

Genomic Insights into Stutzerimonas kunmingensis TFRC-KFRI-1 Isolated from Manila Clam (Ruditapes philippinarum): Functional and Phylogenetic Analysis


Abstract
1. Introduction
2. Materials and Methods
2.1. Isolation and Identification of Stutzerimonas kunmingensis TFRC-KFRI-1
2.2. 16S rRNA Gene Sequencing and Analysis
2.3. Genomic DNA Extraction
2.4. Genome Sequencing
2.5. Library Preparation and Sequencing
2.6. Data Quality Control and Genome Assembly
2.7. Gene Prediction and Annotation
2.8. Phylogenetic Analysis
2.9. Pathogenicity Prediction
3. Results
3.1. Genome Sequencing and Assembly
3.2. Genome Annotation and Comparative Analysis
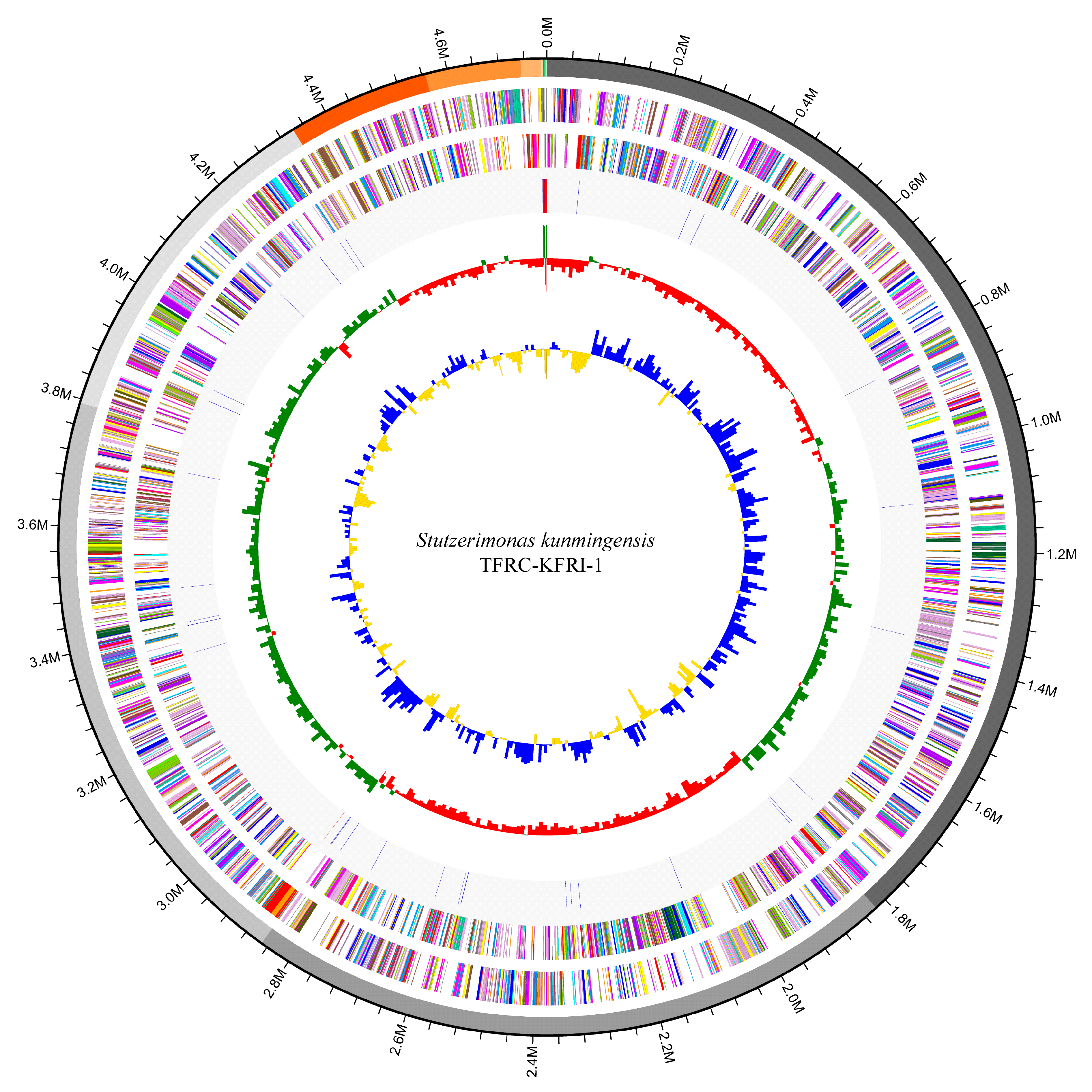
3.3. Genome Map and COG Analysis
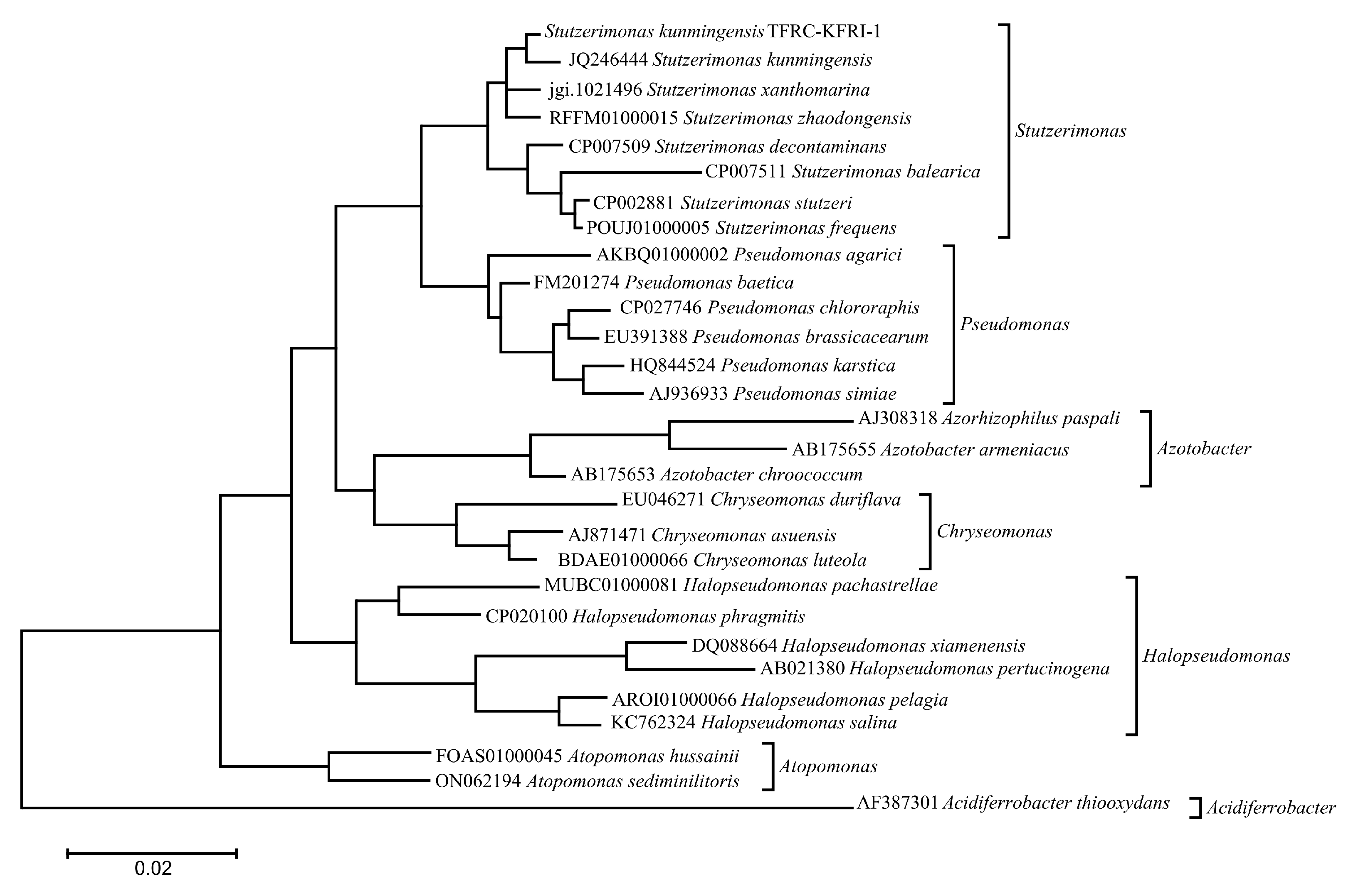
3.4. Phylogenetic Analysis
3.5. Genomic Features
3.6. Pathogenicity Prediction
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Gul, S.; Durante-Mangoni, E. Unraveling the Puzzle: Health Benefits of Probiotics-A Comprehensive Review. J. Clin. Med. 2024, 13, 1436. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Irianto, A.; Austin, B. Probiotics in aquaculture. J. Fish Dis. 2002, 25, 633–642. [Google Scholar] [CrossRef]
- Shreiner, A.B.; Kao, J.Y.; Young, V.B. The gut microbiome in health and in disease. Curr. Opin. Gastroenterol. 2015, 31, 69–75. [Google Scholar] [CrossRef]
- Kesarcodi-Watson, A.; Kaspar, H.; Lategan, M.J.; Gibson, L. Probiotics in aquaculture: The need, principles and mechanisms of action and screening processes. Aquaculture 2008, 274, 1–14. [Google Scholar] [CrossRef]
- Hai, N.V. The use of probiotics in aquaculture. J. Appl. Microbiol. 2015, 119, 917–935. [Google Scholar] [CrossRef]
- Sikorski, J.; Lalucat, J.; Wackernagel, W. Genomovars 11 to 18 of Pseudomonas stutzeri, identified among isolates from soil and marine sediment. Int. J. Syst. Evol. Microbiol. 2005, 55, 1767–1770. [Google Scholar] [CrossRef] [PubMed]
- Lalucat, J.; Gomila, M.; Mulet, M.; Zaruma, A.; García-Valdés, E. Past, present and future of the boundaries of the Pseudomonas genus: Proposal of Stutzerimonas gen. nov. Syst. Appl. Microbiol. 2021, 45, 126289. [Google Scholar] [CrossRef] [PubMed]
- Xie, F.; Ma, H.; Quan, S.; Liu, D.; Chen, G.; Chao, Y.; Qian, S. Pseudomonas kunmingensis sp. nov., an exopolysaccharide-producing bacterium isolated from a phosphate mine. Int. J. Syst. Evol. Microbiol. 2014, 64, 559–564. [Google Scholar] [CrossRef] [PubMed]
- Vieco-Saiz, N.; Belguesmia, Y.; Raspoet, R.; Auclair, E.; Gancel, F.; Kempf, I.; Drider, D. Benefits and Inputs from Lactic Acid Bacteria and Their Bacteriocins as Alternatives to Antibiotic Growth Promoters During Food-Animal Production. Front. Microbiol. 2019, 10, 57. [Google Scholar] [CrossRef]
- Newaj-Fyzul, A.; Austin, B. Probiotics, immunostimulants, plant products and oral vaccines, and their role as feed supplements in the control of bacterial fish diseases. J. Fish Dis. 2015, 38, 937–955. [Google Scholar] [CrossRef]
- Gunaswetha, K.; Sujatha, E.; Anusha, K.; Prathyusha, A.M.; Chandra, M.S.; Berde, C.V.; Reddy, N.N.; Bramhachari, P.V. Understanding the Probiotics and Mechanism of Immunomodulation Interactions with the Gut-Related Immune System. In Human Microbiome in Health, Disease, and Therapy; Springer Nature: Singapore, 2023; pp. 67–79. [Google Scholar] [CrossRef]
- Mulet, M.; García-Valdés, E.; Lalucat, J. Stutzerimonas decontaminans sp. nov. isolated from marine polluted sediments. Syst. Appl. Microbiol. 2023, 46, 126400. [Google Scholar] [CrossRef]
- Gomila, M.; Mulet, M.; García-Valdés, E.; Lalucat, J. Genome-Based Taxonomy of the Genus Stutzerimonas and Proposal of S. frequens sp. nov. and S. degradans sp. nov. and Emended Descriptions of S. perfectomarina and S. chloritidismutans. Microorganisms 2022, 10, 1363. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kozich, J.J.; Westcott, S.L.; Baxter, N.T.; Highlander, S.K.; Schloss, P.D. Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Appl. Environ. Microbiol. 2013, 79, 5112–5120. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Tatusova, T.; DiCuccio, M.; Badretdin, A.; Chetvernin, V.; Nawrocki, E.P.; Zaslavsky, L.; Lomsadze, A.; Pruitt, K.D.; Borodovsky, M.; Ostell, J. NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 2016, 44, 6614–6624. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Hyatt, D.; Chen, G.L.; Locascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 2010, 11, 119. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Huerta-Cepas, J.; Szklarczyk, D.; Forslund, K.; Cook, H.; Heller, D.; Walter, M.C.; Rattei, T.; Mende, D.R.; Sunagawa, S.; Kuhn, M.; et al. eggNOG 4.5: A hierarchical orthology framework with improved functional annotations for eukaryotic, prokaryotic and viral sequences. Nucleic Acids Res. 2016, 44, D286–D293. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Tanabe, M.; Kanehisa, M. Using the KEGG database resource. Curr. Protoc. Bioinform. 2012, 38, 1.12.1–1.12.43. [Google Scholar] [CrossRef] [PubMed]
- Parks, D.H.; Imelfort, M.; Skennerton, C.T.; Hugenholtz, P.; Tyson, G.W. CheckM: Assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015, 25, 1043–1055. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Yoon, S.-H.; Ha, S.-M.; Kwon, S.; Lim, J.; Kim, Y.; Seo, H.; Chun, J. Introducing EzBioCloud: A taxonomically united database of 16S rRNA gene sequences and whole-genome assemblies. Int. J. Syst. Evol. Microbiol. 2017, 67, 1613–1617. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [PubMed]
- Capella-Gutiérrez, S.; Silla-Martínez, J.M.; Gabaldón, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Tamura, K.; Nei, M. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol. Biol. Evol. 1993, 10, 512–526. [Google Scholar] [CrossRef]
- Cosentino, S.; Voldby Larsen, M.; Møller Aarestrup, F.; Lund, O. PathogenFinder—distinguishing friend from foe using bacterial whole genome sequence data. PLoS ONE 2013, 8, e77302. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Chalita, M.; Kim, Y.O.; Park, S.; Oh, H.S.; Cho, J.H.; Moon, J.; Baek, N.; Moon, C.; Lee, K.; Yang, J.; et al. EzBioCloud: A genome-driven database and platform for microbiome identification and discovery. Int. J. Syst. Evol. Microbiol. 2024, 74, 006421. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Grant, J.R.; Stothard, P. The CGView Server: A comparative genomics tool for circular genomes. Nucleic Acids Res. 2008, 1, W181–W184. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Mann, S.; Chen, Y.-P. Bacterial genomic G+C composition-eliciting environmental adaptation. Genomics 2010, 95, 7–15. [Google Scholar] [CrossRef]
- Lombard, V.; Golaconda Ramulu, H.; Drula, E.; Coutinho, P.M.; Henrissat, B. The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 2014, 42, D490–D495. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Lindgren, S.E.; Dobrogosz, W.J. Antagonistic activities of lactic acid bacteria in food and feed fermentations. FEMS Microbiol. Rev. 1990, 7, 149–163. [Google Scholar] [CrossRef] [PubMed]
- Papadimitriou, K.; Alegría, Á.; Bron, P.A.; De Angelis, M.; Gobbetti, M.; Kleerebezem, M.; Lemos, J.A.; Linares, D.M.; Ross, P.; Stanton, C.; et al. Stress physiology of lactic acid bacteria. Microbiol. Mol. Biol. Rev. 2016, 80, 837–890. [Google Scholar] [CrossRef] [PubMed]
- Rucker, R.; Chowanadisai, W.; Nakano, M. Potential physiological importance of pyrroloquinoline quinone. Altern. Med. Rev. 2009, 14, 268–277. [Google Scholar] [PubMed]
- Jobgen, W.S.; Fried, S.K.; Fu, W.J.; Meininger, C.J.; Wu, G. Regulatory role for the arginine-nitric oxide pathway in metabolism of energy substrates. J. Nutr. Biochem. 2006, 17, 571–588. [Google Scholar] [CrossRef] [PubMed]
- Labrie, S.J.; Samson, J.E.; Moineau, S. Bacteriophage resistance mechanisms. Nat. Rev. Microbiol. 2010, 8, 317–327. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| Feature | Value |
|---|---|
| Raw reads | 14,700,428 |
| Filtered reads | 13,069,158 |
| Genome Length (bp) | 4,756,396 |
| GC content (%) | 62.8 |
| Sequencing depth (×) | 231.22 |
| Number of Contigs | 12 |
| N50 (bp) | 1,031,488 |
| tRNA | 56 |
| rRNA (5S, 16S, 23S) | 1, 4, 3 (0, 3, 2) * |
| ncRNA | 4 |
| Total genes | 4519 |
| CDS | 4425 |
| Completeness (%) | 99.67 ** |
| Accession Number | JBGJJB000000000.1. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, M.; Yi, S.; Choi, J.; Park, Y.; Lim, C.; Kim, Y. Genomic Insights into Stutzerimonas kunmingensis TFRC-KFRI-1 Isolated from Manila Clam (Ruditapes philippinarum): Functional and Phylogenetic Analysis. Microorganisms 2024, 12, 2402. https://doi.org/10.3390/microorganisms12122402
Lee M, Yi S, Choi J, Park Y, Lim C, Kim Y. Genomic Insights into Stutzerimonas kunmingensis TFRC-KFRI-1 Isolated from Manila Clam (Ruditapes philippinarum): Functional and Phylogenetic Analysis. Microorganisms. 2024; 12(12):2402. https://doi.org/10.3390/microorganisms12122402
Chicago/Turabian StyleLee, Myunglip, Sunghun Yi, Jiho Choi, Yukyoung Park, Chaehyeon Lim, and Yucheol Kim. 2024. "Genomic Insights into Stutzerimonas kunmingensis TFRC-KFRI-1 Isolated from Manila Clam (Ruditapes philippinarum): Functional and Phylogenetic Analysis" Microorganisms 12, no. 12: 2402. https://doi.org/10.3390/microorganisms12122402
APA StyleLee, M., Yi, S., Choi, J., Park, Y., Lim, C., & Kim, Y. (2024). Genomic Insights into Stutzerimonas kunmingensis TFRC-KFRI-1 Isolated from Manila Clam (Ruditapes philippinarum): Functional and Phylogenetic Analysis. Microorganisms, 12(12), 2402. https://doi.org/10.3390/microorganisms12122402