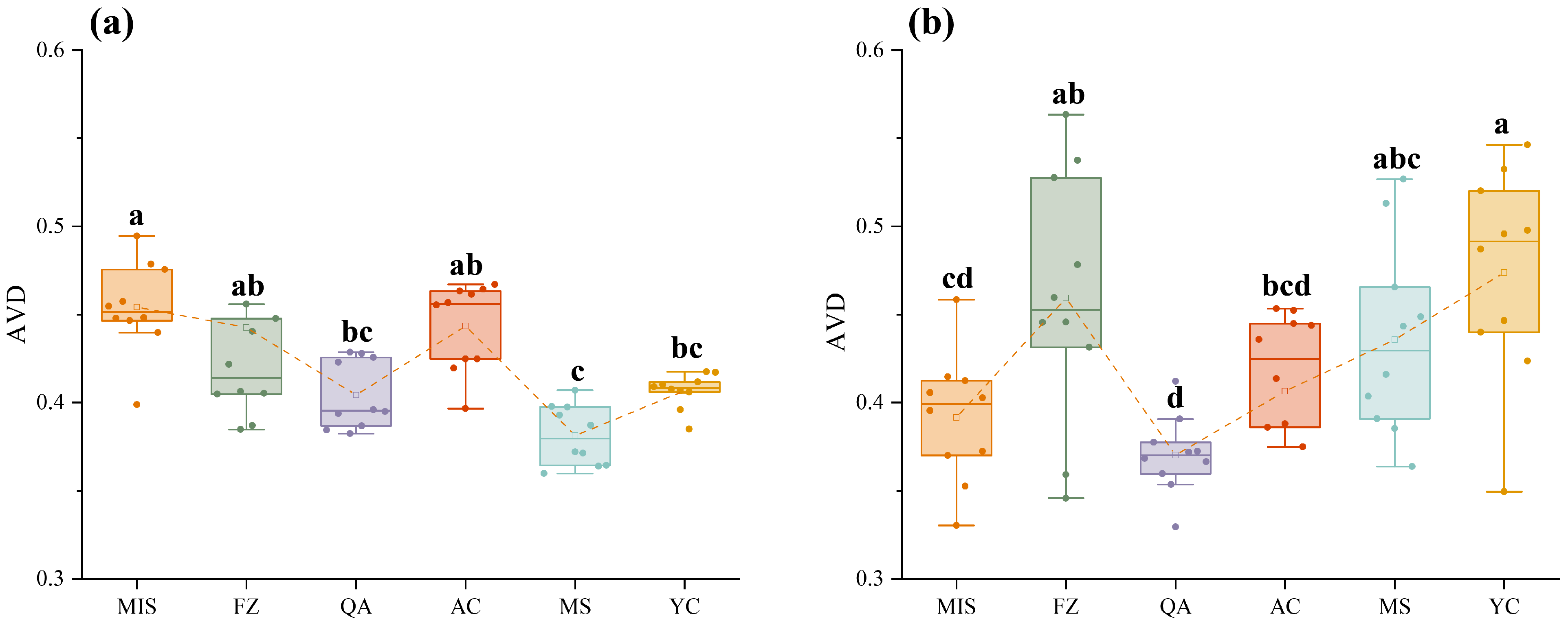
3.1. Soil Chemical Properties, Plant Biomass and Secondary Metabolites
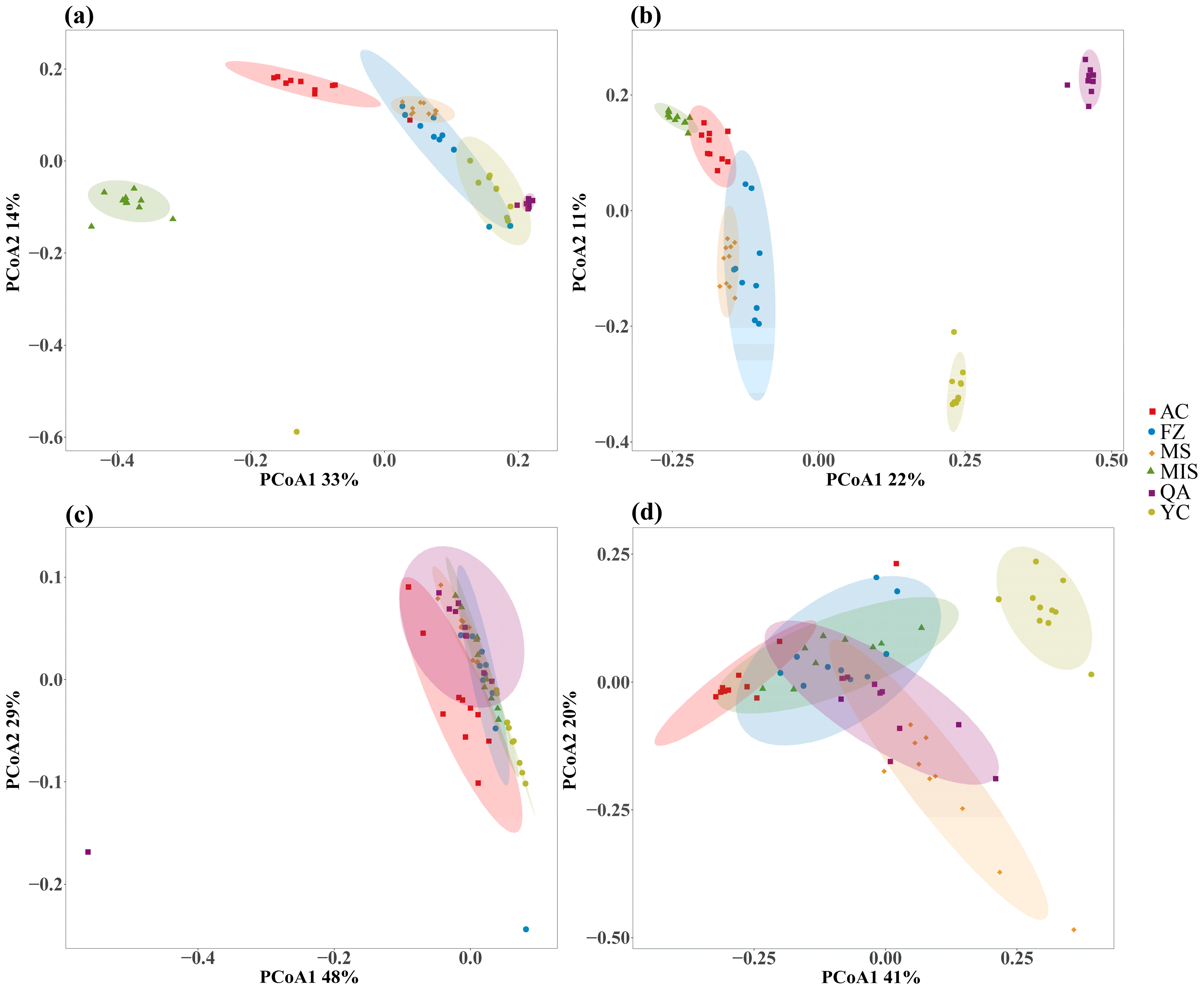
3.2. Changes of Soil Microbial Diversity and Composition in Different Geographic Distributions of Acanthopanax senticosus Rhizosphere Soils
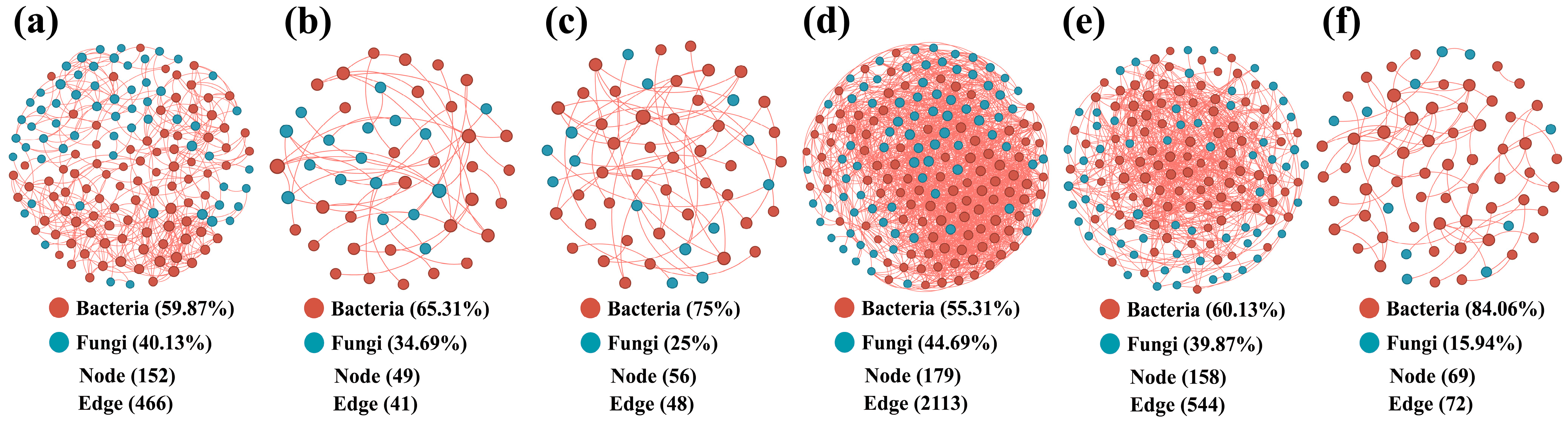
3.3. Soil Microbial Co-Occurrence Networks
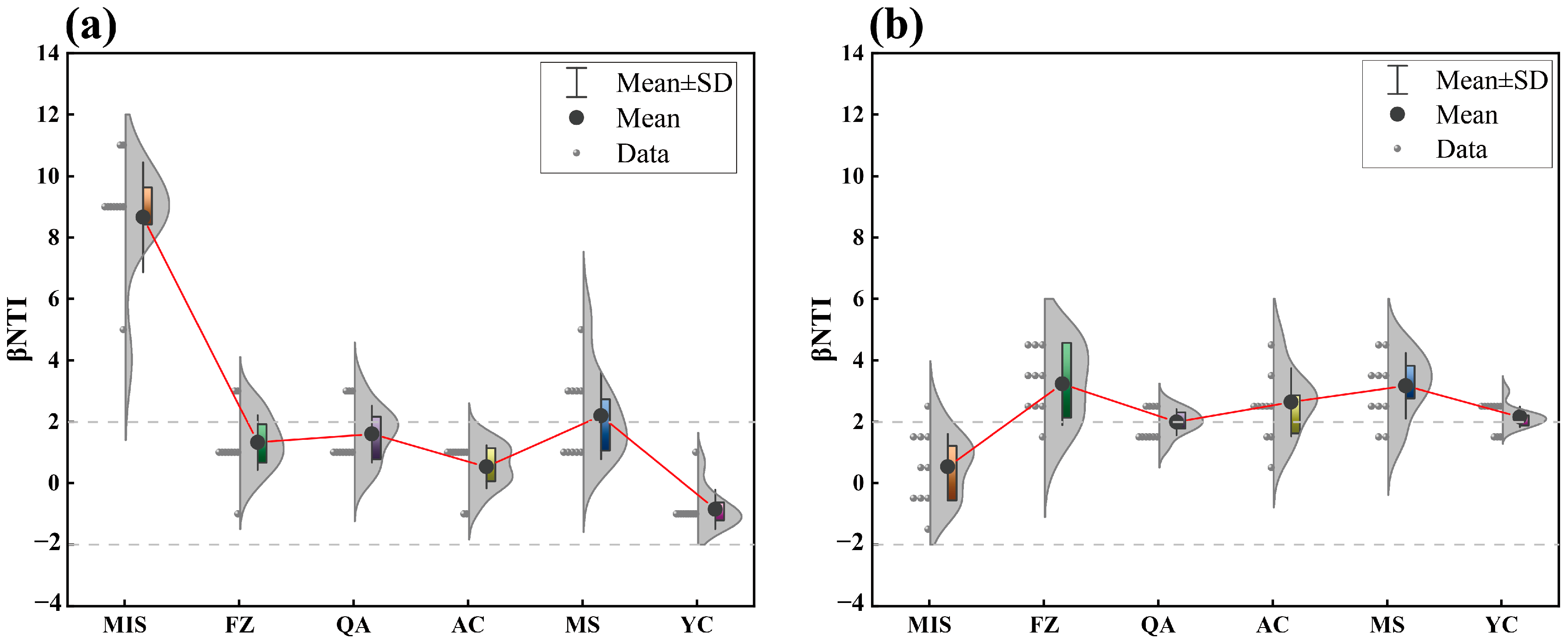
3.4. The Community Assembly of Soil Microbial Communities
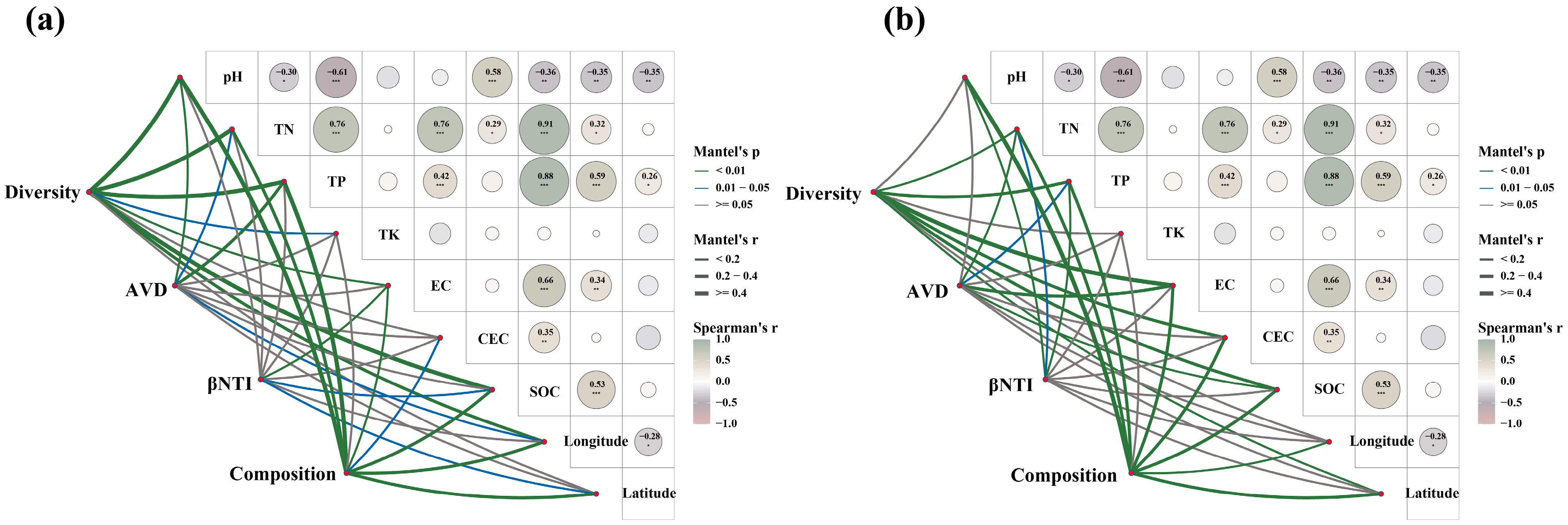
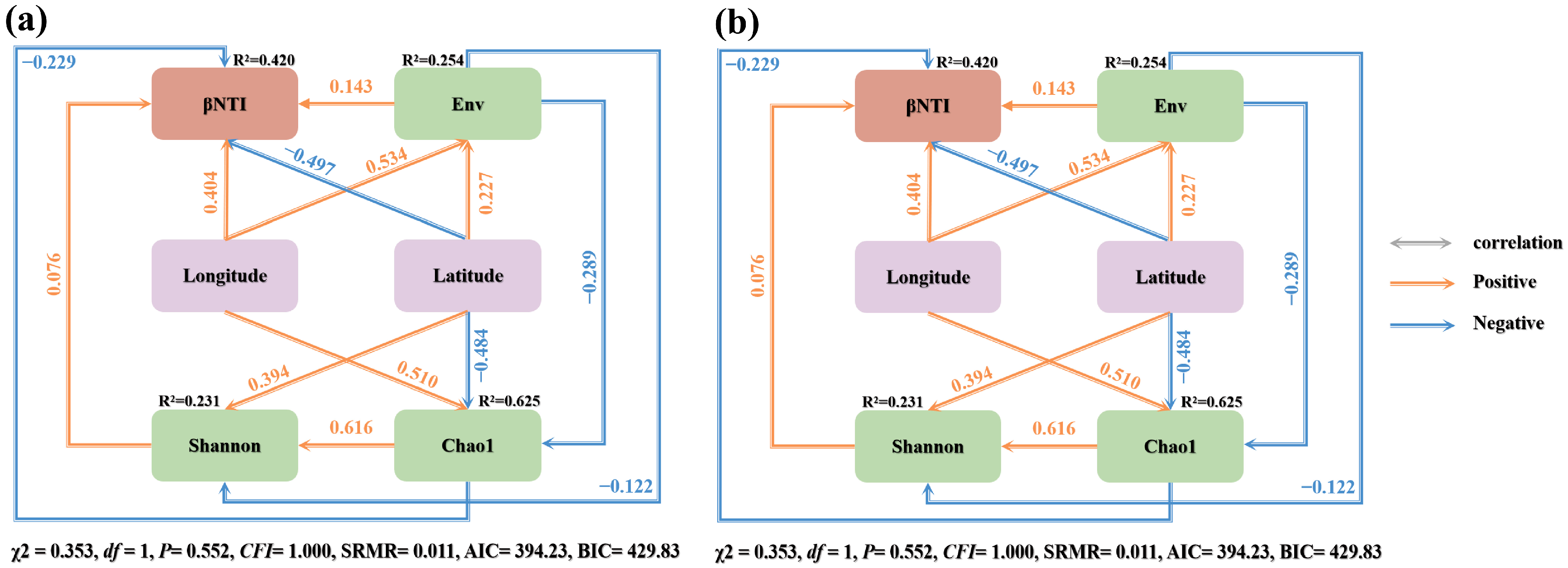
3.5. Factors Influencing Changes in Soil Microbial Communities
3.6. Prediction and Analysis of Soil Microbial Community Function
4. Discussion
4.1. Soil Microbial Diversity and Composition
4.2. Soil Microbial Co-Occurrence Network and Community Assembly
4.3. Key Regulators Influencing Changes in Soil Microbial Communities
4.4. Prediction and Analysis of Soil Nicrobial Community Function
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Coelho, M.T.P.; Barreto, E.; Rangel, T.F.; Diniz-Filho, J.A.F.; Wüest, R.O.; Bach, W.; Skeels, A.; McFadden, I.R.; Roberts, D.W.; Pellissier, L.; et al. The geography of climate and the global patterns of species diversity. Nature 2023, 622, 537–544. [Google Scholar] [CrossRef]
- Loiseau, N.; Mouquet, N.; Casajus, N.; Grenié, M.; Guéguen, M.; Maitner, B.; Mouillot, D.; Ostling, A.; Renaud, J.; Tucker, C.; et al. Global distribution and conservation status of ecologically rare mammal and bird species. Nat. Commun. 2020, 11, 5071. [Google Scholar] [CrossRef]
- Lawlor, J.A.; Comte, L.; Grenouillet, G.; Lenoir, J.; Baecher, J.A.; Bandara, R.M.W.J.; Bertrand, R.; Chen, I.C.; Diamond, S.E.; Lancaster, L.T.; et al. Mechanisms, detection and impacts of species redistributions under climate change. Nat. Rev. Earth Environ. 2024, 5, 351–368. [Google Scholar] [CrossRef]
- Cao, W.; Wu, D.; Huang, L.; Pan, M.; Huhe, T. Determinizing the contributions of human activities and climate change on greening in the Beijing–Tianjin–Hebei Region, China. Sci. Rep. 2021, 11, 21201. [Google Scholar] [CrossRef]
- Hartmann, M.; Six, J. Soil structure and microbiome functions in agroecosystems. Nat. Rev. Earth Environ. 2023, 4, 4–18. [Google Scholar] [CrossRef]
- Nabi, M. Chapter eleven—Role of microorganisms in plant nutrition and soil health. In Sustainable Plant Nutrition; Aftab, T., Hakeem, K.R., Eds.; Academic Press: Cambridge, MA, USA, 2023; pp. 263–282. [Google Scholar]
- Hanson, C.A.; Fuhrman, J.A.; Horner-Devine, M.C.; Martiny, J.B.H. Beyond biogeographic patterns: Processes shaping the microbial landscape. Nat. Rev. Microbiol. 2012, 10, 497–506. [Google Scholar] [CrossRef]
- Martiny, J.B.H.; Bohannan, B.J.M.; Brown, J.H.; Colwell, R.K.; Fuhrman, J.A.; Green, J.L.; Horner-Devine, M.C.; Kane, M.; Krumins, J.A.; Kuske, C.R.; et al. Microbial biogeography: Putting microorganisms on the map. Nat. Rev. Microbiol. 2006, 4, 102–112. [Google Scholar] [CrossRef]
- Liu, N.; Hu, H.; Ma, W.; Deng, Y.; Wang, Q.; Luo, A.; Meng, J.; Feng, X.; Wang, Z. Relative Importance of Deterministic and Stochastic Processes on Soil Microbial Community Assembly in Temperate Grasslands. Microorganisms 2021, 9, 1929. [Google Scholar] [CrossRef]
- Dumbrell, A.J.; Nelson, M.; Helgason, T.; Dytham, C.; Fitter, A.H. Relative roles of niche and neutral process in structuring a soil microbial community. ISME J. 2010, 4, 1078. [Google Scholar] [CrossRef]
- Jiao, S.; Yang, Y.; Xu, Y.; Zhang, J.; Lu, Y. Balance between community assembly processes mediates species coexistence in agricultural soil microbiomes across eastern China. ISME J. 2020, 14, 202–216. [Google Scholar] [CrossRef]
- Gentry, T.J.; Pepper, I.L.; Pierson, L.S. Chapter 19—Microbial Diversity and Interactions in Natural Ecosystems. In Environmental Microbiology, 3rd ed.; Pepper, I.L., Gerba, C.P., Gentry, T.J., Eds.; Academic Press: San Diego, CA, USA, 2015; pp. 441–460. [Google Scholar]
- Aguirre-Liguori, J.A.; Ramírez-Barahona, S.; Gaut, B.S. The evolutionary genomics of species’ responses to climate change. Nat. Ecol. Evol. 2021, 5, 1350–1360. [Google Scholar] [CrossRef] [PubMed]
- Levine, J.M.; Bascompte, J.; Adler, P.B.; Allesina, S. Beyond pairwise mechanisms of species coexistence in complex communities. Nature 2017, 546, 56–64. [Google Scholar] [CrossRef]
- Aqeel, M.; Ran, J.; Hu, W.; Irshad, M.K.; Dong, L.; Akram, M.A.; Eldesoky, G.E.; Aljuwayid, A.M.; Chuah, L.F.; Deng, J. Plant-soil-microbe interactions in maintaining ecosystem stability and coordinated turnover under changing environmental conditions. Chemosphere 2023, 318, 137924. [Google Scholar] [CrossRef]
- Zuo, X.; Sun, S.; Wang, S.; Yue, P.; Hu, Y.; Zhao, S.; Guo, X.; Li, X.; Chen, M.; Ma, X.; et al. Contrasting relationships between plant-soil microbial diversity are driven by geographic and experimental precipitation changes. Sci. Total Environ. 2023, 861, 160654. [Google Scholar] [CrossRef]
- Li, T.; Ferns, K.; Yan, Z.Q.; Yin, S.Y.; Kou, J.J.; Li, D.; Zeng, Z.; Yin, L.; Wang, X.; Bao, H.X.; et al. Acanthopanax senticosus: Photochemistry and Anticancer Potential. Am. J. Chin. Med. 2016, 44, 1543–1558. [Google Scholar] [CrossRef]
- Molefe-Madlaliso, R.; Amoo, A.; Babalola, O. Communication between plant roots and the soil microbiome; involvement in plant growth and development. Symbiosis 2023, 90, 231–239. [Google Scholar] [CrossRef]
- Islam, W.; Noman, A.; Naveed, H.; Huang, Z.; Chen, H.Y.H. Role of environmental factors in shaping the soil microbiome. Environ. Sci. Pollut. Res. Int. 2020, 27, 41225–41247. [Google Scholar] [CrossRef]
- Yu, Y.; Liu, H.; Zhang, L.; Sun, Z.; Lei, B.; Miao, Y.; Chu, H.; Han, S.; Shi, Y.; Zheng, J. Distinct response patterns of plants and soil microorganisms to agronomic practices and seasonal variation in a floodplain ecosystem. Front. Microbiol. 2023, 14, 1094750. [Google Scholar] [CrossRef]
- Yu, Y.; Ru, J.; Lei, B.; Han, S.; Wan, S.; Zheng, J. Distinct response patterns of soil micro-eukaryotic communities to early-season and late-season precipitation in a semiarid grassland. Soil Biol. Biochem. 2024, 194, 109427. [Google Scholar] [CrossRef]
- Zhang, G.; Bai, J.; Zhao, Q.; Jia, J.; Wang, X.; Wang, W.; Wang, X. Soil carbon storage and carbon sources under different Spartina alterniflora invasion periods in a salt marsh ecosystem. Catena 2021, 196, 104831. [Google Scholar] [CrossRef]
- Baradaran Motie, J.; Aghkhani, M.H.; Rohani, A.; Lakzian, A. A soft-computing approach to estimate soil electrical conductivity. Biosyst. Eng. 2021, 205, 105–120. [Google Scholar] [CrossRef]
- Razzaghi, F.; Arthur, E.; Moosavi, A.A. Evaluating models to estimate cation exchange capacity of calcareous soils. Geoderma 2021, 400, 115221. [Google Scholar] [CrossRef]
- Yang, X.; Li, Y.; Li, C.; Li, Q.; Qiao, B.; Shi, S.; Zhao, C. Enhancement of Interplanting of Ficus carica L. with Taxus cuspidata Sieb. et Zucc. on Growth of Two Plants. Agriculture 2021, 11, 1276. [Google Scholar] [CrossRef]
- Arellano, P.; Tansey, K.; Balzter, H.; Boyd, D.S. Field spectroscopy and radiative transfer modelling to assess impacts of petroleum pollution on biophysical and biochemical parameters of the Amazon rainforest. Environ. Earth Sci. 2017, 76, 217. [Google Scholar] [CrossRef]
- Jacquemoud, S.; Baret, F. PROSPECT: A model of leaf optical properties spectra. Remote Sens. Environ. 1990, 34, 75–91. [Google Scholar] [CrossRef]
- Wang, Z.; Tang, C.; Dai, F.; Xiao, G.; Luo, G. HPLC determination of phenolic compounds in different solvent extracts of mulberry leaves and antioxidant capacity of extracts. Int. J. Food Prop. 2021, 24, 544–552. [Google Scholar] [CrossRef]
- Smyrniotakis, C.G.; Archontaki, H.A. C18 columns for the simultaneous determination of oxytetracycline and its related substances by reversed-phase high performance liquid chromatography and UV detection. J. Pharm. Biomed. Anal. 2007, 43, 506–514. [Google Scholar] [CrossRef]
- Huse, S.M.; Dethlefsen, L.; Huber, J.A.; Mark Welch, D.; Relman, D.A.; Sogin, M.L. Exploring microbial diversity and taxonomy using SSU rRNA hypervariable tag sequencing. PLoS Genet. 2008, 4, e1000255. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berg-Lyons, D.; Lozupone, C.A.; Turnbaugh, P.J.; Fierer, N.; Knight, R. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc. Natl. Acad. Sci. USA 2011, 108 (Suppl. S1), 4516–4522. [Google Scholar] [CrossRef]
- Liu, J.; Yu, Y.; Cai, Z.; Bartlam, M.; Wang, Y. Comparison of ITS and 18S rDNA for estimating fungal diversity using PCR-DGGE. World J. Microbiol. Biotechnol. 2015, 31, 1387–1395. [Google Scholar] [CrossRef]
- Ravi, R.K.; Walton, K.; Khosroheidari, M. MiSeq: A Next Generation Sequencing Platform for Genomic Analysis. Methods Mol. Biol. 2018, 1706, 223–232. [Google Scholar] [CrossRef]
- Li, J.; Hua, Z.S.; Liu, T.; Wang, C.; Li, J.; Bai, G.; Lücker, S.; Jetten, M.S.M.; Zheng, M.; Guo, J. Selective enrichment and metagenomic analysis of three novel comammox Nitrospira in a urine-fed membrane bioreactor. ISME Commun. 2021, 1, 7. [Google Scholar] [CrossRef]
- Pruesse, E.; Quast, C.; Knittel, K.; Fuchs, B.M.; Ludwig, W.; Peplies, J.; Glöckner, F.O. SILVA: A comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res. 2007, 35, 7188–7196. [Google Scholar] [CrossRef]
- Větrovský, T.; Morais, D.; Kohout, P.; Lepinay, C.; Algora, C.; Awokunle Hollá, S.; Bahnmann, B.D.; Bílohnědá, K.; Brabcová, V.; D’Alò, F.; et al. GlobalFungi, a global database of fungal occurrences from high-throughput-sequencing metabarcoding studies. Sci. Data 2020, 7, 228. [Google Scholar] [CrossRef]
- Toole, D.R.; Zhao, J.; Martens-Habbena, W.; Strauss, S.L. Bacterial functional prediction tools detect but underestimate metabolic diversity compared to shotgun metagenomics in southwest Florida soils. Appl. Soil Ecol. 2021, 168, 104129. [Google Scholar] [CrossRef]
- Zhang, P.; Guan, P.; Hao, C.; Yang, J.; Xie, Z.; Wu, D. Changes in assembly processes of soil microbial communities in forest-to-cropland conversion in Changbai Mountains, northeastern China. Sci. Total Environ. 2022, 818, 151738. [Google Scholar] [CrossRef]
- Zhang, H.; Pu, R.; Liu, X. A New Image Processing Procedure Integrating PCI-RPC and ArcGIS-Spline Tools to Improve the Orthorectification Accuracy of High-Resolution Satellite Imagery. Remote. Sens. 2016, 8, 827. [Google Scholar] [CrossRef]
- Hishe, S.; Lyimo, J.G.; Bewket, W. Soil and water conservation effects on soil properties in the Middle Silluh Valley, northern Ethiopia. Int. Soil Water Conserv. Res. 2017, 5, 231–240. [Google Scholar] [CrossRef]
- Foster, Z.S.L.; Sharpton, T.; Grünwald, N.J. Metacoder: An R package for visualization and manipulation of community taxonomic diversity data. PLoS Comput. Biol. 2016, 13, e1005404. [Google Scholar] [CrossRef]
- Wang, T. Improved random forest classification model combined with C5.0 algorithm for vegetation feature analysis in non-agricultural environments. Sci. Rep. 2024, 14, 10367. [Google Scholar] [CrossRef]
- Bastian, M.; Heymann, S.; Jacomy, M. Gephi: An Open Source Software for Exploring and Manipulating Networks. In Proceedings of the 3rd International AAAI Conference on Weblogs and Social Media, San Jose, CA, USA, 17–20 May 2009. [Google Scholar]
- Langendorf, R.E.; Burgess, M.G. Empirically classifying network mechanisms. Sci. Rep. 2021, 11, 20501. [Google Scholar] [CrossRef]
- Charmpi, K.; Chokkalingam, M.; Johnen, R.; Beyer, A. Optimizing network propagation for multi-omics data integration. PLoS Comput. Biol. 2021, 17, e1009161. [Google Scholar] [CrossRef]
- Chen, W.; Ren, K.; Isabwe, A.; Chen, H.; Liu, M.; Yang, J. Stochastic processes shape microeukaryotic community assembly in a subtropical river across wet and dry seasons. Microbiome 2019, 7, 138. [Google Scholar] [CrossRef]
- Dilnessa, G.; Zerihun, W.; Sileshi, N.; Enyew, A. Impacts of environmental conditions on woody plant diversity, structure and regeneration in forest patches of Guna Mountain: Este District, South Gondar Zone, Ethiopia. J. Mt. Sci. 2023, 20, 1940–1953. [Google Scholar] [CrossRef]
- Sheng, Y.; Li, G.; Dong, H.; Liu, Y.; Ma, L.; Yang, M.; Liu, Y.; Liu, J.; Deng, S.; Zhang, D. Distinct assembly processes shape bacterial communities along unsaturated, groundwater fluctuated, and saturated zones. Sci. Total Environ. 2021, 761, 143303. [Google Scholar] [CrossRef]
- Friedman, L.M.; Furberg, C.D.; DeMets, D.L. The Randomization Process. In Fundamentals of Clinical Trials; Friedman, L.M., Furberg, C.D., DeMets, D.L., Eds.; Springer New York: New York, NY, USA, 2010; pp. 97–117. [Google Scholar]
- Stegen, J.C.; Lin, X.; Konopka, A.E.; Fredrickson, J.K. Stochastic and deterministic assembly processes in subsurface microbial communities. ISME J. 2012, 6, 1653–1664. [Google Scholar] [CrossRef]
- Skrondal, A.; Rabe-Hesketh, S. Structural Equation Modeling: Categorical Variables. In Encyclopedia of Statistics in Behavioral Science; Wiley: Hoboken, NJ, USA, 2005. [Google Scholar]
- Shi, D.X.; Lee, T.; Terry, R.A. Revisiting the Model Size Effect in Structural Equation Modeling (SEM). Multivar. Behav. Res. 2015, 50, 142. [Google Scholar] [CrossRef]
- Carrasco-Espinosa, K.; Avitia, M.; Santini, N.S.; Escalante, A.E. Nutrient contents and microbial communities as mediators of the effects of land-use in ecosystem functioning in alpine ecosystems from Central Mexico. J. Soils Sediments 2024, 24, 2986–3000. [Google Scholar] [CrossRef]
- Flores-Rentería, D.; Rincón, A.; Morán-López, T.; Heres, A.M.; Pérez-Izquierdo, L.; Valladares, F.; Yuste, J.C. Habitat fragmentation is linked to cascading effects on soil functioning and CO2 emissions in Mediterranean holm-oak-forests. Peerj 2018, 6, e5857. [Google Scholar] [CrossRef]
- Martínez-García, E.A.; Rodriguez, N.A.; Rodriguez Jorge, R.; Mizera-Pietraszko, J.; Sheba, J.K.; Mohan, R.E.; Magid, E. Non Linear Fitting Methods for Machine Learning. In Proceedings of the International Conference on P2P, Parallel, Grid, Cloud and Internet Computing, Barcelona, Spain, 8–10 November 2017. [Google Scholar]
- O’Brien, T.E.; Silcox, J.W. Nonlinear Regression Modelling: A Primer with Applications and Caveats. Bull. Math. Biol. 2024, 86, 40. [Google Scholar] [CrossRef]
- Yang, N.; Li, X.; Liu, D.; Zhang, Y.; Chen, Y.; Wang, B.; Hua, J.; Zhang, J.; Peng, S.; Ge, Z.; et al. Diversity patterns and drivers of soil bacterial and fungal communities along elevational gradients in the Southern Himalayas, China. Appl. Soil Ecol. 2022, 178, 104563. [Google Scholar] [CrossRef]
- Stuart, A.D.; Ilić, M.; Simmons, B.I.; Sutherland, W.J. Sea stack plots: Replacing bar charts with histograms. Ecol. Evol. 2024, 14, e11237. [Google Scholar] [CrossRef]
- El Kurdi, R.; Kumar, K.; Patra, D. Introducing Principal Coordinate Analysis (PCoA) Assisted EEMF Spectroscopic Based Novel Analytical Approach for the Discrimination of Commercial Gasoline Fuels. J. Fluoresc. 2020, 30, 1583–1589. [Google Scholar] [CrossRef]
- Khan, A.; Jiang, H.; Bu, J.; Adnan, M.; Gillani, S.W.; Hussain, M.A.; Zhang, M. Untangling the Rhizosphere Bacterial Community Composition and Response of Soil Physiochemical Properties to Different Nitrogen Applications in Sugarcane Field. Front. Microbiol. 2022, 13, 856078. [Google Scholar] [CrossRef]
- Simpson, G.L.; Solymos, P.; Stevens, M.; Wagner, H. Vegan: Community Ecology Package; R-Project: Vienna, Austria, 2018. [Google Scholar]
- Crupi, V. Measures of Biological Diversity: Overview and Unified Framework. In From Assessing to Conserving Biodiversity; Springer: Cham, Switzerland, 2019. [Google Scholar]
- Louthan, A.M.; DeMarche, M.L.; Shoemaker, L.G. Climate sensitivity across latitude: Scaling physiology to communities. Trends Ecol. Evol. 2021, 36, 931–942. [Google Scholar] [CrossRef]
- Saupe, E.E.; Myers, C.E.; Townsend Peterson, A.; Soberón, J.; Singarayer, J.; Valdes, P.; Qiao, H. Spatio-temporal climate change contributes to latitudinal diversity gradients. Nat. Ecol. Evol. 2019, 3, 1419–1429. [Google Scholar] [CrossRef]
- Bastida, F.; Eldridge, D.J.; García, C.; Kenny Png, G.; Bardgett, R.D.; Delgado-Baquerizo, M. Soil microbial diversity-biomass relationships are driven by soil carbon content across global biomes. ISME J. 2021, 15, 2081–2091. [Google Scholar] [CrossRef]
- Rousk, J.; Bååth, E.; Brookes, P.C.; Lauber, C.L.; Lozupone, C.; Caporaso, J.G.; Knight, R.; Fierer, N. Soil bacterial and fungal communities across a pH gradient in an arable soil. ISME J. 2010, 4, 1340–1351. [Google Scholar] [CrossRef]
- Xun, W.; Liu, Y.; Li, W.; Ren, Y.; Xiong, W.; Xu, Z.; Zhang, N.; Miao, Y.; Shen, Q.; Zhang, R. Specialized metabolic functions of keystone taxa sustain soil microbiome stability. Microbiome 2021, 9, 35. [Google Scholar] [CrossRef]
- Huang, Y.H.; Yang, Y.J.; Li, J.Y.; Lü, H.; Zhao, H.M.; Xiang, L.; Li, H.; Mo, C.H.; Li, Y.W.; Cai, Q.Y.; et al. Root-associated bacteria strengthen their community stability against disturbance of antibiotics on structure and functions. J. Hazard. Mater. 2024, 465, 133317. [Google Scholar] [CrossRef]
- Liu, S.; Yu, H.; Yu, Y.; Huang, J.; Zhou, Z.-X.; Zeng, J.; Chen, P.; Xiao, F.; He, Z.; Yan, Q. Ecological stability of microbial communities in Lake Donghu regulated by keystone taxa. Ecol. Indic. 2022, 136, 108695. [Google Scholar] [CrossRef]
- Routson, C.C.; McKay, N.P.; Kaufman, D.S.; Erb, M.P.; Goosse, H.; Shuman, B.N.; Rodysill, J.R.; Ault, T. Mid-latitude net precipitation decreased with Arctic warming during the Holocene. Nature 2019, 568, 83–87. [Google Scholar] [CrossRef]
- Hernandez, D.J.; David, A.S.; Menges, E.S.; Searcy, C.A.; Afkhami, M.E. Environmental stress destabilizes microbial networks. ISME J. 2021, 15, 1722–1734. [Google Scholar] [CrossRef]
- Wang, C.; Morrissey, E.M.; Mau, R.L.; Hayer, M.; Piñeiro, J.; Mack, M.C.; Marks, J.C.; Bell, S.L.; Miller, S.N.; Schwartz, E.; et al. The temperature sensitivity of soil: Microbial biodiversity, growth, and carbon mineralization. ISME J. 2021, 15, 2738–2747. [Google Scholar] [CrossRef]
- Dai, T.; Wen, D.; Bates, C.T.; Wu, L.; Guo, X.; Liu, S.; Su, Y.; Lei, J.; Zhou, J.; Yang, Y. Nutrient supply controls the linkage between species abundance and ecological interactions in marine bacterial communities. Nat. Commun. 2022, 13, 175. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, J.; Fernandez, V.; Pontrelli, S.; Sauer, U.; Ackermann, M.; Stocker, R. A distinct growth physiology enhances bacterial growth under rapid nutrient fluctuations. Nat. Commun. 2021, 12, 3662. [Google Scholar] [CrossRef]
- Scheuerl, T.; Hopkins, M.; Nowell, R.W.; Rivett, D.W.; Barraclough, T.G.; Bell, T. Bacterial adaptation is constrained in complex communities. Nat. Commun. 2020, 11, 754. [Google Scholar] [CrossRef] [PubMed]
- Yin, Z.; Liang, J.; Zhang, M.; Chen, B.; Yu, Z.; Tian, X.; Deng, X.; Peng, L. Pan-genome insights into adaptive evolution of bacterial symbionts in mixed host-microbe symbioses represented by human gut microbiota Bacteroides cellulosilyticus. Sci. Total Environ. 2024, 927, 172251. [Google Scholar] [CrossRef]
- Kemppinen, J.; Niittynen, P.; Le Roux, P.C.; Momberg, M.; Happonen, K.; Aalto, J.; Rautakoski, H.; Enquist, B.J.; Vandvik, V.; Halbritter, A.H.; et al. Consistent trait-environment relationships within and across tundra plant communities. Nat. Ecol. Evol. 2021, 5, 458–467. [Google Scholar] [CrossRef]
- Egidi, E.; Delgado-Baquerizo, M.; Plett, J.M.; Wang, J.; Eldridge, D.J.; Bardgett, R.D.; Maestre, F.T.; Singh, B.K. A few Ascomycota taxa dominate soil fungal communities worldwide. Nat. Commun. 2019, 10, 2369. [Google Scholar] [CrossRef]
- Willms, I.M.; Rudolph, A.Y.; Göschel, I.; Bolz, S.H.; Schneider, D.; Penone, C.; Poehlein, A.; Schöning, I.; Nacke, H. Globally Abundant “Candidatus Udaeobacter” Benefits from Release of Antibiotics in Soil and Potentially Performs Trace Gas Scavenging. mSphere 2020, 5, 10-1128. [Google Scholar] [CrossRef]
- Spyridonov, I.; Krafft, L.; Schöning, I.; Schrumpf, M.; Nacke, H. The ubiquitous soil verrucomicrobial clade ‘Candidatus Udaeobacter’ shows preferences for acidic pH. Environ. Microbiol. Rep. 2021, 13, 878–883. [Google Scholar] [CrossRef]
- Li, C.; Aluko, O.O.; Yuan, G.; Li, J.; Liu, H. The responses of soil organic carbon and total nitrogen to chemical nitrogen fertilizers reduction base on a meta-analysis. Sci. Rep. 2022, 12, 16326. [Google Scholar] [CrossRef]
- Brewer, T.E.; Handley, K.M.; Carini, P.; Gilbert, J.A.; Fierer, N. Genome reduction in an abundant and ubiquitous soil bacterium ‘Candidatus Udaeobacter copiosus’. Nat. Microbiol. 2016, 2, 16198. [Google Scholar] [CrossRef]
- Patel, K.F.; Fansler, S.J.; Campbell, T.P.; Bond-Lamberty, B.; Smith, A.P.; RoyChowdhury, T.; McCue, L.A.; Varga, T.; Bailey, V.L. Soil texture and environmental conditions influence the biogeochemical responses of soils to drought and flooding. Commun. Earth Environ. 2021, 2, 127. [Google Scholar] [CrossRef]
- Li, F.; Zhang, S.; Wang, Y.; Li, Y.; Li, P.; Chen, L.; Jie, X.; Hu, D.; Feng, B.; Yue, K.; et al. Rare fungus, Mortierella capitata, promotes crop growth by stimulating primary metabolisms related genes and reshaping rhizosphere bacterial community. Soil Biol. Biochem. 2020, 151, 108017. [Google Scholar] [CrossRef]
- Ozimek, E.; Hanaka, A. Mortierella Species as the Plant Growth-Promoting Fungi Present in the Agricultural Soils. Agriculture 2021, 11, 7. [Google Scholar] [CrossRef]
- Biggin, R. Environment and Site-Specificity: Space, Place and Immersion. In Immersive Theatre and Audience Experience: Space, Game and Story in the Work of Punchdrunk; Biggin, R., Ed.; Springer International Publishing: Cham, Switzerland, 2017; pp. 177–205. [Google Scholar]
- Le, V.V.; Kang, M.; Ko, S.R.; Jeong, S.; Park, C.Y.; Lee, J.J.; Choi, I.C.; Oh, H.M.; Ahn, C.Y. Dynamic response of bacterial communities to Microcystis blooms: A three-year study. Sci. Total Environ. 2023, 902, 165888. [Google Scholar] [CrossRef]
- Bai, X.; Dinkla, I.J.T.; Muyzer, G. Shedding light on the total and active core microbiomes in slow sand filters for drinking water production. Water Res. 2023, 243, 120404. [Google Scholar] [CrossRef]
- Menkis, A.; Urbina, H.; James, T.Y.; Rosling, A. Archaeorhizomyces borealis sp. nov. and a sequence-based classification of related soil fungal species. Fungal Biol. 2014, 118, 943–955. [Google Scholar] [CrossRef]
- Yan, J.-Q.; Li, G.-W.; Liu, W.-H.; Chen, Z.-H.; Zhang, P. Updated taxonomy of Chinese Clavaria subg. Syncoryne (Clavariaceae, Agaricales): Description of two new species and one newly recorded species. Mycol. Prog. 2022, 21, 67. [Google Scholar] [CrossRef]
- Bickel, S.; Or, D. Soil bacterial diversity mediated by microscale aqueous-phase processes across biomes. Nat. Commun. 2020, 11, 116. [Google Scholar] [CrossRef]
- Braga, R.M.; Dourado, M.N.; Araújo, W.L. Microbial interactions: Ecology in a molecular perspective. Braz. J. Microbiol. 2016, 47 (Suppl. S1), 86–98. [Google Scholar] [CrossRef]
- Wu, H.; Cui, H.; Fu, C.; Li, R.; Qi, F.; Liu, Z.; Yang, G.; Xiao, K.; Qiao, M. Unveiling the crucial role of soil microorganisms in carbon cycling: A review. Sci. Total Environ. 2024, 909, 168627. [Google Scholar] [CrossRef]
- Zheng, Q.; Hu, Y.; Zhang, S.; Noll, L.; Böckle, T.; Dietrich, M.; Herbold, C.W.; Eichorst, S.A.; Woebken, D.; Richter, A.; et al. Soil multifunctionality is affected by the soil environment and by microbial community composition and diversity. Soil Biol. Biochem. 2019, 136, 107521. [Google Scholar] [CrossRef]
- Hibbing, M.E.; Fuqua, C.; Parsek, M.R.; Peterson, S.B. Bacterial competition: Surviving and thriving in the microbial jungle. Nat. Rev. Microbiol. 2010, 8, 15–25. [Google Scholar] [CrossRef]
- Bajic, D.; Sanchez, A. The ecology and evolution of microbial metabolic strategies. Curr. Opin. Biotechnol. 2020, 62, 123–128. [Google Scholar] [CrossRef]
- Lin, Q.; Li, L.; Adams, J.M.; Heděnec, P.; Tu, B.; Li, C.; Li, T.; Li, X. Nutrient resource availability mediates niche differentiation and temporal co-occurrence of soil bacterial communities. Appl. Soil Ecol. 2021, 163, 103965. [Google Scholar] [CrossRef]
- Tan, W.; Wang, J.; Bai, W.; Qi, J.; Chen, W. Soil bacterial diversity correlates with precipitation and soil pH in long-term maize cropping systems. Sci. Rep. 2020, 10, 6012. [Google Scholar] [CrossRef]
- Manlick, P.J.; Perryman, N.L.; Koltz, A.M.; Cook, J.A.; Newsome, S.D. Climate warming restructures food webs and carbon flow in high-latitude ecosystems. Nat. Clim. Change 2024, 14, 184–189. [Google Scholar] [CrossRef]
- Zhang, W.; Xu, Q.; Hao, Q.; Peng, S.; Zhang, H.; Ding, M.; Zhao, Q. Tectonic and high-latitude climate controls on Quaternary sedimentary processes on the northern coast of Bohai Bay. Palaeogeogr. Palaeoclimatol. Palaeoecol. 2024, 642, 112169. [Google Scholar] [CrossRef]
- Dal Bello, M.; Lee, H.; Goyal, A.; Gore, J. Resource-diversity relationships in bacterial communities reflect the network structure of microbial metabolism. Nat. Ecol. Evol. 2021, 5, 1424–1434. [Google Scholar] [CrossRef]
- Funk, J.L.; Vitousek, P.M. Resource-use efficiency and plant invasion in low-resource systems. Nature 2007, 446, 1079–1081. [Google Scholar] [CrossRef]
- Nicolás, C.; Martin-Bertelsen, T.; Floudas, D.; Bentzer, J.; Smits, M.; Johansson, T.; Troein, C.; Persson, P.; Tunlid, A. The soil organic matter decomposition mechanisms in ectomycorrhizal fungi are tuned for liberating soil organic nitrogen. ISME J. 2019, 13, 977–988. [Google Scholar] [CrossRef]
- Drew, G.C.; Stevens, E.J.; King, K.C. Microbial evolution and transitions along the parasite–mutualist continuum. Nat. Rev. Microbiol. 2021, 19, 623–638. [Google Scholar] [CrossRef]
- Crits-Christoph, A.; Olm, M.R.; Diamond, S.; Bouma-Gregson, K.; Banfield, J.F. Soil bacterial populations are shaped by recombination and gene-specific selection across a grassland meadow. ISME J. 2020, 14, 1834–1846. [Google Scholar] [CrossRef]
- Xue, P.P.; Carrillo, Y.; Pino, V.; Minasny, B.; McBratney, A.B. Soil Properties Drive Microbial Community Structure in a Large Scale Transect in South Eastern Australia. Sci. Rep. 2018, 8, 11725. [Google Scholar] [CrossRef]
- Zhao, S.; Liu, J.-J.; Banerjee, S.; Zhou, N.; Zhao, Z.-Y.; Zhang, K.; Tian, C.-Y. Soil pH is equally important as salinity in shaping bacterial communities in saline soils under halophytic vegetation. Sci. Rep. 2018, 8, 4550. [Google Scholar] [CrossRef]
- Ruan, B.; Wu, P.-x.; Lai, X.; Wang, H.; Li, L.; Chen, L.; Kang, C.; Zhu, N.; Dang, Z.; Lu, G. Effects of Sphingomonas sp. GY2B on the structure and physicochemical properties of stearic acid-modified montmorillonite in the biodegradation of phenanthrene. Appl. Clay Sci. 2018, 156, 36–44. [Google Scholar] [CrossRef]
- Cai, F.; Gao, R.; Zhao, Z.; Ding, M.; Jiang, S.; Yagtu, C.; Zhu, H.; Zhang, J.; Ebner, T.; Mayrhofer-Reinhartshuber, M.; et al. Evolutionary compromises in fungal fitness: Hydrophobins can hinder the adverse dispersal of conidiospores and challenge their survival. ISME J. 2020, 14, 2610–2624. [Google Scholar] [CrossRef]
- Calhim, S.; Halme, P.; Petersen, J.H.; Læssøe, T.; Bässler, C.; Heilmann-Clausen, J. Fungal spore diversity reflects substrate-specific deposition challenges. Sci. Rep. 2018, 8, 5356. [Google Scholar] [CrossRef]
- Yang, B.; Liang, Y.; Schmid, B.; Baruffol, M.; Li, Y.; He, L.-j.; Salmon, Y.; Tian, Q.; Niklaus, P.A.; Ma, K. Soil Fungi Promote Biodiversity–Productivity Relationships in Experimental Communities of Young Trees. Ecosystems 2021, 25, 858–871. [Google Scholar] [CrossRef]
- Balami, S.; Vašutová, M.; Košnar, J.; Karki, R.; Khadka, C.; Tripathi, G.; Cudlín, P. Soil fungal communities in abandoned agricultural land has not yet moved towards the seminatural forest. For. Ecol. Manag. 2021, 491, 119181. [Google Scholar] [CrossRef]
- Ham, L.; Coomer, M.A.; Öcal, K.; Grima, R.; Stumpf, M.P.H. A stochastic vs deterministic perspective on the timing of cellular events. Nat. Commun. 2024, 15, 5286. [Google Scholar] [CrossRef]
- Chen, W.; Gao, Y.; Yang, J.; Fan, F.; Zhang, W.; Li, J.; Zhou, C.; Shi, G.; Tong, F.; Fan, G. Taxonomical and functional bacterial community selection in the rhizosphere of the rice genotypes with different nitrogen use efficiencies. Plant Soil 2022, 470, 111–125. [Google Scholar] [CrossRef]
- Asaf, S.; Numan, M.; Khan, A.L.; Al-Harrasi, A. Sphingomonas: From diversity and genomics to functional role in environmental remediation and plant growth. Crit. Rev. Biotechnol. 2020, 40, 138–152. [Google Scholar] [CrossRef]
- White, D.C.; Sutton, S.D.; Ringelberg, D.B. The genus Sphingomonas: Physiology and ecology. Curr. Opin. Biotechnol. 1996, 7, 301–306. [Google Scholar] [CrossRef]
- Waigi, M.G.; Kang, F.; Goikavi, C.; Ling, W.; Gao, Y. Phenanthrene biodegradation by sphingomonads and its application in the contaminated soils and sediments: A review. Int. Biodeterior. Biodegrad. 2015, 104, 333–349. [Google Scholar] [CrossRef]
- Duan, H.; He, P.; Shao, L.; Lü, F. Functional genome-centric view of the CO-driven anaerobic microbiome. ISME J. 2021, 15, 2906–2919. [Google Scholar] [CrossRef]
- Leiva, A.M.; Pardo, J.M.; Arinaitwe, W.; Newby, J.; Vongphachanh, P.; Chittarath, K.; Oeurn, S.; Thi Hang, L.; Gil-Ordóñez, A.; Rodriguez, R.; et al. Ceratobasidium sp. is associated with cassava witches’ broom disease, a re-emerging threat to cassava cultivation in Southeast Asia. Sci. Rep. 2023, 13, 22500. [Google Scholar] [CrossRef]
- Mosquera-Espinosa, A.T.; Bayman, P.; Prado, G.A.; Gómez-Carabalí, A.; Otero, J.T. The double life of Ceratobasidium: Orchid mycorrhizal fungi and their potential for biocontrol of Rhizoctonia solani sheath blight of rice. Mycologia 2013, 105, 141–150. [Google Scholar] [CrossRef] [PubMed]
- Oberwinkler, F.; Riess, K.; Bauer, R.; Kirschner, R.; Garnica, S. Taxonomic re-evaluation of the Ceratobasidium-Rhizoctonia complex and Rhizoctonia butinii, a new species attacking spruce. Mycol. Prog. 2013, 12, 763–776. [Google Scholar] [CrossRef]
- Liang, Y.; Rillig, M.C.; Chen, H.Y.H.; Shan, R.; Ma, Z. Soil pH drives the relationship between the vertical distribution of soil microbial biomass and soil organic carbon across terrestrial ecosystems: A global synthesis. Catena 2024, 238, 107873. [Google Scholar] [CrossRef]
- Liu, T.; Wu, X.; Li, H.-Y.; Alharbi, H.A.; Wang, J.; Dang, P.; Chen, X.; Kuzyakov, Y.; Yan, W. Soil organic matter, nitrogen and pH driven change in bacterial community following forest conversion. For. Ecol. Manag. 2020, 477, 118473. [Google Scholar] [CrossRef]
- Queiroz, M.E.F.d.; Monteiro, J.S.; Viana-Júnior, A.B.; Praxedes, C.d.L.B.; Lavelle, P.; Vasconcelos, S.S. Litter thickness and soil pH influence the diversity of saprotrophic fungi in primary forest fragments in the Amazon. Pedobiologia 2021, 89, 150771. [Google Scholar] [CrossRef]
- Guo, X.; Li, H.; Yu, H.; Li, W.; Ye, Y.; Biswas, A. Drivers of spatio-temporal changes in paddy soil pH in Jiangxi Province, China from 1980 to 2010. Sci. Rep. 2018, 8, 2702. [Google Scholar] [CrossRef]
- Puissant, J.; Jones, B.A.; Goodall, T.; Mang, D.; Blaud, A.; Gweon, H.S.; Malik, A.A.; Jones, D.L.; Clark, I.M.; Hirsch, P.R.; et al. The pH optimum of soil exoenzymes adapt to long term changes in soil pH. Soil Biol. Biochem. 2019, 138, 107601. [Google Scholar] [CrossRef]
- Wu, Z.; Sun, X.; Sun, Y.; Yan, J.; Zhao, Y.; Chen, J. Soil acidification and factors controlling topsoil pH shift of cropland in central China from 2008 to 2018. Geoderma 2022, 408, 115586. [Google Scholar] [CrossRef]
- Chen, L.; Zhou, S.; Zhang, Q.; Zou, M.; Yin, Q.; Qiu, Y.; Qin, W. Effect of organic material addition on active soil organic carbon and microbial diversity: A meta-analysis. Soil Tillage Res. 2024, 241, 106128. [Google Scholar] [CrossRef]
- Lehmann, J.; Kleber, M. The contentious nature of soil organic matter. Nature 2015, 528, 60–68. [Google Scholar] [CrossRef]
- Yang, H.; Yao, B.; Lian, J.; Su, Y.; Li, Y. Tree species-dependent effects of afforestation on soil fungal diversity, functional guilds and co-occurrence networks in northern China. Environ. Res. 2024, 263, 120258. [Google Scholar] [CrossRef] [PubMed]
- Kuang, J.; Han, S.; Chen, Y.; Bates, C.T.; Wang, P.; Shu, W. Root-associated fungal community reflects host spatial co-occurrence patterns in a subtropical forest. ISME Commun. 2021, 1, 65. [Google Scholar] [CrossRef]
- Pastore, A.I.; Barabás, G.; Bimler, M.D.; Mayfield, M.M.; Miller, T.E. The evolution of niche overlap and competitive differences. Nat. Ecol. Evol. 2021, 5, 330–337. [Google Scholar] [CrossRef]
- Song, Z.; Yang, H.; Huang, X.; Yu, W.; Huang, J.; Ma, M. The spatiotemporal pattern and influencing factors of land surface temperature change in China from 2003 to 2019. Int. J. Appl. Earth Obs. Geoinf. 2021, 104, 102537. [Google Scholar] [CrossRef]
- Yang, Y.; Qiu, K.; Xie, Y.; Li, X.; Zhang, S.; Liu, W.; Huang, Y.; Cui, L.; Wang, S.; Bao, P. Geographical, climatic, and soil factors control the altitudinal pattern of rhizosphere microbial diversity and its driving effect on root zone soil multifunctionality in mountain ecosystems. Sci. Total Environ. 2023, 904, 166932. [Google Scholar] [CrossRef]
- Li, T.; Zhou, Q. The key role of Geobacter in regulating emissions and biogeochemical cycling of soil-derived greenhouse gases. Environ. Pollut. 2020, 266, 115135. [Google Scholar] [CrossRef]
- Chen, X.; Feng, J.; Ding, Z.; Tang, M.; Zhu, B. Changes in soil total, microbial and enzymatic C-N-P contents and stoichiometry with depth and latitude in forest ecosystems. Sci. Total Environ. 2022, 816, 151583. [Google Scholar] [CrossRef]
- Wu, J.; Wang, H.; Li, G.; Ma, W.; Wu, J.; Gong, Y.; Xu, G. Vegetation degradation impacts soil nutrients and enzyme activities in wet meadow on the Qinghai-Tibet Plateau. Sci. Rep. 2020, 10, 21271. [Google Scholar] [CrossRef]
- Georgiou, K.; Koven, C.D.; Wieder, W.R.; Hartman, M.D.; Riley, W.J.; Pett-Ridge, J.; Bouskill, N.J.; Abramoff, R.Z.; Slessarev, E.W.; Ahlström, A.; et al. Emergent temperature sensitivity of soil organic carbon driven by mineral associations. Nat. Geosci. 2024, 17, 205–212. [Google Scholar] [CrossRef]
- Datta, R. Enzymatic degradation of cellulose in soil: A review. Heliyon 2024, 10, e24022. [Google Scholar] [CrossRef]
- Husson, O. Redox potential (Eh) and pH as drivers of soil/plant/microorganism systems: A transdisciplinary overview pointing to integrative opportunities for agronomy. Plant Soil 2013, 362, 389–417. [Google Scholar] [CrossRef]
- Mattila, T.J. Redox potential as a soil health indicator—How does it compare to microbial activity and soil structure? Plant Soil 2024, 494, 617–625. [Google Scholar] [CrossRef]
- Chen, S.; Ai, X.; Dong, T.; Li, B.; Luo, R.; Ai, Y.; Chen, Z.; Li, C. The physico-chemical properties and structural characteristics of artificial soil for cut slope restoration in Southwestern China. Sci. Rep. 2016, 6, 20565. [Google Scholar] [CrossRef] [PubMed]
- Delgado, A.; Gómez, J.A. The Soil. Physical, Chemical and Biological Properties. In Principles of Agronomy for Sustainable Agriculture; Villalobos, F.J., Fereres, E., Eds.; Springer International Publishing: Cham, Switzerland, 2016; pp. 15–26. [Google Scholar]
- Gao, X.-s.; Xiao, Y.; Deng, L.-j.; Li, Q.-q.; Wang, C.-q.; Li, B.; Deng, O.-p.; Zeng, M. Spatial variability of soil total nitrogen, phosphorus and potassium in Renshou County of Sichuan Basin, China. J. Integr. Agric. 2019, 18, 279–289. [Google Scholar] [CrossRef]
- Beillouin, D.; Corbeels, M.; Demenois, J.; Berre, D.; Boyer, A.; Fallot, A.; Feder, F.; Cardinael, R. A global meta-analysis of soil organic carbon in the Anthropocene. Nat. Commun. 2023, 14, 3700. [Google Scholar] [CrossRef]
- Liu, Y.; Sun, Q.; Li, J.; Lian, B. Bacterial diversity among the fruit bodies of ectomycorrhizal and saprophytic fungi and their corresponding hyphosphere soils. Sci. Rep. 2018, 8, 11672. [Google Scholar] [CrossRef]
- Wang, N.Q.; Kong, C.H.; Wang, P.; Meiners, S.J. Root exudate signals in plant-plant interactions. Plant Cell Environ. 2021, 44, 1044–1058. [Google Scholar] [CrossRef]
- Zhang, H.; Wang, L.; Jin, X.; Bian, L.; Ge, Y. High-throughput phenotyping of plant leaf morphological, physiological, and biochemical traits on multiple scales using optical sensing. Crop J. 2023, 11, 1303–1318. [Google Scholar] [CrossRef]
- Rawat, M.; Arunachalam, K.; Arunachalam, A.; Alatalo, J.M.; Pandey, R. Assessment of leaf morphological, physiological, chemical and stoichiometry functional traits for understanding the functioning of Himalayan temperate forest ecosystem. Sci. Rep. 2021, 11, 23807. [Google Scholar] [CrossRef]
- Hodge, A.; Berta, G.; Doussan, C.; Merchan, F.; Crespi, M. Plant root growth, architecture and function. Plant Soil 2009, 321, 153–187. [Google Scholar] [CrossRef]
- Éva, C.; Oszvald, M.; Tamás, L. Current and possible approaches for improving photosynthetic efficiency. Plant Sci. 2019, 280, 433–440. [Google Scholar] [CrossRef]
- Hornyák, M.; Dziurka, M.; Kula-Maximenko, M.; Pastuszak, J.; Szczerba, A.; Szklarczyk, M.; Płażek, A. Photosynthetic efficiency, growth and secondary metabolism of common buckwheat (Fagopyrum esculentum Moench) in different controlled-environment production systems. Sci. Rep. 2022, 12, 257. [Google Scholar] [CrossRef]
- Upadhyay, S.K.; Srivastava, A.K.; Rajput, V.D.; Chauhan, P.K.; Bhojiya, A.A.; Jain, D.; Chaubey, G.; Dwivedi, P.; Sharma, B.; Minkina, T. Root Exudates: Mechanistic Insight of Plant Growth Promoting Rhizobacteria for Sustainable Crop Production. Front. Microbiol. 2022, 13, 916488. [Google Scholar] [CrossRef]
- Sheng, M.; Hu, W.; Liu, C.-Q.; Niu, M.; Jin, R.; Deng, J.; Wu, L.; Li, P.; Yan, Z.; Zhu, Y.-G.; et al. Characteristics and assembly mechanisms of bacterial and fungal communities in soils from Chinese forests across different climatic zones. Catena 2024, 245, 108306. [Google Scholar] [CrossRef]
- Pérez-Izquierdo, L.; Rincón, A.; Lindahl, B.D.; Buée, M. Chapter 13—Fungal community of forest soil: Diversity, functions, and services. In Forest Microbiology; Asiegbu, F.O., Kovalchuk, A., Eds.; Academic Press: Cambridge, MA, USA, 2021; pp. 231–255. [Google Scholar]
- Liu, C.; Li, Y.; Xu, L.; Chen, Z.; He, N. Variation in leaf morphological, stomatal, and anatomical traits and their relationships in temperate and subtropical forests. Sci. Rep. 2019, 9, 5803. [Google Scholar] [CrossRef]
- Li, Y.; He, N.; Hou, J.; Xu, L.; Liu, C.; Zhang, J.; Wang, Q.-F.; Zhang, X.; Wu, X. Factors Influencing Leaf Chlorophyll Content in Natural Forests at the Biome Scale. Front. Ecol. Evol. 2018, 6, 64. [Google Scholar] [CrossRef]
- Lei, J.; Duan, A.; Guo, W.; Zhang, J. Effects of tree species mixing and soil depth on the soil bacterial and fungal communities in Chinese fir (Cunninghamia lanceolata) plantations. Appl. Soil Ecol. 2024, 195, 105270. [Google Scholar] [CrossRef]
- Sritongon, N.; Sarin, P.; Theerakulpisut, P.; Riddech, N. The effect of salinity on soil chemical characteristics, enzyme activity and bacterial community composition in rice rhizospheres in Northeastern Thailand. Sci. Rep. 2022, 12, 20360. [Google Scholar] [CrossRef]
- Luo, X.; Keenan, T.F.; Chen, J.M.; Croft, H.; Colin Prentice, I.; Smith, N.G.; Walker, A.P.; Wang, H.; Wang, R.; Xu, C.; et al. Global variation in the fraction of leaf nitrogen allocated to photosynthesis. Nat. Commun. 2021, 12, 4866. [Google Scholar] [CrossRef]
- Witzgall, K.; Vidal, A.; Schubert, D.I.; Höschen, C.; Schweizer, S.A.; Buegger, F.; Pouteau, V.; Chenu, C.; Mueller, C.W. Particulate organic matter as a functional soil component for persistent soil organic carbon. Nat. Commun. 2021, 12, 4115. [Google Scholar] [CrossRef]
- Fabian, J.; Zlatanovic, S.; Mutz, M.; Premke, K. Fungal–bacterial dynamics and their contribution to terrigenous carbon turnover in relation to organic matter quality. ISME J. 2017, 11, 415–425. [Google Scholar] [CrossRef] [PubMed]
- Wilson, D.N. Ribosome-targeting antibiotics and mechanisms of bacterial resistance. Nat. Rev. Microbiol. 2014, 12, 35–48. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Site | SOC (g kg−1) | pH | TN (mg kg−1) | TP (mg kg−1) | TK (g kg−1) | EC (mS m−1) | CEC (cmol+ kg−1) |
|---|---|---|---|---|---|---|---|
| AC | 55.1 ± 0.57 c | 6.44 ± 0.05 b | 0.0059 ± 0.00006 b | 795.0 ± 66.45 d | 17.0 ± 0.211 ab | 159.3 ± 5.63 b | 31.7 ± 0.52 b |
| FZ | 29.1 ± 0.57 e | 5.53 ± 0.09 d | 0.0028 ± 0.00010 e | 723.5 ± 50.70 e | 16.8 ± 0.395 b | 95.9 ± 5.11 c | 18.8 ± 0.29 f |
| MS | 60.3 ± 0.73 b | 5.79 ± 0.10 c | 0.0052 ± 0.00015 c | 1060.4 ± 67.04 b | 17.1 ± 0.357 ab | 89.1 ± 3.00 d | 33.1 ± 0.55 a |
| MIS | 19.3 ± 0.50 f | 6.94 ± 0.11 a | 0.0020 ± 0.00003 f | 508.4 ± 26.24 f | 16.9 ± 0.543 ab | 70.4 ± 4.82 e | 30.7 ± 0.42 c |
| QA | 45.9 ± 0.56 d | 5.07 ± 0.08 f | 0.0039 ± 0.00015 d | 932.2 ± 26.21 c | 17.3 ± 0.293 a | 69.1 ± 6.04 e | 24.8 ± 0.26 e |
| YC | 76.5 ± 0.75 a | 5.31 ± 0.05 e | 0.0061 ± 0.00008 a | 1156.0 ± 53.58 a | 16.9 ± 0.391 ab | 175.5 ± 5.14 a | 26.46 ± 0.52 d |
| Microbial Community | Sample | Shannon Index | Simpson Index | Richness Index | Chao1 Index |
|---|---|---|---|---|---|
| Bacteria | AC | 7.146 ± 0.2045 a | 0.9953 ± 0.0025 a | 6568 ± 385.9283 ab | 8274.0958 ± 387.4743 a |
| FZ | 6.989 ± 0.1787 a | 0.9961 ± 0.0016 a | 6633 ± 633.0136 a | 8455.0776 ± 591.7943 a | |
| MS | 6.6361 ± 0.2716 a | 0.9826 ± 0.0092 a | 6102 ± 393.0759 ab | 8089.7406 ± 436.9896 a | |
| MIS | 6.8555 ± 0.203 a | 0.9923 ± 0.0058 a | 5929 ± 411.8709 ab | 7511.4929 ± 440.4228 a | |
| QA | 6.478 ± 0.0821 a | 0.9892 ± 0.0016 a | 6008.6 ± 293.6454 ab | 7871.2933 ± 245.61 a | |
| YC | 6.5037 ± 1.2603 a | 0.9753 ± 0.0608 a | 5505.8 ± 1798.4429 b | 7169.7982 ± 2385.0721 a | |
| Fungi | AC | 4.3871 ± 1.2121 a | 0.8774 ± 0.2139 a | 2156.3 ± 193.8889 bc | 2195.0035 ± 233.3214 e |
| FZ | 4.889 ± 0.7767 a | 0.9463 ± 0.055 a | 2320.8 ± 194.5975 ab | 2800.6151 ± 205.7821 c | |
| MS | 4.8615 ± 0.6511 a | 0.9388 ± 0.0779 a | 2433.3 ± 95.7695 a | 2931.2043 ± 201.4482 c | |
| MIS | 4.8285 ± 0.4692 a | 0.9564 ± 0.0338 a | 1901.5 ± 125.2945 de | 2575.2015 ± 155.2967 d | |
| QA | 4.3839 ± 0.1917 a | 0.9571 ± 0.0125 a | 1842.7 ± 95.2378 e | 2541.2489 ± 163.4956 d | |
| YC | 4.5497 ± 0.4523 a | 0.9392 ± 0.033 a | 2025.9 ± 259.6486 cd | 2329.837 ± 126.5806 e |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, M.; Xing, X.; Zhang, Y.; Sui, X.; Zheng, C. Geographic Distribution Pattern Determines Soil Microbial Community Assembly Process in Acanthopanax senticosus Rhizosphere Soil. Microorganisms 2024, 12, 2506. https://doi.org/10.3390/microorganisms12122506
Wang M, Xing X, Zhang Y, Sui X, Zheng C. Geographic Distribution Pattern Determines Soil Microbial Community Assembly Process in Acanthopanax senticosus Rhizosphere Soil. Microorganisms. 2024; 12(12):2506. https://doi.org/10.3390/microorganisms12122506
Chicago/Turabian StyleWang, Mingyu, Xiangyu Xing, Youjia Zhang, Xin Sui, and Chunying Zheng. 2024. "Geographic Distribution Pattern Determines Soil Microbial Community Assembly Process in Acanthopanax senticosus Rhizosphere Soil" Microorganisms 12, no. 12: 2506. https://doi.org/10.3390/microorganisms12122506
APA StyleWang, M., Xing, X., Zhang, Y., Sui, X., & Zheng, C. (2024). Geographic Distribution Pattern Determines Soil Microbial Community Assembly Process in Acanthopanax senticosus Rhizosphere Soil. Microorganisms, 12(12), 2506. https://doi.org/10.3390/microorganisms12122506