
Nasopharyngeal Bacterial Prevalence and Microbial Diversity at First Treatment for Bovine Respiratory Disease (BRD) and Its Associations with Health and Mortality Outcomes in Feedyard Cattle


 , , , ,
, , , , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals and Nasopharyngeal Swab Collection
2.2. Bacterial Isolation and Identification
2.3. Antimicrobial Susceptibility Determinations
2.4. DNA Extraction from Nasopharyngeal Samples
2.5. Library Preparation and Sequencing
2.6. Bioinformatics Analysis
3. Results
3.1. Bacterial Prevalence
3.2. Antimicrobial Minimal Inhibitory Concentrations
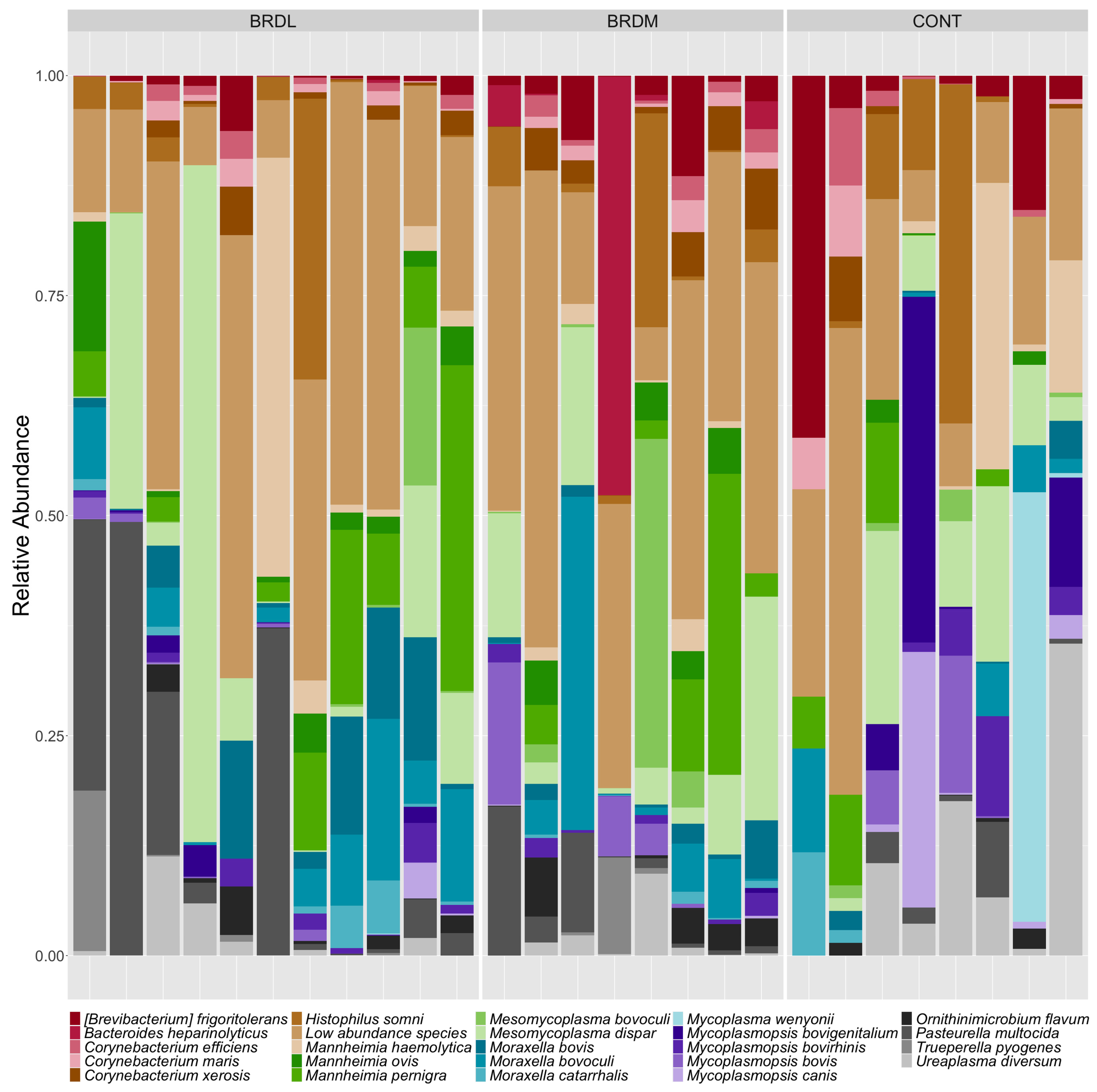
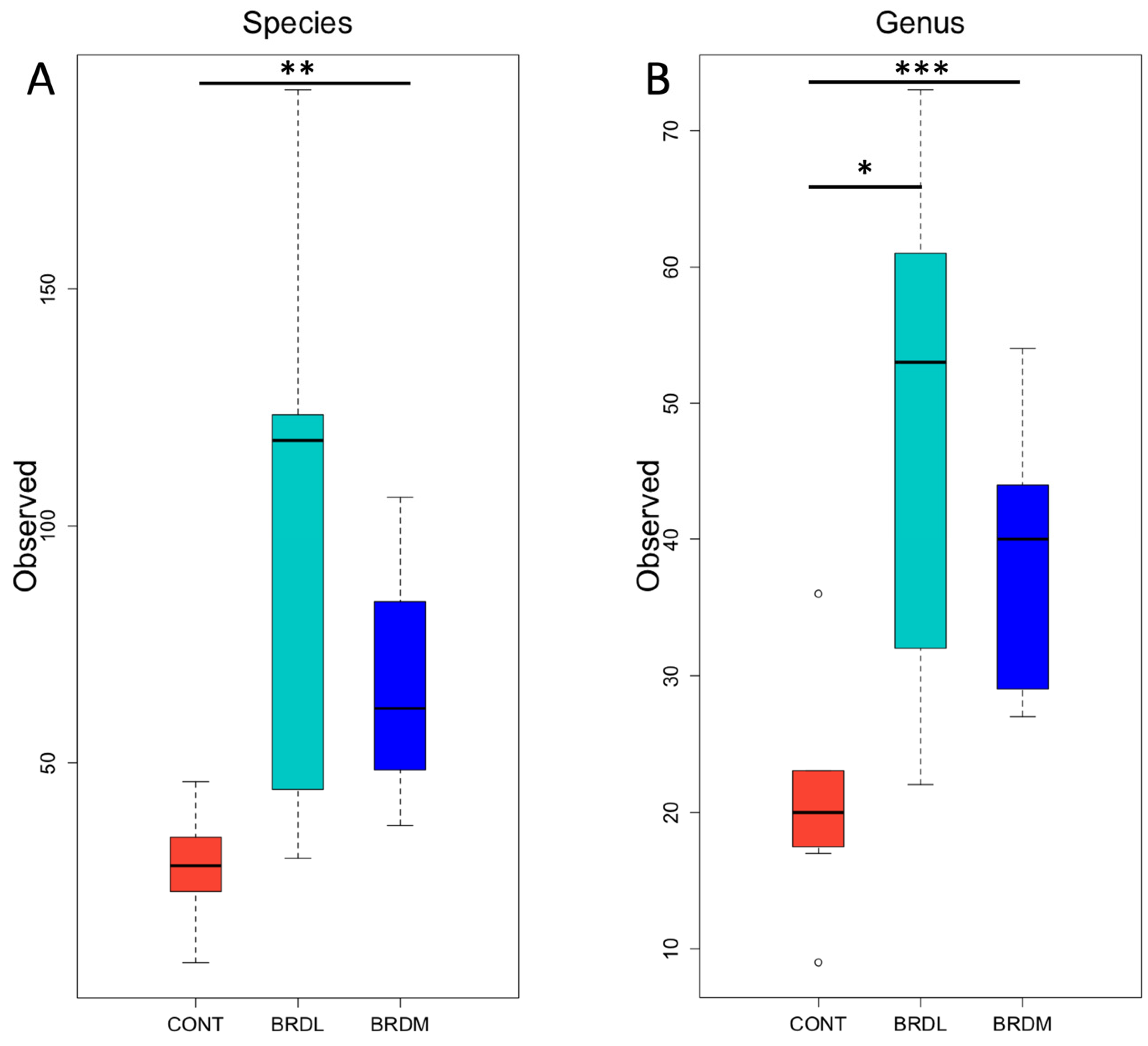
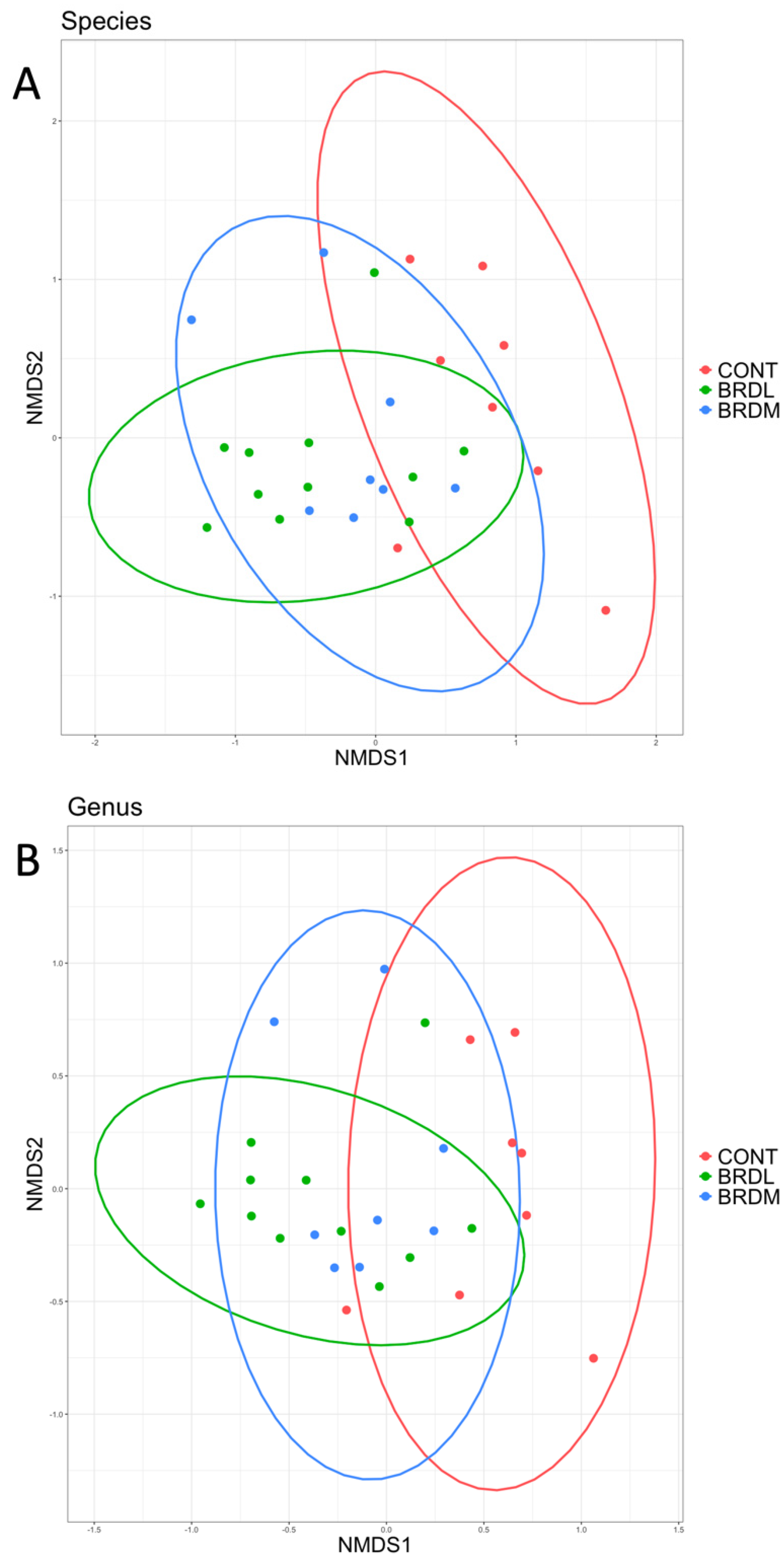
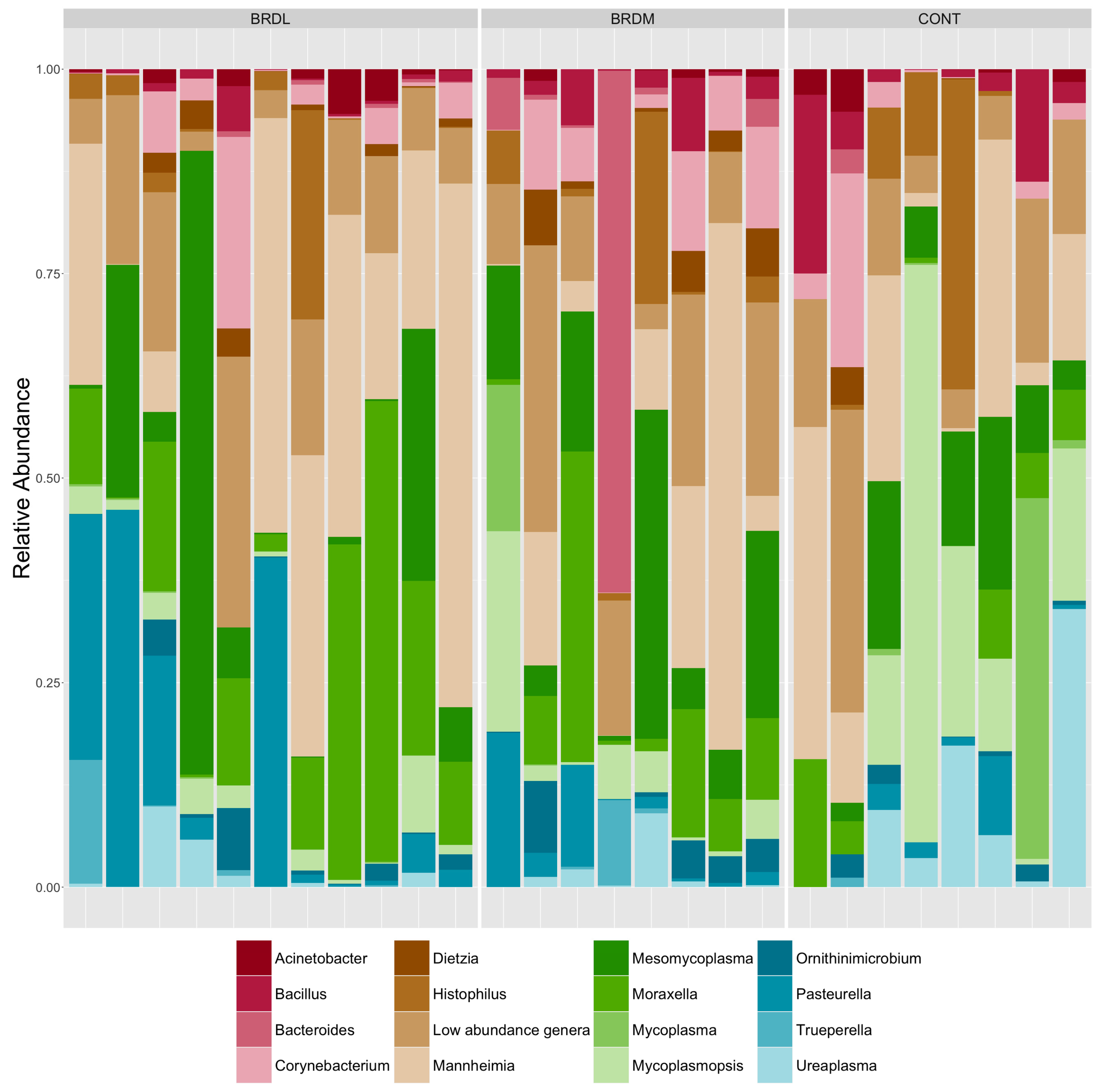
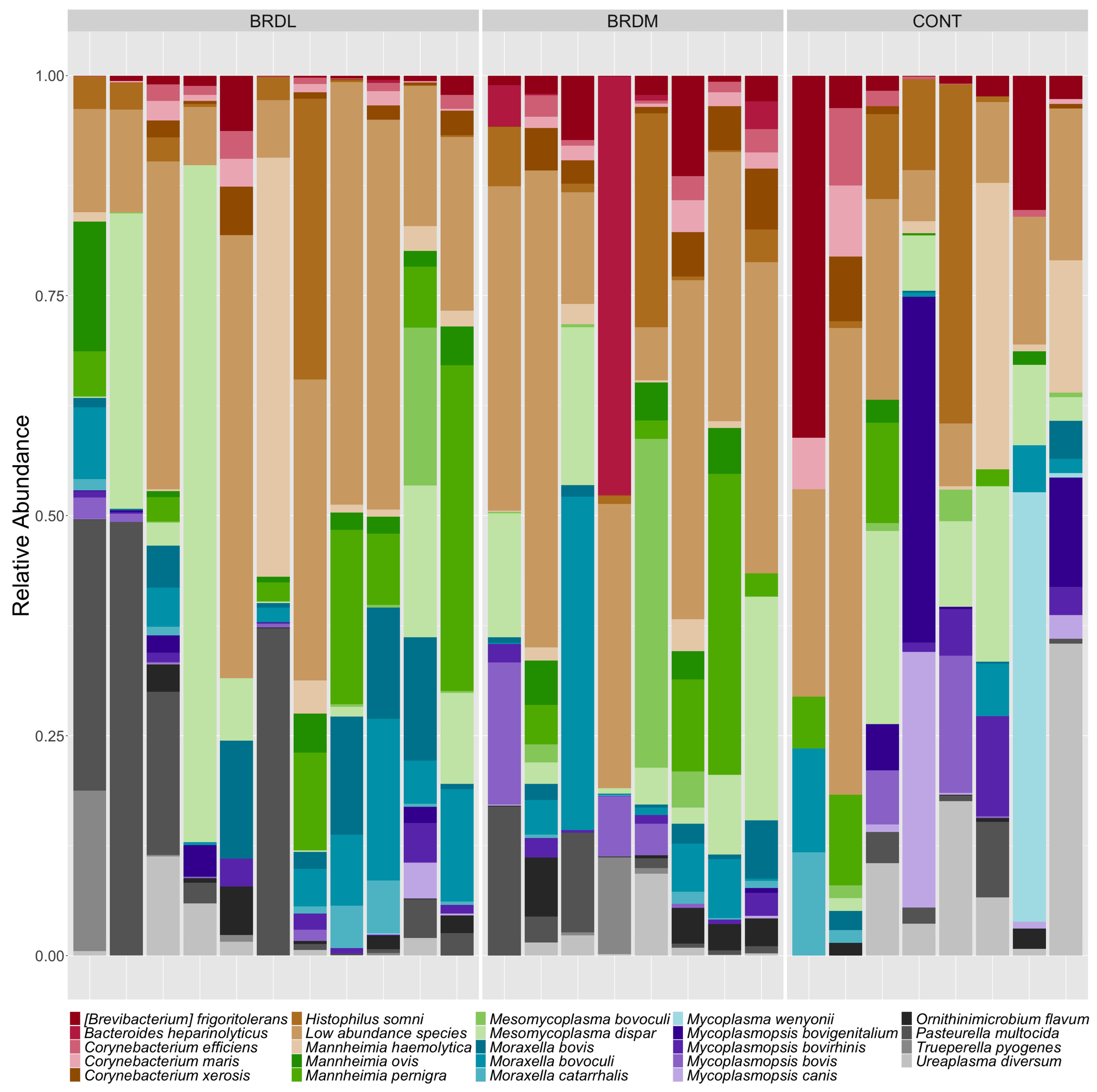
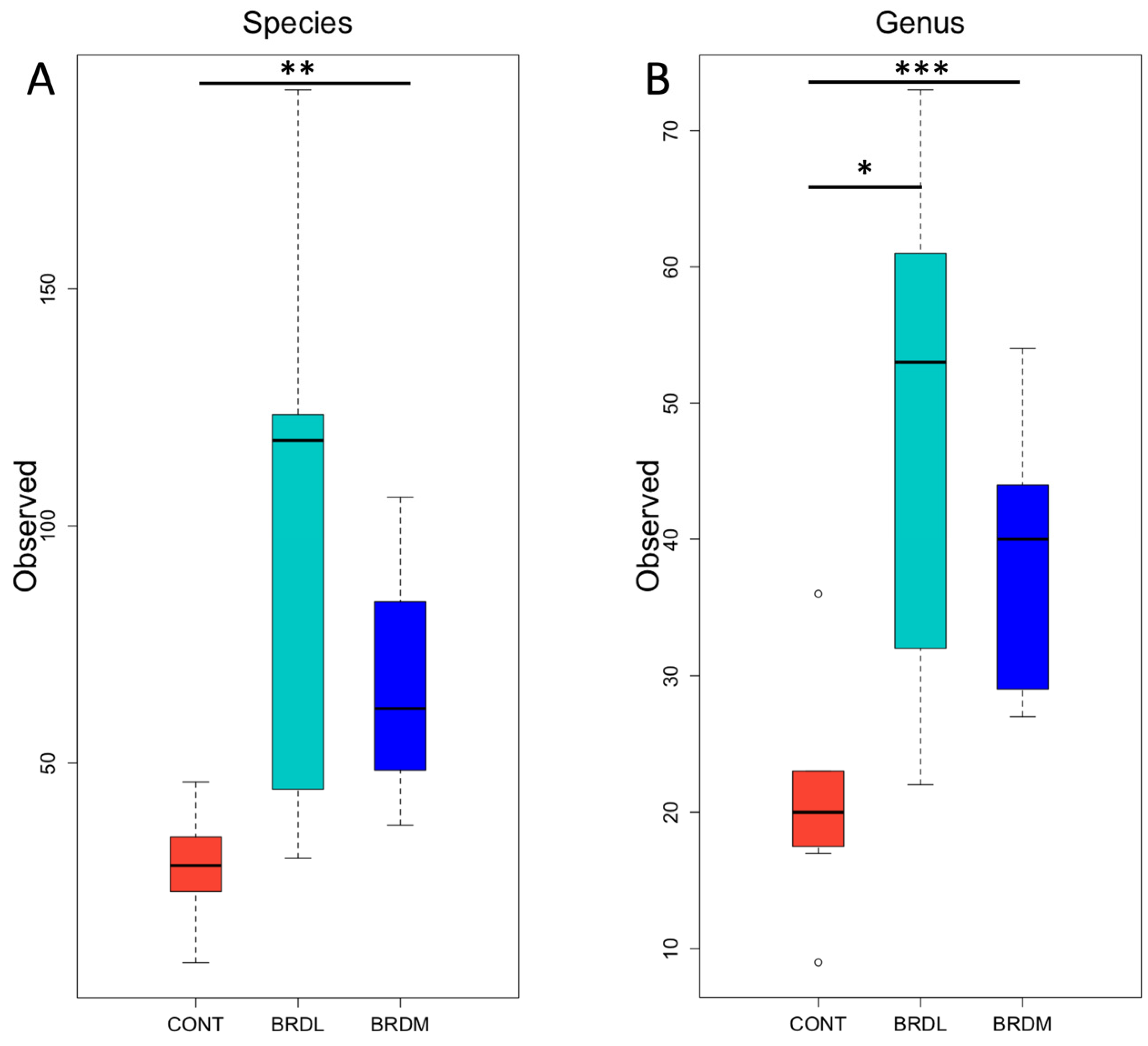
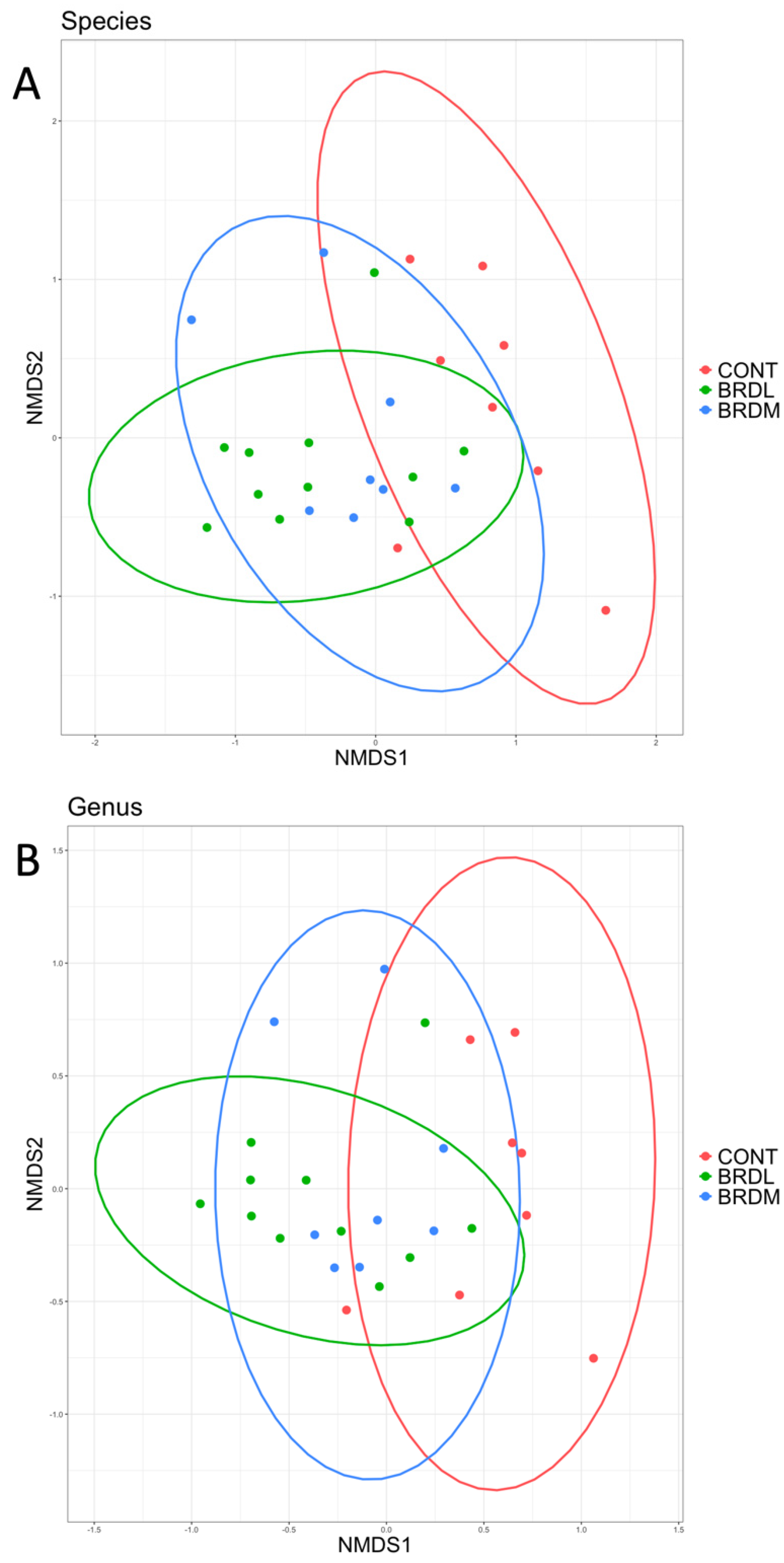
3.3. Shotgun Metagenomics
3.4. AMR Genes
4. Discussion
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Schneider, M.J.; Tait, R.G.; Busby, W.D.; Reecy, J.M. An evaluation of bovine respiratory disease complex in feedlot cattle: Impact on performance and carcass traits using treatment records and lung lesion scores 1, 2. J. Anim. Sci. 2009, 87, 1821–1827. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.A. North American cattle marketing and bovine respiratory disease (BRD). Anim. Health Res. Rev. 2009, 10, 105–108. [Google Scholar] [CrossRef] [PubMed]
- Babcock, A.H.; Cernicchiaro, N.; White, B.; Dubnicka, S.R.; Thomson, D.U.; Ives, S.E.; Scott, H.M.; Milliken, G.A.; Renter, D.G. A multivariable assessment quantifying effects of cohort-level factors associated with combined mortality and culling risk in cohorts of U.S. commercial feedlot cattle. Prev. Vet. Med. 2013, 108, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Blakebrough-Hall, C.; Hick, P.; González, L.A. Predicting bovine respiratory disease outcome in feedlot cattle using latent class analysis. J. Anim. Sci. 2020, 98, skaa381. [Google Scholar] [CrossRef] [PubMed]
- White, B.J.; Larson, B.L. Impact of bovine respiratory disease in U.S. beef cattle. Anim. Health Res. Rev. 2020, 21, 132–134. [Google Scholar] [CrossRef]
- Griffin, D. Economic Impact Associated with Respiratory Disease in Beef Cattle. Vet. Clin. N. Am. Food Anim. Pract. 1997, 13, 367–377. [Google Scholar] [CrossRef]
- Cernicchiaro, N.; White, B.J.; Renter, D.G.; Babcock, A.H. Evaluation of economic and performance outcomes associated with the number of treatments after an initial diagnosis of bovine respiratory disease in commercial feeder cattle. Am. J. Vet. Res. 2013, 74, 300–309. [Google Scholar] [CrossRef]
- Johnson, K.K.; Pendell, D.L. Market Impacts of Reducing the Prevalence of Bovine Respiratory Disease in United States Beef Cattle Feedlots. Front. Vet. Sci. 2017, 4, 189. [Google Scholar] [CrossRef]
- Chai, J.; Liu, X.; Usdrowski, H.; Deng, F.; Li, Y.; Zhao, J. Geography, niches, and transportation influence bovine respiratory microbiome and health. Front. Cell Infect. Microbiol. 2022, 12, 961644. [Google Scholar] [CrossRef]
- Confer, A.W. Update on bacterial pathogenesis in BRD. Anim. Health Res. Rev. 2009, 10, 145–148. [Google Scholar] [CrossRef]
- Griffin, D.; Chengappa, M.M.; Kuszak, J.; McVey, D.S. Bacterial Pathogens of the Bovine Respiratory Disease Complex. Vet. Clin. North Am. Food Anim. Pract. 2010, 26, 381–394. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Hill, J.E.; Godson, D.L.; Ngeleka, M.; Fernando, C.; Huang, Y. The pulmonary virome, bacteriological and histopathological findings in bovine respiratory disease from western Canada. Transbound. Emerg. Dis. 2020, 67, 924–934. [Google Scholar] [CrossRef] [PubMed]
- Nobrega, D.; Andres-Lasheras, S.; Zaheer, R.; McAllister, T.; Homerosky, E.; Anholt, R.M.; Dorin, C. Prevalence, Risk Factors, and Antimicrobial Resistance Profile of Respiratory Pathogens Isolated from Suckling Beef Calves to Reprocessing at the Feedlot: A Longitudinal Study. Front. Vet. Sci. 2021, 8, 764701. [Google Scholar] [CrossRef] [PubMed]
- Holman, D.B.; Yang, W.; Alexander, T.W. Antibiotic treatment in feedlot cattle: A longitudinal study of the effect of oxytetracycline and tulathromycin on the fecal and nasopharyngeal microbiota. Microbiome 2019, 7, 86. [Google Scholar] [CrossRef] [PubMed]
- Zeineldin, M.; Lowe, J.; de Godoy, M.; Maradiaga, N.; Ramirez, C.; Ghanem, M.; Abd El-Raof, Y.; Aldridge, B. Disparity in the nasopharyngeal microbiota between healthy cattle on feed, at entry processing and with respiratory disease. Vet. Microbiol. 2017, 208, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Jobman, E.; Hagenmaier, J.; Meyer, N.; Harper, L.B.; Taylor, L.; Lukasiewicz, K.; Thomson, D.; Lowe, J.; Terrell, S. Cross-Section Observational Study to Assess Antimicrobial Resistance Prevalence among Bovine Respiratory Disease Bacterial Isolates from Commercial US Feedlots. Antibiotics 2023, 12, 215. [Google Scholar] [CrossRef] [PubMed]
- DeRosa, D.C.; Mechor, G.D.; Staats, J.J.; Chengappa, M.M.; Shryock, T.R. Comparison of Pasteurella spp. simultaneously isolated from nasal and transtracheal swabs from cattle with clinical signs of bovine respiratory disease. J. Clin. Microbiol. 2000, 38, 327–332. [Google Scholar] [CrossRef]
- Zaheer, R.; Cook, S.R.; Klima, C.L.; Stanford, K.; Alexander, T.; Topp, E.; Read, R.R.; McAllister, T.A. Effect of subtherapeutic vs. therapeutic administration of macrolides on antimicrobial resistance in Mannheimia haemolytica and enterococci isolated from beef cattle. Front. Microbiol. 2013, 4, 133. [Google Scholar] [CrossRef]
- Deressa, A.; Asfaw, Y.; Lubke, B.; Kyule, M.; Tefera, G.; Zessin, K. Molecular Detection of Pasteurella multocida and Mannheimia haemolytica in Sheep Respiratory Infections in Ethiopia. Int. J. Appl. Res. Vet. Med. 2010, 8, 101–108. [Google Scholar]
- Clinical and Laboratory Standard Institute (CLSI). Performance Standard for Antimicrobial Disk and Dilution Susceptibility Tests for Bacteria Isolated from Animals, 6th ed.; CLSI Supplement VET01S: Wayne, PA, USA, 2023. [Google Scholar]
- Doster, E.; Lakin, S.M.; Dean, C.J.; Wolfe, C.; Young, J.G.; Boucher, C.; Belk, K.E.; Noyes, N.R.; Morley, P.S. MEGARes 2.0: A database for classification of antimicrobial drug, biocide and metal resistance determinants in metagenomic sequence data. Nucleic Acids Res. 2020, 48, 561–569. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [PubMed]
- Wood, D.E.; Lu, J.; Langmead, B. Improved metagenomic analysis with Kraken 2. Genome Biol 2019, 20, 257. [Google Scholar] [CrossRef] [PubMed]
- Bonin, N.; Doster, E.; Worley, H.; Pinnell, L.J.; Bravo, J.E.; Ferm, P.; Marini, S.; Prosperi, M.; Noyes, N.; Morley, P.S. MEGARes and AMR++, v3.0: An updated comprehensive database of antimicrobial resistance determinants and an improved software pipeline for classification using high-throughput sequencing. Nucleic Acids Res. 2023, 51, D744–D752. [Google Scholar] [CrossRef]
- Hunt, M.; Mather, A.E.; Sánchez-Busó, L.; Page, A.J.; Parkhill, J.; Keane, J.A.; Harris, S.R. ARIBA: Rapid antimicrobial resistance genotyping directly from sequencing reads. Microb. Genomics 2017, 3, 10. [Google Scholar] [CrossRef] [PubMed]
- Alcock, B.P.; Raphenya, A.R.; Lau, T.T.Y.; Tsang, K.K.; Bouchard, M.; Edalatmand, A.; Huynh, W.; Nguyen, A.L.V.; Cheng, A.A.; Liu, S. CARD 2020: Antibiotic resistome surveillance with the comprehensive antibiotic resistance database. Nucleic Acids Res. 2020, 48, D517–D525. [Google Scholar] [CrossRef]
- Paulson, J.N.; Colin Stine, O.; Bravo, H.C.; Pop, M. Differential abundance analysis for microbial marker-gene surveys. Nat. Methods 2013, 10, 1200–1202. [Google Scholar] [CrossRef]
- McMurdie, P.J.; Holmes, S. phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef]
- Wickham, H. ggplot2 Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016. [Google Scholar]
- Oksanen, J.; Simpson, G.; Blanchet, G.; Kindt, R.; Legendre, P.; Minchin, P.; O’Hara, R.B.; Solymos, P.; Stevens, M.H.H.; Szoecs, E. Community Ecology Package “Vegan”, Version 2.6-2. 2022. Available online: https://github.com/vegandevs/vegan (accessed on 6 November 2023).
- Anderson, M.J. Permutational Multivariate Analysis of Variance (PERMANOVA). Wiley StatsRef: Stat. Ref Online 2017, 1–15. [Google Scholar] [CrossRef]
- Lin, H.; Peddada, S. Das. Analysis of compositions of microbiomes with bias correction. Nat. Commun. 2020, 11, 3514. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Eggesbø, M.; Das Peddada, S. Linear and nonlinear correlation estimators unveil undescribed taxa interactions in microbiome data. Nat. Commun. 2022, 13, 4946. [Google Scholar] [CrossRef] [PubMed]
- Mikryukov, V. metagMisc: Miscellaneous Functions for Metagenomic Analysis. 2023. Available online: https://github.com/vmikk/metagMisc (accessed on 6 November 2023).
- Booker, C.W.; Abutarbush, S.M.; Morley, P.S.; Jim, G.K.; Pittman, T.J.; Schunicht, O.C.; Perrett, T.; Wildman, B.K.; Fenton, R.K.; Guichon, P.T.; et al. Microbiological and histopathological findings in cases of fatal bovine respiratory disease of feedlot cattle in western Canada. Can. Vet. J. 2008, 49, 473–481. [Google Scholar] [PubMed]
- Fulton, R.W.; Blood, K.S.; Panciera, R.J.; Payton, M.E.; Ridpath, J.F.; Confer, A.W.; Saliki, J.T.; Burge, L.T.; Welsh, R.D.; Johnson, B.J.; et al. Lung Pathology and Infectious Agents in Fatal Feedlot Pneumonias and Relationship with Mortality, Disease Onset, and Treatments. J. Vet. Diagn. Invest. 2009, 21, 464–477. [Google Scholar] [CrossRef] [PubMed]
- Roier, S.; Fenniger, J.C.; Leitner, D.R.; Rechberger, G.N.; Reidl, J.; Schild, S. Immunogenicity of Pasteurella multocida and Mannheimia haemolytica outer membrane vesicles. Int J Med Microbiology 2013, 305, 247–256. [Google Scholar] [CrossRef]
- Wilson, B.A.; Ho, M. Pasteurella multocida: From zoonosis to cellular microbiology. Clin Microbiol Rev 2013, 26, 631–655. [Google Scholar] [CrossRef]
- McMullen, C.; Alexander, T.W.; Orsel, K.; Timsit, E. Progression of nasopharyngeal and tracheal bacterial microbiotas of feedlot cattle during development of bovine respiratory disease. Vet. Microbiol. 2020, 248, 108826. [Google Scholar] [CrossRef]
- McMullen, C.; Orsel, K.; Alexander, T.W.; van der Meer, F.; Plastow, G.; Timsit, E. Comparison of the nasopharyngeal bacterial microbiota of beef calves raised without the use of antimicrobials between healthy calves and those diagnosed with bovine respiratory disease. Vet. Microbiol. 2019, 231, 56–62. [Google Scholar] [CrossRef]
- Li, J.; Mukiibi, R.; Jiminez, J.; Wang, Z.; Akanno, E.C.; Timsit, E.; Plastow, G.S. Applying multi-omics data to study the genetic background of bovine respiratory disease infection in feedlot crossbred cattle. Front. Genet. 2022, 13, 1046192. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antimicrobial | MIC Susceptible | MIC Intermediate | MIC Resistant | Sample 1 BRDM | Sample 2 BRDL | Sample 3 CONT | Sample 4 BRDL | Sample 5 BRDL | Sample 6 CONT | Sample 7 CONT | Sample 8 BRDM | Sample 9 BRDL |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Ampicillin | ≤0.03 | 0.06–0.12 | ≥0.25 | <0.25 | <0.25 | <0.25 | <0.25 | <0.25 | <0.25 | <0.25 | <0.25 | <0.25 |
| Ceftiofur | ≤2 | 4 | ≥8 | <0.25 | <0.25 | <0.25 | <0.25 | <0.25 | <0.25 | <0.25 | <0.25 | <0.25 |
| Chlortetracycline | ≤2 | 4 | ≥8 | 2 | <0.5 | <0.5 | <0.5 | 1 | 4 | 4 | 2 | 2 |
| Danofloxacin | ≤0.25 | 0.5 | ≥1 | <0.12 | <0.12 | <0.12 | <0.12 | <0.12 | <0.12 | 0.25 | <0.12 | <0.12 |
| Enrofloxacin | ≤0.25 | 0.5–1 | ≥2 | <0.12 | <0.12 | <0.12 | <0.12 | <0.12 | <0.12 | <0.12 | <0.12 | <0.12 |
| Florfenicol | ≤2 | 4 | ≥8 | <0.25 | 0.5 | 1 | 1 | 0.5 | 0.5 | 0.5 | 0.5 | 1 |
| Oxytetracycline | ≤2 | 4 | ≥8 | <0.5 | <0.5 | <0.5 | <0.5 | >8 | >8 | >8 | >8 | >8 |
| Penicillin | ≤0.25 | 0.5 | ≥1 | <0.12 | <0.12 | 0.5 | 0.5 | <0.12 | <0.12 | 0.25 | <0.12 | 0.5 |
| Spectinomycin | ≤32 | 64 | ≥128 | <8 | 16 | 32 | 32 | 16 | 16 | 32 | >64 | 32 |
| Tulathromycin | ≤16 | 32 | ≥64 | 2 | <1 | 16 | 16 | <1 | 2 | 16 | <1 | 2 |
| MEGARes Database Accession | Sequenced Samples | |||||
|---|---|---|---|---|---|---|
| 94 CONT | 102 BRDL | 111 BRDL | 112 BRDL | 113 BRDL | 114 BRDL | |
| MEG_2860|Drugs|MLS| 23S_rRNA_methyltransferases|ERMX | 0 | 13/13 | 0 | 0 | 0 | 0 |
| MEG_3977|Drugs|MLS| Macrolide-resistant_23S_rRNA_mutation|MLS23S| RequiresSNPConfirmation | 0 | 0 | 0/341 | 0 | 0 | 0 |
| MEG_3978|Drugs|MLS| Macrolide-resistant_23S_rRNA_mutation MLS23S|RequiresSNPConfirmation | 0 | 0/219 | 0 | 0 | 0/290 | 0/64 |
| MEG_3979|Drugs|MLS| Macrolide-resistant_23S_rRNA_mutation|MLS23S |RequiresSNPConfirmation | 0 | 17/158 | 0 | 0 | 0 | 0 |
| MEG_3983|Drugs|MLS| Macrolide-resistant_23S_rRNA_mutation|MLS23S| RequiresSNPConfirmation | 4/37 | 0 | 0 | 0 | 0 | 0 |
| MEG_6|Drugs|Aminoglycosides| Aminoglycoside-resistant_16S_ribosomal_subunit_protein|A16S| RequiresSNPConfirmation | 0 | 0/319 | 0/401 | 0/36 | 0/63 | 0 |
| MEG_7310|Drugs|Elfamycins|EF-Tu_inhibition|TUFAB|RequiresSNPConfirmation | 0 | 4/55 | 11/102 | 0 | 0 | 0 |
| MEG_8249|Drugs|MLS| Macrolide-resistant_23S_rRNA_mutation|MLS23S| RequiresSNPConfirmation | 0 | 196/196 | 248/248 | 0 | 0 | 0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Neal, K.; Amachawadi, R.G.; White, B.J.; Shippy, T.D.; Theurer, M.E.; Larson, R.L.; Lubbers, B.V.; Kleinhenz, M. Nasopharyngeal Bacterial Prevalence and Microbial Diversity at First Treatment for Bovine Respiratory Disease (BRD) and Its Associations with Health and Mortality Outcomes in Feedyard Cattle. Microorganisms 2024, 12, 33. https://doi.org/10.3390/microorganisms12010033
Neal K, Amachawadi RG, White BJ, Shippy TD, Theurer ME, Larson RL, Lubbers BV, Kleinhenz M. Nasopharyngeal Bacterial Prevalence and Microbial Diversity at First Treatment for Bovine Respiratory Disease (BRD) and Its Associations with Health and Mortality Outcomes in Feedyard Cattle. Microorganisms. 2024; 12(1):33. https://doi.org/10.3390/microorganisms12010033
Chicago/Turabian StyleNeal, Kyndall, Raghavendra G. Amachawadi, Brad J. White, Teresa D. Shippy, Miles E. Theurer, Robert L. Larson, Brian V. Lubbers, and Michael Kleinhenz. 2024. "Nasopharyngeal Bacterial Prevalence and Microbial Diversity at First Treatment for Bovine Respiratory Disease (BRD) and Its Associations with Health and Mortality Outcomes in Feedyard Cattle" Microorganisms 12, no. 1: 33. https://doi.org/10.3390/microorganisms12010033
APA StyleNeal, K., Amachawadi, R. G., White, B. J., Shippy, T. D., Theurer, M. E., Larson, R. L., Lubbers, B. V., & Kleinhenz, M. (2024). Nasopharyngeal Bacterial Prevalence and Microbial Diversity at First Treatment for Bovine Respiratory Disease (BRD) and Its Associations with Health and Mortality Outcomes in Feedyard Cattle. Microorganisms, 12(1), 33. https://doi.org/10.3390/microorganisms12010033