
Whole-Genome Sequencing of Peribacillus frigoritolerans Strain d21.2 Isolated in the Republic of Dagestan, Russia

 , , ,
, , ,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Isolation of the Strain from a Soil Sample
2.2. Morphological Description of the Strain
2.3. DNA Extraction, Quality Control, and Whole-Genome Sequencing
2.4. Whole-Genome Sequencing, De Novo Genome Assembly and Annotation
2.5. Scaffolding Genome Assembly and Detecting MGEs
2.6. Comparative Genome Analysis Procedure
3. Results
3.1. Isolation and Characterization of the Strain According to Morphology
3.2. Analysis of Whole-Genome Sequencing (WGS) Results
3.3. Connections Between MGEs and Functional Loci in Raw and Refined Genome Assembly
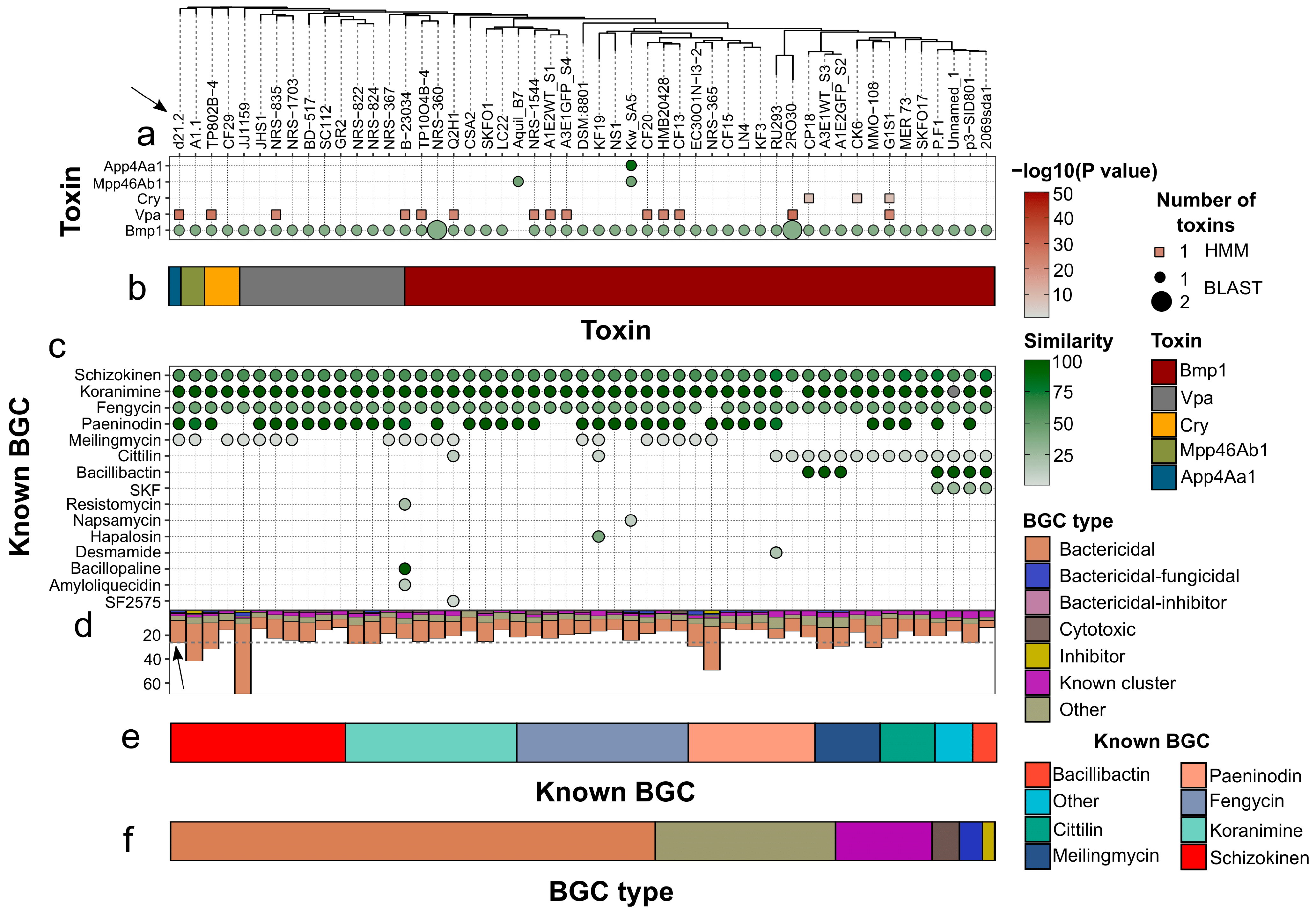
3.4. Comparative Genomic Analysis with P. frigoritolerans Strains
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Montecillo, J.A.V.; Bae, H. Reclassification of Brevibacterium frigoritolerans as Peribacillus frigoritolerans comb. nov. Based on Phylogenomics and Multiple Molecular Synapomorphies. Int. J. Syst. Evol. Microbiol. 2022, 72, 005389. [Google Scholar] [CrossRef]
- Świątczak, J.; Kalwasińska, A.; Brzezinska, M.S. Plant Growth–Promoting Rhizobacteria: Peribacillus frigoritolerans 2RO30 and Pseudomonas sivasensis 2RO45 for Their Effect on Canola Growth under Controlled as Well as Natural Conditions. Front. Plant Sci. 2024, 14, 1233237. [Google Scholar] [CrossRef]
- Selvakumar, G.; Sushil, S.N.; Stanley, J.; Mohan, M.; Deol, A.; Rai, D.; Ramkewal; Bhatt, J.C.; Gupta, H.S. Brevibacterium frigoritolerans a Novel Entomopathogen of Anomala dimidiata and Holotrichia longipennis (Scarabaeidae: Coleoptera). Biocontrol Sci. Technol. 2011, 21, 821–827. [Google Scholar] [CrossRef]
- Marik, D.; Sharma, P.; Chauhan, N.S.; Jangir, N.; Shekhawat, R.S.; Verma, D.; Mukherjee, M.; Abiala, M.; Roy, C.; Yadav, P.; et al. Peribacillus frigoritolerans T7-IITJ, a Potential Biofertilizer, Induces Plant Growth-Promoting Genes of Arabidopsis thaliana. J. Appl. Microbiol. 2024, 135, lxae066. [Google Scholar] [CrossRef]
- Chacón-López, A.; Guardado-Valdivia, L.; Bañuelos-González, M.; López-García, U.; Montalvo-González, E.; Arvizu-Gómez, J.; Stoll, A.; Aguilera, S. Effect of Metabolites Produced by Bacillus atrophaeus and Brevibacterium frigoritolerans Strains on Postharvest Biocontrol of Alternaria alternata in Tomato (Solanum lycopersicum L.). Biocontrol Sci. 2021, 26, 67–74. [Google Scholar] [CrossRef]
- Jariyal, M.; Gupta, V.K.; Mandal, K.; Jindal, V. Brevibacterium frigoritolerans as a Novel Organism for the Bioremediation of Phorate. Bull. Environ. Contam. Toxicol. 2015, 95, 680–686. [Google Scholar] [CrossRef]
- Delaporte, B.; Sasson, A. [Study of Bacteria from Arid Soils of Morocco: Brevibacterium haloterans n. sp. and Brevibacterium frigoritolerans n. sp]. C. R. Acad. Hebd. Seances Acad. Sci. D 1967, 264, 2257–2260. [Google Scholar]
- Choe, Y.-H.; Lee, J.I.; Kim, M. Complete Genome Sequence of Brevibacterium frigoritolerans Ant232, Isolated from Antarctic Snow. Microbiol. Resour. Announc. 2022, 11, e0015222. [Google Scholar] [CrossRef]
- Choi, E.J.; Lee, S.H.; Jung, J.Y.; Jeon, C.O. Brevibacterium jeotgali sp. nov., Isolated from Jeotgal, a Traditional Korean Fermented Seafood. Int. J. Syst. Evol. Microbiol. 2013, 63, 3430–3436. [Google Scholar] [CrossRef]
- Wufuer, R.; Li, W.; Wang, S.; Duo, J. Isolation and Degradation Characteristics of PBAT Film Degrading Bacteria. Int. J. Environ. Res. Public Health 2022, 19, 17087. [Google Scholar] [CrossRef]
- McLoon, A.L.; Awad, T.T.; Bogardus, M.F.; Buono, M.G.; Devine, K.A.; Draper, R.M.; Femenella, B.; Gallagher, H.M.; Morelock, L.A.; Razi, M.; et al. Draft Genome Sequences for 6 Isolates of Endospore-Forming Class Bacilli Species Isolated from Soil from a Suburban, Wooded, Developed Space. Microbiol. Resour. Announc. 2022, 11, e0087422. [Google Scholar] [CrossRef]
- Janakiev, T.; Milošević, Đ.; Petrović, M.; Miljković, J.; Stanković, N.; Zdravković, D.S.; Dimkić, I. Chironomus riparius Larval Gut Bacteriobiota and Its Potential in Microplastic Degradation. Microb. Ecol. 2023, 86, 1909–1922. [Google Scholar] [CrossRef]
- Lu, P.; Jiang, K.; Hao, Y.-Q.; Chu, W.-Y.; Xu, Y.-D.; Yang, J.-Y.; Chen, J.-L.; Zeng, G.-H.; Gu, Z.-H.; Zhao, H.-X. Profiles of Bacillus Spp. Isolated from the Rhizosphere of Suaeda glauca and Their Potential to Promote Plant Growth and Suppress Fungal Phytopathogens. J. Microbiol. Biotechnol. 2021, 31, 1231–1240. [Google Scholar] [CrossRef]
- Gupta, A.; Singh, A.N.; Tiwari, R.K.; Sahu, P.K.; Yadav, J.; Srivastava, A.K.; Kumar, S. Salinity Alleviation and Reduction in Oxidative Stress by Endophytic and Rhizospheric Microbes in Two Rice Cultivars. Plants 2023, 12, 976. [Google Scholar] [CrossRef]
- Gupta, A.; Tiwari, R.K.; Shukla, R.; Singh, A.N.; Sahu, P.K. Salinity Alleviator Bacteria in Rice (Oryza sativa L.), Their Colonization Efficacy, and Synergism with Melatonin. Front. Plant Sci. 2023, 13, 1060287. [Google Scholar] [CrossRef]
- Jiang, X.; Li, W.-W.; Han, M.; Chen, G.; Wu, J.; Lai, S.; Fu, Z.; Zhang, S.; Deng, W.-W.; Gao, L.; et al. Aluminum-Tolerant, Growth-Promoting Endophytic Bacteria as Contributors in Promoting Tea Plant Growth and Alleviating Aluminum Stress. Tree Physiol. 2022, 42, 1043–1058. [Google Scholar] [CrossRef]
- Shurigin, V.; Alimov, J.; Davranov, K.; Gulyamova, T.; Egamberdieva, D. The Diversity of Bacterial Endophytes from Iris pseudacorus L. and Their Plant Beneficial Traits. Curr. Res. Microb. Sci. 2022, 3, 100133. [Google Scholar] [CrossRef]
- Montecillo, J.A.V.; Bae, H. In Silico Analysis of Koranimine, a Cyclic Imine Compound from Peribacillus frigoritolerans Reveals Potential Nematicidal Activity. Sci. Rep. 2022, 12, 18883. [Google Scholar] [CrossRef]
- Zhang, C.; Li, X.; Yin, L.; Liu, C.; Zou, H.; Wu, Z.; Zhang, Z. Analysis of the Complete Genome Sequence of Brevibacterium frigoritolerans ZB201705 Isolated from Drought- and Salt-Stressed Rhizosphere Soil of Maize. Ann. Microbiol. 2019, 69, 1489–1496. [Google Scholar] [CrossRef]
- Liu, G.-H.; Liu, B.; Wang, J.-P.; Che, J.-M.; Li, P.-F. Reclassification of Brevibacterium frigoritolerans DSM 8801T as Bacillus frigoritolerans Comb. Nov. Based on Genome Analysis. Curr. Microbiol. 2020, 77, 1916–1923. [Google Scholar] [CrossRef]
- Senchenkov, V.Y.; Lyakhovchenko, N.S.; Nikishin, I.A.; Myagkov, D.A.; Chepurina, A.A.; Polivtseva, V.N.; Abashina, T.N.; Delegan, Y.A.; Nikulicheva, T.B.; Nikulin, I.S.; et al. Whole-Genome Sequencing and Biotechnological Potential Assessment of Two Bacterial Strains Isolated from Poultry Farms in Belgorod, Russia. Microorganisms 2023, 11, 2235. [Google Scholar] [CrossRef]
- González-Reguero, D.; Robas-Mora, M.; Alonso, M.R.; Fernández-Pastrana, V.M.; Lobo, A.P.; Gómez, P.A.J. Induction of Phytoextraction, Phytoprotection and Growth Promotion Activities in Lupinus albus under Mercury Abiotic Stress Conditions by Peribacillus frigoritolerans subsp. mercuritolerans subsp. nov. Ecotoxicol. Environ. Saf. 2024, 285, 117139. [Google Scholar] [CrossRef]
- Jin, M.; Zhao, Q.; Zhou, Z.; Zhu, L.; Zhang, Z.; Jiang, L. Draft Genome Sequence of a Potential Organic Phosphorus-Degrading Bacterium Brevibacterium frigoritolerans GD44, Isolated from Radioactive Soil in Xinjiang, China. Curr. Microbiol. 2020, 77, 2896–2903. [Google Scholar] [CrossRef]
- Kapanadze, K.; Magalashvili, A.; Imnadze, P. Distribution of Natural Radionuclides in the Soils and Assessment of Radiation Hazards in the Khrami Late Variscan Crystal Massif (Georgia). Heliyon 2019, 5, e01377. [Google Scholar] [CrossRef]
- Travers, R.S.; Martin, P.A.W.; Reichelderfer, C.F. Selective Process for Efficient Isolation of Soil Bacillus spp. Appl. Environ. Microbiol. 1987, 53, 1263–1266. [Google Scholar] [CrossRef]
- Stewart, G.S.; Johnstone, K.; Hagelberg, E.; Ellar, D.J. Commitment of Bacterial Spores to Germinate A Measure of the Trigger Reaction. Biochem. J. 1981, 198, 101–106. [Google Scholar] [CrossRef]
- Romanenko, M.N.; Nesterenko, M.A.; Shikov, A.E.; Nizhnikov, A.A.; Antonets, K.S. Draft Genome Sequence Data of Lysinibacillus sphaericus Strain 1795 with Insecticidal Properties. Data 2023, 8, 167. [Google Scholar] [CrossRef]
- Saleem, F.; Shakoori, A. The First Cry2Ac-Type Protein Toxic to Helicoverpa armigera: Cloning and Overexpression of cry2ac7 Gene from SBS-BT1 Strain of Bacillus thuringiensis. Toxins 2017, 9, 358. [Google Scholar] [CrossRef]
- Spizizen, J. Transformation of Biochemically Deficient Strains of Bacillus subtilis by Deoxyribonucleate. Proc. Natl. Acad. Sci. USA 1958, 44, 1072–1078. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data 2010. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 12 September 2024).
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. Fastp: An Ultra-Fast All-in-One FASTQ Preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality Assessment Tool for Genome Assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef]
- Manni, M.; Berkeley, M.R.; Seppey, M.; Simão, F.A.; Zdobnov, E.M. BUSCO Update: Novel and Streamlined Workflows along with Broader and Deeper Phylogenetic Coverage for Scoring of Eukaryotic, Prokaryotic, and Viral Genomes. Mol. Biol. Evol. 2021, 38, 4647–4654. [Google Scholar] [CrossRef]
- Parks, D.H.; Imelfort, M.; Skennerton, C.T.; Hugenholtz, P.; Tyson, G.W. CheckM: Assessing the Quality of Microbial Genomes Recovered from Isolates, Single Cells, and Metagenomes. Genome Res. 2015, 25, 1043–1055. [Google Scholar] [CrossRef]
- Jain, C.; Rodriguez-R, L.M.; Phillippy, A.M.; Konstantinidis, K.T.; Aluru, S. High Throughput ANI Analysis of 90K Prokaryotic Genomes Reveals Clear Species Boundaries. Nat. Commun. 2018, 9, 5114. [Google Scholar] [CrossRef]
- Seemann, T. Prokka: Rapid Prokaryotic Genome Annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef]
- Liu, H.; Zheng, J.; Bo, D.; Yu, Y.; Ye, W.; Peng, D.; Sun, M. BtToxin_Digger: A Comprehensive and High-Throughput Pipeline for Mining Toxin Protein Genes from Bacillus thuringiensis. Bioinformatics 2021, 38, 250–251. [Google Scholar] [CrossRef]
- Shikov, A.E.; Malovichko, Y.V.; Skitchenko, R.K.; Nizhnikov, A.A.; Antonets, K.S. No More Tears: Mining Sequencing Data for Novel Bt Cry Toxins with CryProcessor. Toxins 2020, 12, 204. [Google Scholar] [CrossRef]
- Hannigan, G.D.; Prihoda, D.; Palicka, A.; Soukup, J.; Klempir, O.; Rampula, L.; Durcak, J.; Wurst, M.; Kotowski, J.; Chang, D.; et al. A Deep Learning Genome-Mining Strategy for Biosynthetic Gene Cluster Prediction. Nucleic Acids Res. 2019, 47, e110. [Google Scholar] [CrossRef]
- Blin, K.; Shaw, S.; Augustijn, H.E.; Reitz, Z.L.; Biermann, F.; Alanjary, M.; Fetter, A.; Terlouw, B.R.; Metcalf, W.W.; Helfrich, E.J.N.; et al. AntiSMASH 7.0: New and Improved Predictions for Detection, Regulation, Chemical Structures and Visualisation. Nucleic Acids Res. 2023, 51, W46–W50. [Google Scholar] [CrossRef]
- Carroll, L.M.; Cheng, R.A.; Kovac, J. No Assembly Required: Using BTyper3 to Assess the Congruency of a Proposed Taxonomic Framework for the Bacillus cereus Group with Historical Typing Methods. Front. Microbiol. 2020, 11, 580691. [Google Scholar] [CrossRef]
- Alonge, M.; Lebeigle, L.; Kirsche, M.; Jenike, K.; Ou, S.; Aganezov, S.; Wang, X.; Lippman, Z.B.; Schatz, M.C.; Soyk, S. Automated Assembly Scaffolding Using RagTag Elevates a New Tomato System for High-Throughput Genome Editing. Genome Biol. 2022, 23, 258. [Google Scholar] [CrossRef]
- Bertelli, C.; Brinkman, F.S.L. Improved Genomic Island Predictions with IslandPath-DIMOB. Bioinformatics 2018, 34, 2161–2167. [Google Scholar] [CrossRef]
- Waack, S.; Keller, O.; Asper, R.; Brodag, T.; Damm, C.; Fricke, W.F.; Surovcik, K.; Meinicke, P.; Merkl, R. Score-Based Prediction of Genomic Islands in Prokaryotic Genomes Using Hidden Markov Models. BMC Bioinform. 2006, 7, 142. [Google Scholar] [CrossRef]
- Gan, R.; Zhou, F.; Si, Y.; Yang, H.; Chen, C.; Ren, C.; Wu, J.; Zhang, F. DBSCAN-SWA: An Integrated Tool for Rapid Prophage Detection and Annotation. Front. Genet. 2022, 13, 885048. [Google Scholar] [CrossRef]
- Akhter, S.; Aziz, R.K.; Edwards, R.A. PhiSpy: A Novel Algorithm for Finding Prophages in Bacterial Genomes That Combines Similarity- and Composition-Based Strategies. Nucleic Acids Res. 2012, 40, e126. [Google Scholar] [CrossRef]
- Xie, Z.; Tang, H. ISEScan: Automated Identification of Insertion Sequence Elements in Prokaryotic Genomes. Bioinformatics 2017, 33, 3340–3347. [Google Scholar] [CrossRef]
- Stothard, P.; Wishart, D.S. Circular Genome Visualization and Exploration Using CGView. Bioinformatics 2005, 21, 537–539. [Google Scholar] [CrossRef]
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016; ISBN 978-3-319-24277-4. [Google Scholar]
- Chen, H.; Boutros, P.C. VennDiagram: A Package for the Generation of Highly-Customizable Venn and Euler Diagrams in R. BMC Bioinform. 2011, 12, 35. [Google Scholar] [CrossRef]
- O’Leary, N.A.; Wright, M.W.; Brister, J.R.; Ciufo, S.; Haddad, D.; McVeigh, R.; Rajput, B.; Robbertse, B.; Smith-White, B.; Ako-Adjei, D.; et al. Reference Sequence (RefSeq) Database at NCBI: Current Status, Taxonomic Expansion, and Functional Annotation. Nucleic Acids Res. 2016, 44, D733–D745. [Google Scholar] [CrossRef]
- Tonkin-Hill, G.; MacAlasdair, N.; Ruis, C.; Weimann, A.; Horesh, G.; Lees, J.A.; Gladstone, R.A.; Lo, S.; Beaudoin, C.; Floto, R.A.; et al. Producing Polished Prokaryotic Pangenomes with the Panaroo Pipeline. Genome Biol. 2020, 21, 180. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Page, A.J.; Taylor, B.; Delaney, A.J.; Soares, J.; Seemann, T.; Keane, J.A.; Harris, S.R. SNP-Sites: Rapid Efficient Extraction of SNPs from Multi-FASTA Alignments. Microb. Genom. 2016, 2, e000056. [Google Scholar] [CrossRef]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2—Approximately Maximum-Likelihood Trees for Large Alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef]
- Syed, T.; Askari, M.; Meng, Z.; Li, Y.; Abid, M.; Wei, Y.; Guo, S.; Liang, C.; Zhang, R. Current Insights on Vegetative Insecticidal Proteins (Vip) as Next Generation Pest Killers. Toxins 2020, 12, 522. [Google Scholar] [CrossRef]
- Ricker, N.; Qian, H.; Fulthorpe, R.R. The Limitations of Draft Assemblies for Understanding Prokaryotic Adaptation and Evolution. Genomics 2012, 100, 167–175. [Google Scholar] [CrossRef]
- Patel, S.; Gupta, R.S. A Phylogenomic and Comparative Genomic Framework for Resolving the Polyphyly of the Genus Bacillus: Proposal for Six New Genera of Bacillus Species, Peribacillus gen. nov., Cytobacillus gen. nov., Mesobacillus gen. nov., Neobacillus gen. nov., Metabacillus gen. nov. and Alkalihalobacillus gen. nov. Int. J. Syst. Evol. Microbiol. 2020, 70, 406–438. [Google Scholar] [CrossRef]
- Hoffmaster, A.R.; Ravel, J.; Rasko, D.A.; Chapman, G.D.; Chute, M.D.; Marston, C.K.; De, B.K.; Sacchi, C.T.; Fitzgerald, C.; Mayer, L.W.; et al. Identification of Anthrax Toxin Genes in a Bacillus cereus Associated with an Illness Resembling Inhalation Anthrax. Proc. Natl. Acad. Sci. USA 2004, 101, 8449–8454. [Google Scholar] [CrossRef]
- Sánchez-Chica, J.; Correa, M.M.; Aceves-Diez, A.E.; Castañeda-Sandoval, L.M. Enterotoxin Gene Distribution and Genotypes of Bacillus cereus sensu lato Isolated from Cassava Starch. Toxins 2021, 13, 131. [Google Scholar] [CrossRef]
- Friesema, I.H.; Slegers-Fitz-James, I.A.; Wit, B.; Franz, E. Surveillance and Characteristics of Food-Borne Outbreaks in the Netherlands, 2006 to 2019. Eurosurveillance 2022, 27, 2100071. [Google Scholar] [CrossRef]
- Bulgari, D.; Filisetti, S.; Montagna, M.; Gobbi, E.; Faoro, F. Pathogenic Potential of Bacteria Isolated from Commercial Biostimulants. Arch. Microbiol. 2022, 204, 162. [Google Scholar] [CrossRef]
- Sampson, K.S.; Tomso, D.J.; Agarwal, S.; McNulty, B.; Campbell, C. Toxin Genes and Methods for Their Use. U.S. Patent US8461421B2, 11 June 2013. [Google Scholar]
- Zhu, S.; Hegemann, J.D.; Fage, C.D.; Zimmermann, M.; Xie, X.; Linne, U.; Marahiel, M.A. Insights into the Unique Phosphorylation of the Lasso Peptide Paeninodin. J. Biol. Chem. 2016, 291, 13662–13678. [Google Scholar] [CrossRef]
- Fan, H.; Ru, J.; Zhang, Y.; Wang, Q.; Li, Y. Fengycin Produced by Bacillus subtilis 9407 Plays a Major Role in the Biocontrol of Apple Ring Rot Disease. Microbiol. Res. 2017, 199, 89–97. [Google Scholar] [CrossRef]
- Medeot, D.B.; Fernandez, M.; Morales, G.M.; Jofré, E. Fengycins from Bacillus amyloliquefaciens MEP218 Exhibit Antibacterial Activity by Producing Alterations on the Cell Surface of the Pathogens Xanthomonas axonopodis pv. vesicatoria and Pseudomonas aeruginosa PA01. Front. Microbiol. 2020, 10, 3107. [Google Scholar] [CrossRef]
- Plowman, J.E.; Loehr, T.M.; Goldman, S.J.; Sanders-Loehr, J. Structure and Siderophore Activity of Ferric Schizokinen. J. Inorg. Biochem. 1984, 20, 183–197. [Google Scholar] [CrossRef]
- Khan, A.; Singh, P.; Srivastava, A. Synthesis, Nature and Utility of Universal Iron Chelator—Siderophore: A Review. Microbiol. Res. 2018, 212–213, 103–111. [Google Scholar] [CrossRef]
- Mullis, K.B.; Pollack, J.R.; Neilands, J.B. Structure of Schizokinen, An Iron-Transport Compound from Bacillus megaterium. Biochemistry 1971, 10, 4894–4898. [Google Scholar] [CrossRef]
- Crosa, J.H.; Walsh, C.T. Genetics and Assembly Line Enzymology of Siderophore Biosynthesis in Bacteria. Microbiol. Mol. Biol. Rev. 2002, 66, 223–249. [Google Scholar] [CrossRef]
- Kaeberlein, T.; Lewis, K.; Epstein, S.S. Isolating “Uncultivable” Microorganisms in Pure Culture in a Simulated Natural Environment. Science 2002, 296, 1127–1129. [Google Scholar] [CrossRef]
- Lewis, K.; Epstein, S.; D’Onofrio, A.; Ling, L.L. Uncultured Microorganisms as a Source of Secondary Metabolites. J. Antibiot. 2010, 63, 468–476. [Google Scholar] [CrossRef]
- Saha, M.; Sarkar, S.; Sarkar, B.; Sharma, B.K.; Bhattacharjee, S.; Tribedi, P. Microbial Siderophores and Their Potential Applications: A Review. Environ. Sci. Pollut. Res. 2016, 23, 3984–3999. [Google Scholar] [CrossRef]
- Rungin, S.; Indananda, C.; Suttiviriya, P.; Kruasuwan, W.; Jaemsaeng, R.; Thamchaipenet, A. Plant Growth Enhancing Effects by a Siderophore-Producing Endophytic Streptomycete Isolated from a Thai Jasmine Rice Plant (Oryza sativa L. Cv. KDML105). Antonie Leeuwenhoek 2012, 102, 463–472. [Google Scholar] [CrossRef]
- Masalha, J.; Kosegarten, H.; Elmaci, Ö.; Mengel, K. The Central Role of Microbial Activity for Iron Acquisition in Maize and Sunflower. Biol. Fertil. Soils 2000, 30, 433–439. [Google Scholar] [CrossRef]
- Beneduzi, A.; Ambrosini, A.; Passaglia, L.M.P. Plant Growth-Promoting Rhizobacteria (PGPR): Their Potential as Antagonists and Biocontrol Agents. Genet. Mol. Biol. 2012, 35, 1044–1051. [Google Scholar] [CrossRef]
- Ahmed, E.; Holmström, S.J.M. Siderophores in Environmental Research: Roles and Applications. Microb. Biotechnol. 2014, 7, 196–208. [Google Scholar] [CrossRef]
- O’Brien, S.; Hodgson, D.J.; Buckling, A. Social Evolution of Toxic Metal Bioremediation in Pseudomonas aeruginosa. Proc. R. Soc. B Biol. Sci. 2014, 281, 20140858. [Google Scholar] [CrossRef]
- Rajkumar, M.; Ae, N.; Prasad, M.N.V.; Freitas, H. Potential of Siderophore-Producing Bacteria for Improving Heavy Metal Phytoextraction. Trends Biotechnol. 2010, 28, 142–149. [Google Scholar] [CrossRef]
- Nair, A.; Juwarkar, A.A.; Singh, S.K. Production and Characterization of Siderophores and Its Application in Arsenic Removal from Contaminated Soil. Water Air Soil. Pollut. 2007, 180, 199–212. [Google Scholar] [CrossRef]
- Möllmann, U.; Heinisch, L.; Bauernfeind, A.; Köhler, T.; Ankel-Fuchs, D. Siderophores as Drug Delivery Agents: Application of the “Trojan Horse” Strategy. BioMetals 2009, 22, 615–624. [Google Scholar] [CrossRef]
- Rasool, A.; Imran Mir, M.; Zulfajri, M.; Hanafiah, M.M.; Azeem Unnisa, S.; Mahboob, M. Plant Growth Promoting and Antifungal Asset of Indigenous Rhizobacteria Secluded from Saffron (Crocus sativus L.) Rhizosphere. Microb. Pathog. 2021, 150, 104734. [Google Scholar] [CrossRef]
- Singh, P.; Kim, Y.J.; Yang, D.-C.; Singh, H.; Wang, C.; Farh, M.E.-A.; Hwang, K.H. Biosynthesis, Characterization, and Antimicrobial Applications of Silver Nanoparticles. Int. J. Nanomed. 2015, 10, 2567–2577. [Google Scholar] [CrossRef]
- Shurigin, V.; Li, L.; Alaylar, B.; Egamberdieva, D.; Liu, Y.-H.; Li, W.-J. Plant Beneficial Traits of Endophytic Bacteria Associated with Fennel (Foeniculum vulgare Mill.). AIMS Microbiol. 2024, 10, 449–467. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Feature | Value |
|---|---|
| Total amount of contigs | 48 |
| Largest contig (number of nucleotides) | 592,496 |
| N50 value | 432,533 |
| N90 value | 75,728 |
| L50 value | 6 |
| L90 value | 17 |
| Number of properly paired reads (%) | 99.06 |
| Assembly completeness (%) | 98.91 |
| Suspected contamination (%) | 1.82 |
| Database | Bacillales_odb10 | Bacilli_odb10 |
|---|---|---|
| Single-copy orthologues assembled completely | 449 out of 450 (99.8%) | 302 out of 302 (100.0%) |
| Orthologues present in one copy | 444 out of 450 (98.7%) | 301 out of 302 (99.7%) |
| Multi-copies orthologues | 5 out of 450 (1.1%) | 1 out of 302 (0.3%) |
| Fragmented sequences | 0 out of 450 (0.0%) | 0 out of 302 (0.0%) |
| Orthologues missing from the assembly | 1 out of 450 (0.2%) | 0 out of 302 (0.0%) |
| Total number of single-copy orthologues in the database | 450 | 302 |
| NCBI RefSeq Assembly | Taxon | Strain | ANI |
|---|---|---|---|
| GCF_029625965 | P. frigoritolerans | A1.1 | 98.78 |
| GCF_007828935 | P. frigoritolerans | VIVO01 | 98.66 |
| GCF_030122925 | P. frigoritolerans | CF29 | 98.56 |
| GCF_026794285 | P. frigoritolerans | TP802B-4 | 98.52 |
| GCF_037027045 | P. frigoritolerans | JJ1159 | 98.41 |
| GCF_036220485 | P. frigoritolerans | NRS-835 | 98.38 |
| GCF_036219585 | P. frigoritolerans | NRS-1703 | 98.33 |
| GCF_026891895 | Peribacillus sp. | AS_2 | 98.26 |
| GCF_022394675 | P. frigoritolerans | JHS1 | 98.25 |
| GCF_036220565 | P. frigoritolerans | NRS-822 | 98.18 |
| Contig | Region Product | Type | Number of CDS | Location (Relative Coordinate, b.p.) | Most Similar Known Cluster | Similarity, % |
|---|---|---|---|---|---|---|
| 1 | T3PKS 1 | – | 39 | 375,631–416,719 (total: 41,088) | – | – |
| Terpene | – | 21 | 551,614–572,432 (total: 20,818) | – | – | |
| 2 | Terpene | – | 18 | 189,404–211,299 (total: 21,895) | – | – |
| NI-siderophore 2 | Other 5 | 10 | 513,534–529,046 (total: 15,512) | Schizokinen | 60 | |
| 3 | LAP 3 | – | 17 | 102,897–126,432 (total: 23,535) | – | – |
| 5 | Lassopeptide | RiPP 6 | 23 | 190,546–214,519 (total: 23,973) | Paeninodin | 100 |
| 6 | NRPS 4 | Polyketide | 38 | 63,960–108,027 (total: 44,067) | Meilingmycin | 2 |
| 7 | NRPS | NRP | 46 | 237,872–298,196 (total: 60,324) | Koranimine | 87 |
| 8 | Betalactone | NRP | 25 | 184,667–208,836 (total: 24,169) | Fengycin | 46.67 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Romanenko, M.N.; Shikov, A.E.; Savina, I.A.; Nizhnikov, A.A.; Antonets, K.S. Whole-Genome Sequencing of Peribacillus frigoritolerans Strain d21.2 Isolated in the Republic of Dagestan, Russia. Microorganisms 2024, 12, 2410. https://doi.org/10.3390/microorganisms12122410
Romanenko MN, Shikov AE, Savina IA, Nizhnikov AA, Antonets KS. Whole-Genome Sequencing of Peribacillus frigoritolerans Strain d21.2 Isolated in the Republic of Dagestan, Russia. Microorganisms. 2024; 12(12):2410. https://doi.org/10.3390/microorganisms12122410
Chicago/Turabian StyleRomanenko, Maria N., Anton E. Shikov, Iuliia A. Savina, Anton A. Nizhnikov, and Kirill S. Antonets. 2024. "Whole-Genome Sequencing of Peribacillus frigoritolerans Strain d21.2 Isolated in the Republic of Dagestan, Russia" Microorganisms 12, no. 12: 2410. https://doi.org/10.3390/microorganisms12122410
APA StyleRomanenko, M. N., Shikov, A. E., Savina, I. A., Nizhnikov, A. A., & Antonets, K. S. (2024). Whole-Genome Sequencing of Peribacillus frigoritolerans Strain d21.2 Isolated in the Republic of Dagestan, Russia. Microorganisms, 12(12), 2410. https://doi.org/10.3390/microorganisms12122410