
Whole-Genome Sequencing-Based Characterization of Clostridioides difficile Infection Cases at the University Hospital Centre Zagreb
 , , , ,
, , , ,  and
and
Abstract
1. Introduction
2. Materials and Methods
3. Results
3.1. Patient Characteristics
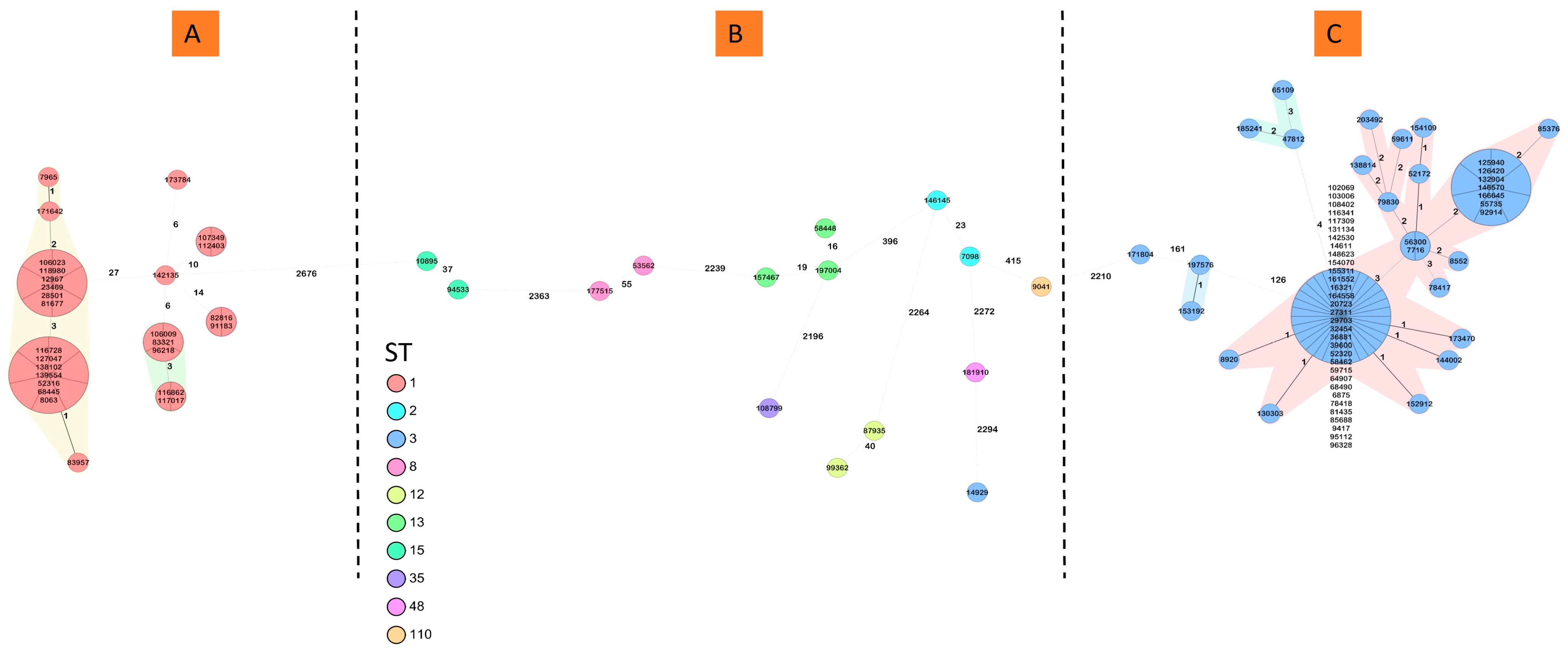
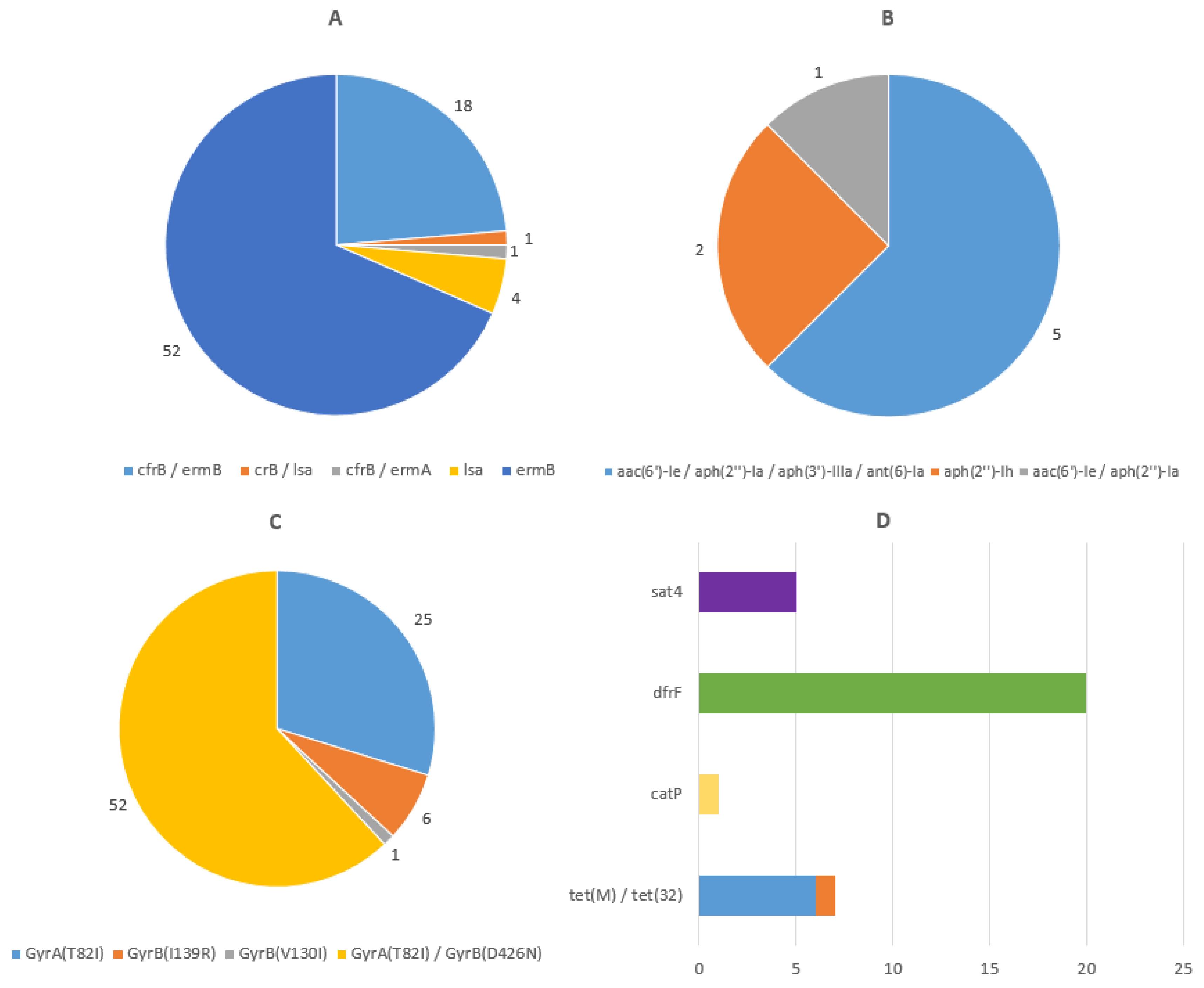
3.2. Characterization of C. difficile Isolates by WGS
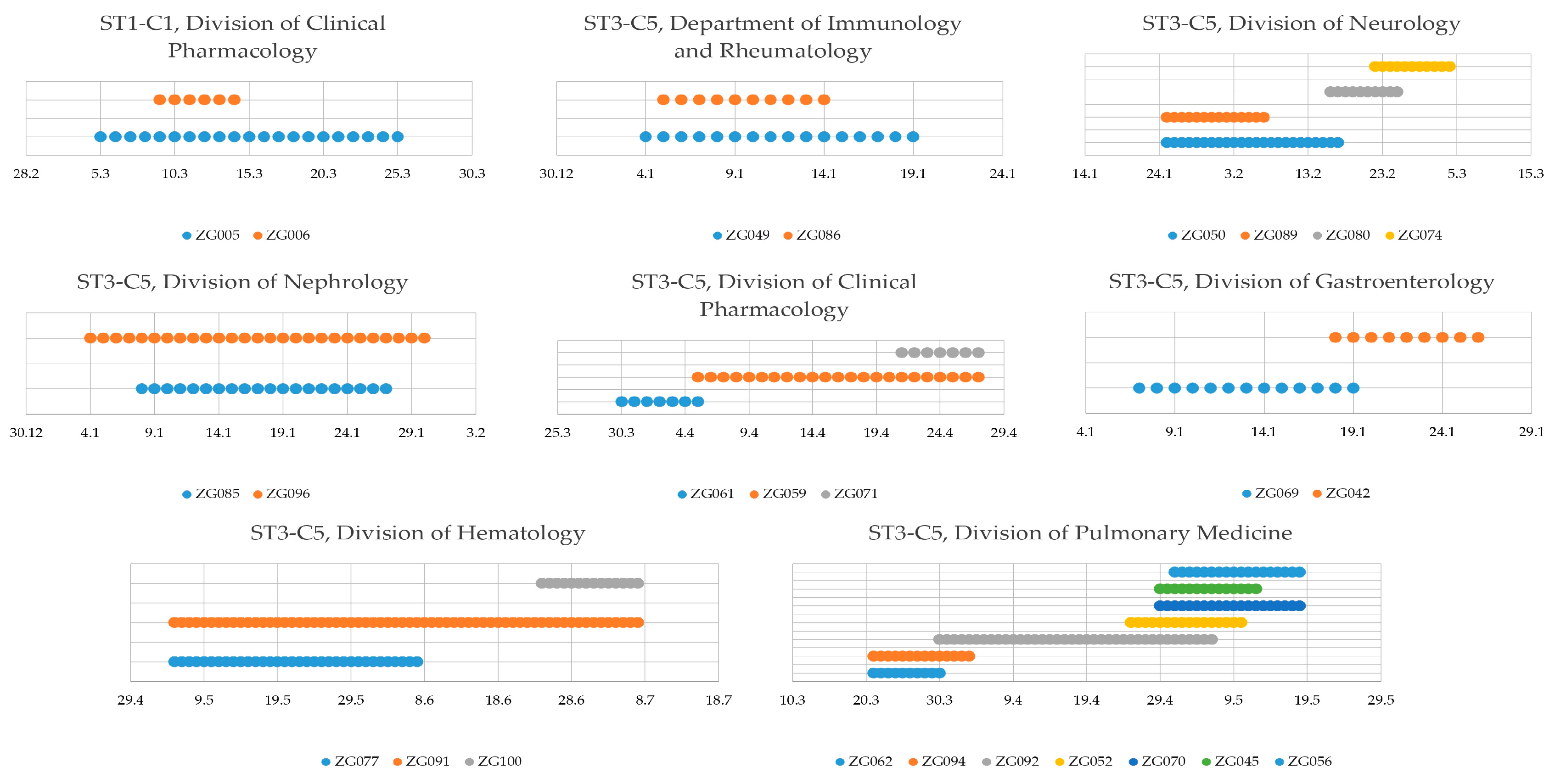
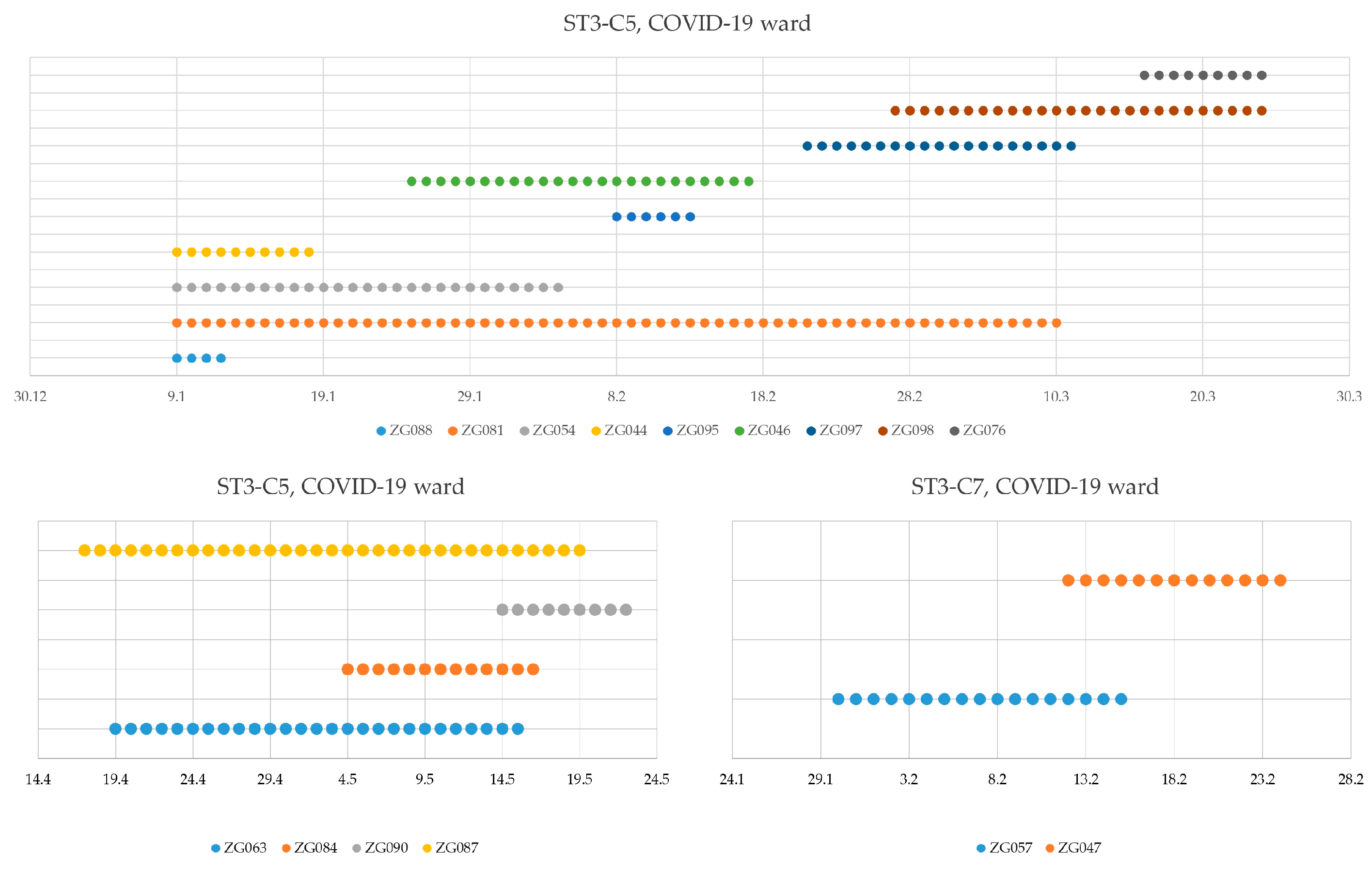
3.3. Nosocomial Transmission
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Lawson, P.A.; Citron, D.M.; Tyrrell, K.L.; Finegold, S.M. Reclassification of Clostridium difficile as Clostridioides difficile (Hall and O’Toole 1935) Prévot 1938. Anaerobe 2016, 40, 95–99. [Google Scholar] [CrossRef] [PubMed]
- Finn, E.; Andersson, F.L.; Madin-Warburton, M. Burden of Clostridioides difficile infection (CDI)—A systematic review of the epidemiology of primary and recurrent CDI. BMC Infect. Dis. 2021, 21, 456. [Google Scholar] [CrossRef]
- Son, K.J.; Kim, Y.A.; Park, Y.S. Economic burden attributable to Clostridioides difficile infections in South Korea: A nationwide propensity score-matched study. J. Hosp. Infect. 2022, 120, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Wingen-Heimann, S.M.; Davies, K.; Viprey, V.F.; Davis, G.; Wilcox, M.H.; Vehreschild, M.J.G.T.; Lurienne, L.; Bandinelli, P.-A.; Cornely, O.A.; Vilken, T.; et al. COMBACTE-CDI consortium. Clostridioides difficile infection (CDI): A pan-European multicenter cost and resource utilization study, results from the Combatting Bacterial Resistance in Europe CDI (COMBACTE-CDI). Clin. Microbiol. Infect. 2023, 29, 651.e1–651.e8. [Google Scholar] [CrossRef] [PubMed]
- Braae, U.C.; Møller, F.T.; Ibsen, R.; Ethelberg, S.; Kjellberg, J.; Mølbak, K. The Economic Burden of Clostridioides difficile in Denmark: A Retrospective Cohort Study. Front. Public Health 2020, 8, 562957. [Google Scholar] [CrossRef]
- Pereira, J.A.; McGeer, A.; Tomovici, A.; Selmani, A.; Chit, A. Incidence and economic burden of Clostridioides difficile infection in Ontario: A retrospective population-based study. CMAJ Open 2020, 8, E16–E25. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Luo, Y.; Grinspan, A.M. Epidemiology of community-acquired and recurrent Clostridioides difficile infection. Ther. Adv. Gastroenterol. 2021, 14, 17562848211016248. [Google Scholar] [CrossRef]
- Viprey, V.F.; Davis, G.L.; Benson, A.D.; Ewin, D.; Spittal, W.; Vernon, J.J.; Rupnik, M.; Banz, A.; Allantaz, F.; Cleuziat, P.; et al. COMBACTE-CDI National Coordinators; Wilcox MH, Davies KA.; COMBACTE-CDI consortium; Members of the COMBACTE-CDI National coordinators. A point-prevalence study on community and inpatient Clostridioides difficile infections (CDI): Results from Combatting Bacterial Resistance in Europe CDI (COMBACTE-CDI), July to November 2018. Euro Surveill. 2022, 27, 2100704. [Google Scholar]
- Buddle, J.E.; Fagan, R.P. Pathogenicity and virulence of Clostridioides difficile. Virulence 2023, 14, 2150452. [Google Scholar] [CrossRef]
- Zhao, H.; Nickle, D.C.; Zeng, Z.; Law, P.Y.T.; Wilcox, M.H.; Chen, L.; Peng, Y.; Meng, J.; Deng, Z.; Albright, A.; et al. Global Landscape of Clostridioides Difficile Phylogeography, Antibiotic Susceptibility, and Toxin Polymorphisms by Post-Hoc Whole-Genome Sequencing from the MODIFY I/II Studies. Infect. Dis. Ther. 2021, 10, 853–870. [Google Scholar] [CrossRef]
- Sood, G.; Truelove, S.; Dougherty, G.; Landrum, B.M.; Qasba, S.; Patel, M.; Miller, A.; Wilson, C.; Martin, J.; Sears, C.; et al. Clostridioides difficile infection (CDI) in a previous room occupant predicts CDI in subsequent room occupants across different hospital settings. Am. J. Infect. Control. 2022, 50, 1352–1354. [Google Scholar] [CrossRef] [PubMed]
- Spigaglia, P. Recent advances in the understanding of antibiotic resistance in Clostridium difficile infection. Ther. Adv. Infect. Dis. 2016, 3, 23–42. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Xie, X.; Ge, Q.; He, X.; Sun, Z.; Li, Y.; Guo, Y.; Geng, C.; Li, X.; Wang, C. The global burden and trend of Clostridioides difficile and its association with world antibiotic consumption, 1990–2019. J. Glob. Health 2024, 14, 04135. [Google Scholar] [CrossRef] [PubMed]
- Balsells, E.; Shi, T.; Leese, C.; Lyell, I.; Burrows, J.; Wiuff, C.; Campbell, H.; Kyaw, M.H.; Nair, H. Global burden of Clostridium difficile infections: A systematic review and meta-analysis. J. Glob. Health 2019, 9, 010407. [Google Scholar] [CrossRef]
- European Centre for Disease Prevention and Control. Clostridioides (Clostridium) Difficile Infections. Annual Epidemiological Report for 2016–2017; ECDC: Stockholm, Sweden, 2022; Available online: https://www.ecdc.europa.eu/en/publications-data/clostridiodes-difficile-infections-annual-epidemiological-report-2016-2017 (accessed on 4 September 2024).
- European Centre for Disease Prevention and Control. Clostridioides Difficile Infections. Annual Epidemiological Report for 2018–2020; ECDC: Stockholm, Sweden, 2024; Available online: https://www.ecdc.europa.eu/en/publications-data/clostridioides-difficile-infections-annual-epidemiological-report-2018-2020 (accessed on 6 October 2024).
- European Centre for Disease Prevention and Control. Study Protocol for a Survey of Whole Genome Sequencing of Clostridioides Difficile Isolates from Tertiary Acute Care Hospitals, EU/EEA, 2022–2023; ECDC: Stockholm, Sweden, 2024; Available online: https://www.ecdc.europa.eu/en/publications-data/survey-protocol-whole-genome-sequencing-clostridioides-difficile-isolates (accessed on 16 November 2024).
- Martínez-Meléndez, A.; Morfin-Otero, R.; Villarreal-Treviño, L.; Baines, S.D.; Camacho-Ortíz, A.; Garza-González, E. Molecular epidemiology of predominant and emerging Clostridioides difficile ribotypes. J. Microbiol. Methods 2020, 175, 105974. [Google Scholar] [CrossRef]
- Abad-Fau, A.; Sevilla, E.; Martín-Burriel, I.; Moreno, B.; Bolea, R. Update on Commonly Used Molecular Typing Methods for Clostridioides difficile. Microorganisms 2023, 11, 1752. [Google Scholar] [CrossRef]
- Budimir, A.; Mareković, I.; Mijač, M.; Bosnjak, Z.; Payerl-Pal, M.; Susic, E.; Matas, I.; Novak, A.; Harmanus, C.; Kuijper, E. Epidemiology of Clostridium difficile Infections in Croatia-National Study. CROSBI. 2018. Available online: https://www.bib.irb.hr:8443/1238985 (accessed on 16 November 2024).
- Paradžik, M. The First Evidence of Epidemic Strain Clostridium Difficile (027/NAP1/BI) in Eastern Croatia. J. Clin. Microbiol. Biochem. Technol. 2017, 3, 14–16. [Google Scholar] [CrossRef]
- Novak, A.; Spigaglia, P.; Barbanti, F.; Goic-Barisic, I.; Tonkic, M. First clinical and microbiological characterization of Clostridium difficile infection in a Croatian University Hospital. Anaerobe 2014, 30, 18–23. [Google Scholar] [CrossRef]
- Rupnik, M.; Andrasevic, A.T.; Dokic, E.T.; Matas, I.; Jovanovic, M.; Pasic, S.; Kocuvan, A.; Janezic, S. Distribution of Clostridium difficile PCR ribotypes and high proportion of 027 and 176 in some hospitals in four South Eastern European countries. Anaerobe 2016, 42, 142–144. [Google Scholar] [CrossRef]
- Baktash, A.; Corver, J.; Harmanus, C.; Smits, W.K.; Fawley, W.; Wilcox, M.H.; Kumar, N.; Eyre, D.W.; Indra, A.; Mellmann, A.; et al. Comparison of Whole-Genome Sequence-Based Methods and PCR Ribotyping for Subtyping of Clostridioides difficile. J. Clin. Microbiol. 2022, 60, e0173721. [Google Scholar] [CrossRef]
- Zankari, E.; Allesøe, R.; Joensen, K.G.; Cavaco, L.M.; Lund, O.; Aarestrup, F.M. PointFinder: A novel web tool for WGS-based detection of antimicrobial resistance associated with chromosomal point mutations in bacterial pathogens. J. Antimicrob. Chemother. 2020, 72, 2764–2768. [Google Scholar] [CrossRef] [PubMed]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef] [PubMed]
- Alcock, B.P.; Raphenya, A.R.; Lau, T.T.Y.; Tsang, K.K.; Bouchard, M.; Edalatmand, A.; Huynh, W.; Nguyen, A.-L.V.; Cheng, A.A.; Liu, S.; et al. CARD 2020: Antibiotic resistome surveillance with the comprehensive antibiotic resistance database. Nucleic Acids Res. 2020, 48, D517–D525. [Google Scholar] [CrossRef]
- Wen, X.; Shen, C.; Xia, J.; Zhong, L.-L.; Wu, Z.; Ahmed, M.A.E.-G.E.-S.; Long, N.; Ma, F.; Zhang, G.; Wu, W.; et al. Whole-Genome Sequencing Reveals the High Nosocomial Transmission and Antimicrobial Resistance of Clostridioides difficile in a Single Center in China, a Four-Year Retrospective Study. Microbiol. Spectr. 2022, 10, e0132221. [Google Scholar] [CrossRef] [PubMed]
- Reigadas, E.; Vázquez-Cuesta, S.; Bouza, E. Economic Burden of Clostridioides difficile Infection in European Countries. Adv. Exp. Med. Biol. 2024, 1435, 1–12. [Google Scholar]
- Rupnik, M.; Viprey, V.; Janezic, S.; Tkalec, V.; Davis, G.; Sente, B.; Devos, N.; Muller, B.H.; Santiago-Allexant, E.; Cleuziat, P.; et al. Distribution of Clostridioides difficile ribotypes and sequence types across humans, animals and food in thirteen European countries. Emerg. Microbes Infect. 2024, 13, 2427804. [Google Scholar] [CrossRef]
- Azimirad, M.; Noori, M.; Raeisi, H.; Yadegar, A.; Shahrokh, S.; Asadzadeh Aghdaei, H.; Bentivegna, E.; Martelletti, P.; Petrosillo, N.; Zali, M.R. How Does COVID-19 Pandemic Impact on Incidence of Clostridioides difficile Infection and Exacerbation of Its Gastrointestinal Symptoms? Front. Med. 2021, 8, 775063. [Google Scholar] [CrossRef]
- Leffler, D.A.; Lamont, J.T. Clostridium difficile infection. N. Engl. J. Med. 2015, 373, 287–288. [Google Scholar] [CrossRef]
- Enoch, D.A.; Butler, M.J.; Pai, S.; Aliyu, S.H.; Karas, J.A. Clostridium difficile in children: Colonisation and disease. J. Infect. 2011, 63, 105–113. [Google Scholar] [CrossRef]
- Freeman, J.; Bauer, M.P.; Baines, S.D.; Corver, J.; Fawley, W.N.; Goorhuis, B.; Kuijper, E.J.; Wilcox, M.H. The changing epidemiology of Clostridium difficile infections. Clin. Microbiol. Rev. 2010, 23, 529–549. [Google Scholar] [CrossRef]
- Liu, X.S.; Li, W.G.; Zhang, W.Z.; Wu, Y.; Lu, J.X. Molecular Characterization of Clostridium difficile Isolates in China From 2010 to 2015. Front. Microbiol. 2018, 9, 845. [Google Scholar] [CrossRef] [PubMed]
- Dingle, K.E.; Griffiths, D.; Didelot, X.; Evans, J.; Vaughan, A.; Kachrimanidou, M.; Stoesser, N.; Jolley, K.A.; Golubchik, T.; Harding, R.M.; et al. Clinical Clostridium difficile: Clonality and pathogenicity locus diversity. PLoS ONE 2011, 6, e19993. [Google Scholar] [CrossRef] [PubMed]
- Lanis, J.M.; Heinlen, L.D.; James, J.A.; Ballard, J.D. Clostridium difficile 027/BI/NAP1 encodes a hypertoxic and antigenically variable form of TcdB. PLoS Pathog. 2013, 9, e1003523. [Google Scholar] [CrossRef] [PubMed]
- Shen, E.; Zhu, K.; Li, D.; Pan, Z.; Luo, Y.; Bian, Q.; He, L.; Song, X.; Zhen, Y.; Jin, D.; et al. Subtyping analysis reveals new variants accelerated evolution of Clostridioides difficile toxin, B. Commun. Biol. 2020, 3, 347. [Google Scholar] [CrossRef] [PubMed]
- Zaiss, N.H.; Witte, W.; Nübel, U. Fluoroquinolone resistance and Clostridium difficile, Germany. Emerg. Infect. Dis. 2010, 16, 675–677. [Google Scholar] [CrossRef]
- Li, H.; Li, W.-G.; Zhang, W.-Z.; Yu, S.-B.; Liu, Z.-J.; Zhang, X.; Wu, Y.; Lu, J.-X. Antibiotic resistance of clinical isolates of Clostridioides difficile in China and its association with geographical regions and patient age. Anaerobe 2019, 60, 102094. [Google Scholar] [CrossRef]
- Bishop, E.J.; Tiruvoipati, R. Management of Clostridioides difficile Infection in Adults and Challenges in Clinical Practice: Review and Comparison of Current IDSA/SHEA, ESCMID and ASID Guidelines. J. Antimicrob. Chemother. 2022, 78, 21–30. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Total CD Positive | TCD Positive | |
|---|---|---|
| Number of cases, N (%) | 103 | 100 (97.1%) |
| Age, years Mean ± SD Range Median [quartiles] | 68.76 ± 21.03 0–93 73 [64.5, 83] | 70.02 ± 18.99 0–93 73 [65.5, 83] |
| Sex, N (%) Female Male Wards, N (%) | 52 51 | 50 (96.1%) 50 (98.0%) |
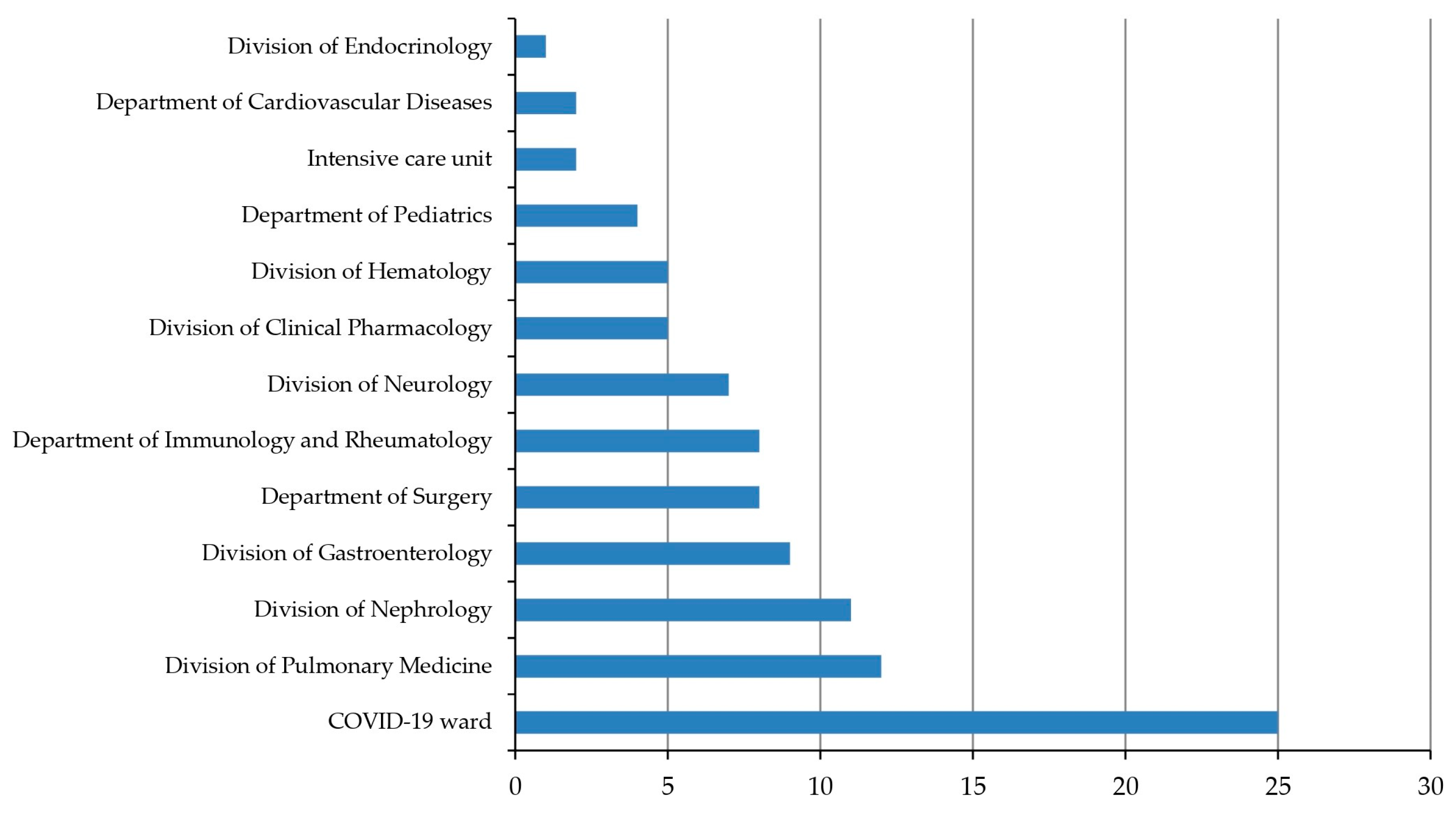
| COVID-19 ward | 25 | 25 (100.0%) |
| Division of Pulmonary Medicine | 12 | 12 (100.0%) |
| Division of Nephrology | 11 | 11 (100.0%) |
| Division of Gastroenterology | 9 | 9 (100.0%) |
| Department of Surgery | 8 | 8 (100.0%) |
| Immunology and Rheumatology | 8 | 7 (87.5%) |
| Division of Neurology | 7 | 7 (100.0%) |
| Division of Clinical Pharmacology | 5 | 5 (100.0%) |
| Division of Hematology | 5 | 5 (100.0%) |
| Department of Pediatrics | 4 | 2 (50.0%) |
| Other wards | 9 | 9 (100.0%) |
| Recent previous hospitalizations | 94 | 91 (96.8%) |
| Recent antibiotic use | 81 | 80 (98.8%) |
| Presence of comorbidities, N(%) | ||
| Psychiatric disorders | 17 | 17 (100.0%) |
| Gastroesophageal reflux disease | 5 | 5 (100.0%) |
| Chronic obstructive pulmonary disease | 12 | 12 (100.0%) |
| Cardiovascular disease | 31 | 30 (96.8%) |
| Cerebrovascular disease | 12 | 12 (100.0%) |
| Diabetes | 21 | 21 (100.0%) |
| Solid tumor | 19 | 19 (100.0%) |
| Renal impairment or failure | 20 | 20 (100.0%) |
| Ulcerative colitis | 1 | 1 (100.0%) |
| Immunocompromised status | 34 | 33 (97.1%) |
| Charlson comorbidity index (≥5) | 58 | 57 (98.3%) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Siroglavic, M.; Higgins, P.G.; Kanizaj, L.; Ferencak, I.; Juric, D.; Augustin, G.; Budimir, A. Whole-Genome Sequencing-Based Characterization of Clostridioides difficile Infection Cases at the University Hospital Centre Zagreb. Microorganisms 2024, 12, 2434. https://doi.org/10.3390/microorganisms12122434
Siroglavic M, Higgins PG, Kanizaj L, Ferencak I, Juric D, Augustin G, Budimir A. Whole-Genome Sequencing-Based Characterization of Clostridioides difficile Infection Cases at the University Hospital Centre Zagreb. Microorganisms. 2024; 12(12):2434. https://doi.org/10.3390/microorganisms12122434
Chicago/Turabian StyleSiroglavic, Marko, Paul G. Higgins, Lucija Kanizaj, Ivana Ferencak, Dragan Juric, Goran Augustin, and Ana Budimir. 2024. "Whole-Genome Sequencing-Based Characterization of Clostridioides difficile Infection Cases at the University Hospital Centre Zagreb" Microorganisms 12, no. 12: 2434. https://doi.org/10.3390/microorganisms12122434
APA StyleSiroglavic, M., Higgins, P. G., Kanizaj, L., Ferencak, I., Juric, D., Augustin, G., & Budimir, A. (2024). Whole-Genome Sequencing-Based Characterization of Clostridioides difficile Infection Cases at the University Hospital Centre Zagreb. Microorganisms, 12(12), 2434. https://doi.org/10.3390/microorganisms12122434