
Microbiota Profiling on Veterinary Faculty Restroom Surfaces and Source Tracking

 , , , and
, , , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Sampled Surfaces
2.2. Total DNA Extraction and Sequencing Library Preparation
2.3. Bioinformatic Analysis
2.3.1. Microbial Profiling
2.3.2. Source Tracker Analysis
- Data collection stored in our Database
- Database design
- Analysis of restroom data
2.4. Statistical and Ecological Analysis
3. Results
3.1. Microbial Profiling
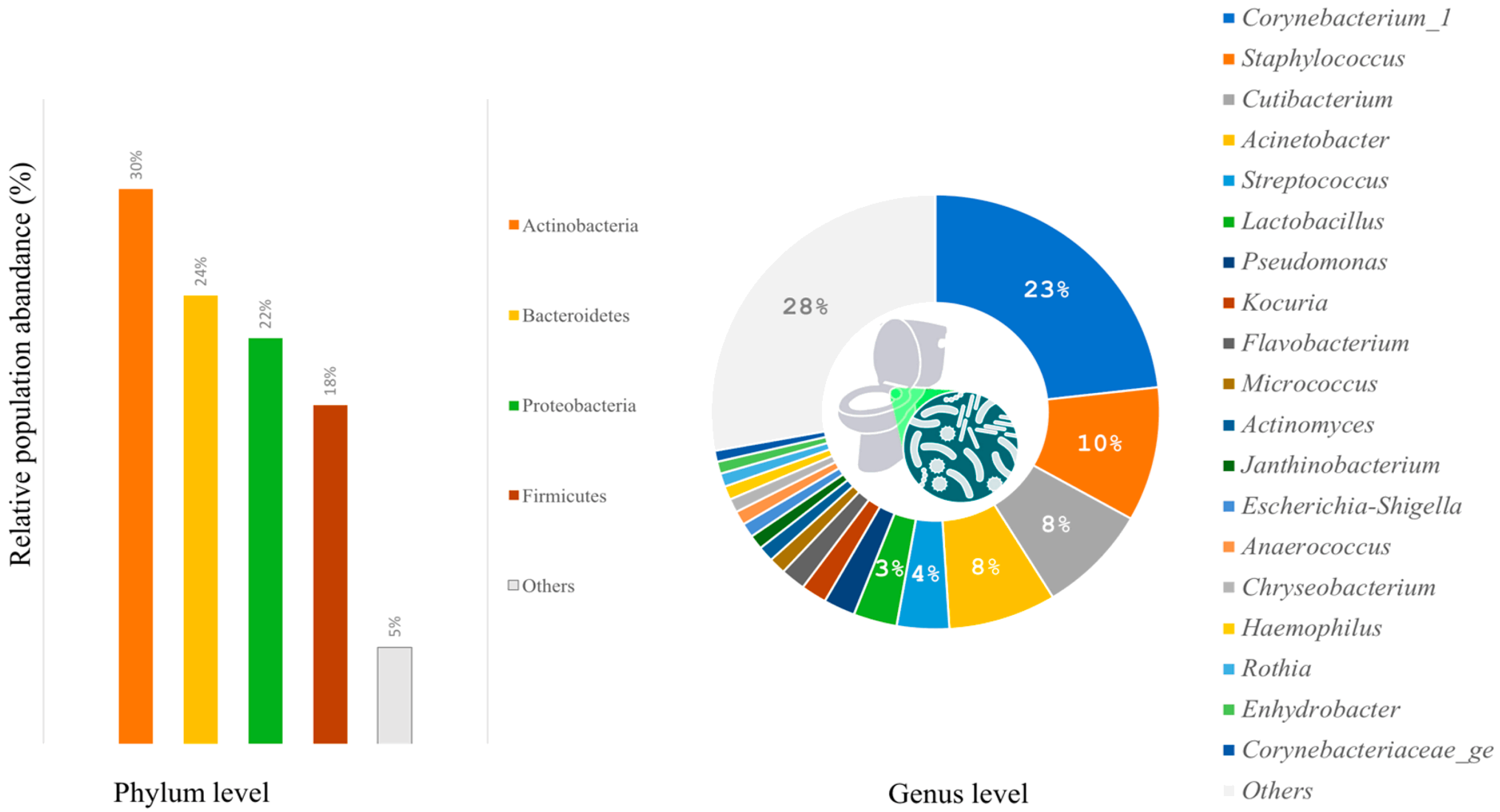
3.1.1. General Characteristics of Microbial Communities
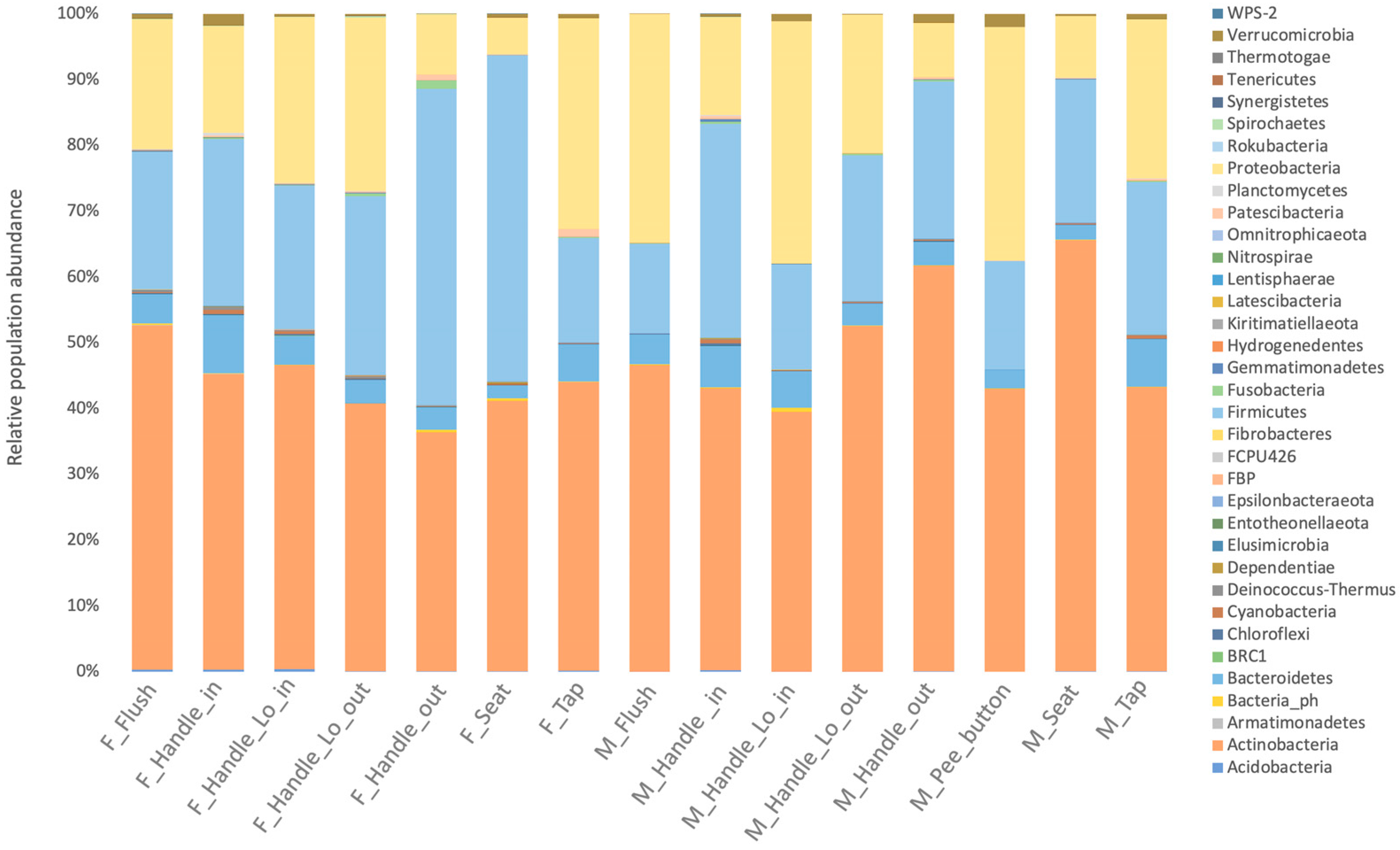
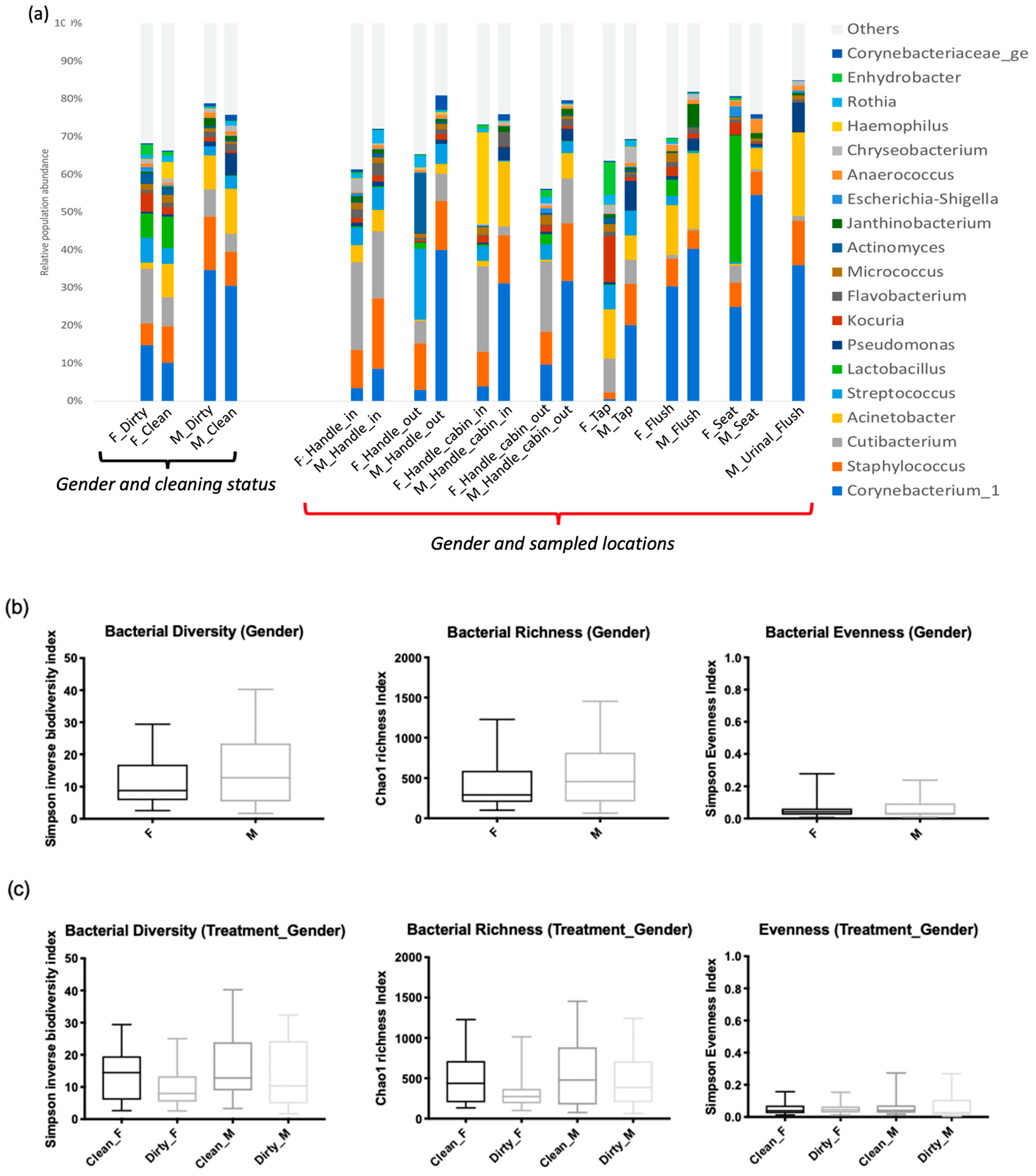
3.1.2. Characterization of Microbial Communities on Different Surfaces of Restrooms
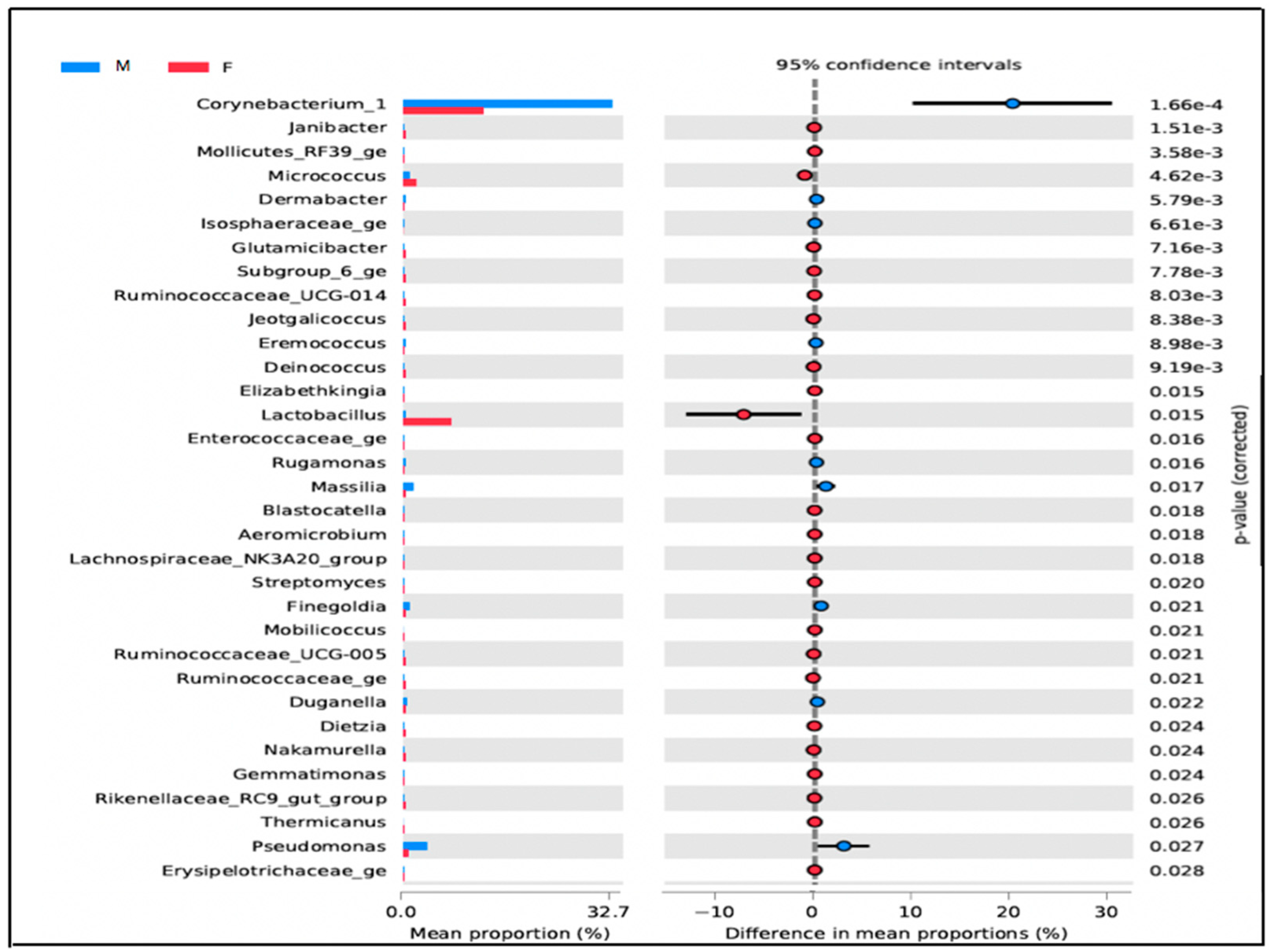
3.1.3. Microbial Diversity between Men’s and Women’s Users
3.1.4. Bacteria Associated with Animals
3.2. Source Tracker Analysis
3.2.1. General Characteristics Sources of the Microbial Community in Restrooms
3.2.2. Bacterial Sources Associated with Animal Hosts
4. Discussion
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Burow, E.; Rostalski, A.; Harlizius, J.; Gangl, A.; Simoneit, C.; Grobbel, M.; Kollas, C.; Tenhagen, B.-A.; Käsbohrer, A. Antibiotic resistance in Escherichia coli from pigs from birth to slaughter and its association with antibiotic treatment. Prev. Vet. Med. 2019, 165, 52–62. [Google Scholar] [CrossRef]
- van Duijkeren, E.; Box, A.; Heck, M.; Wannet, W.; Fluit, A. Methicillin-resistant staphylococci isolated from animals. Vet. Microbiol. 2004, 103, 91–97. [Google Scholar] [CrossRef]
- Seguin, J.C.; Walker, R.D.; Caron, J.P.; Kloos, W.E.; George, C.G.; Hollis, R.J.; Jones, R.N.; Pfaller, M.A. Methicillin-Resistant Staphylococcus aureus Outbreak in a Veterinary Teaching Hospital: Potential Human-to-Animal Transmission. J. Clin. Microbiol. 1999, 37, 1459–1463. [Google Scholar] [CrossRef] [PubMed]
- Juhász-Kaszanyitzky, É.; Jánosi, S.; Somogyi, P.; Dán, A.; Bloois, L.V.D.G.-V.; Van Duijkeren, E.; Wagenaar, J.A. MRSA Transmission between Cows and Humans. Emerg. Infect. Dis. 2007, 13, 630–632. [Google Scholar] [CrossRef]
- Bojanova, D.P.; Bordenstein, S.R. Fecal Transplants: What Is Being Transferred? PLoS Biol. 2016, 14, e1002503. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, K.; Senay, C.; Young, S.; Nayak, B.; Lobos, A.; Conrad, J.; Harwood, V. Determination of wild animal sources of fecal indicator bacteria by microbial source tracking (MST) influences regulatory decisions. Water Res. 2018, 144, 424–434. [Google Scholar] [CrossRef] [PubMed]
- Knights, D.; Kuczynski, J.; Charlson, E.S.; Zaneveld, J.; Mozer, M.C.; Collman, R.G.; Bushman, F.D.; Knight, R.T.; Kelley, S.T. Bayesian community-wide culture-independent microbial source tracking. Nat. Methods 2011, 8, 761–763. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA Ribosomal RNA Gene Database Project: Improved Data Processing and Web-Based Tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef]
- Christenfeld, N. Choices from Identical Options. Psychol. Sci. 1995, 6, 50–55. [Google Scholar] [CrossRef]
- Ceugniez, A.; Taminiau, B.; Coucheney, F.; Jacques, P.; Delcenserie, V.; Daube, G.; Drider, D. Use of a metagenetic approach to monitor the bacterial microbiota of “Tomme d’Orchies” cheese during the ripening process. Int. J. Food Microbiol. 2017, 247, 65–69. [Google Scholar] [CrossRef]
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahé, F. VSEARCH: A versatile open source tool for metagenomics. PeerJ 2016, 2016, e2584. [Google Scholar] [CrossRef]
- Pruesse, E.; Quast, C.; Knittel, K.; Fuchs, B.M.; Ludwig, W.; Peplies, J.; Glöckner, F.O. SILVA: A comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res. 2007, 35, 7188–7196. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, C.; Taminiau, B.; Brévers, B.; Avesani, V.; Van Broeck, J.; Leroux, A.; Gallot, M.; Bruwier, A.; Amory, H.; Delmée, M.; et al. Faecal microbiota characterisation of horses using 16 rdna barcoded pyrosequencing, and carriage rate of clostridium difficile at hospital admission. BMC Microbiol. 2015, 15, 181. [Google Scholar] [CrossRef]
- Elmasri, R.; Navathe, S.B. Database Systems, 6th ed.; Hirsch, M., Ed.; Addison-Wesley: Melbourne, FL, USA, 2017. [Google Scholar]
- Chao, A.; Bunge, J. Estimating the Number of Species in a Stochastic Abundance Model. Biometrics 2002, 58, 531–539. [Google Scholar] [CrossRef]
- Chao, A.; Shen, T.-J. Nonparametric estimation of Shannon’s index of diversity when there are unseen species in sample. Environ. Ecol. Stat. 2003, 10, 429–443. [Google Scholar] [CrossRef]
- Simpson, E.H. Measurement of diversity. Nature 1949, 163, 668. [Google Scholar] [CrossRef]
- Clarke, K.; Ainsworth, M. A method of linking multivariate community structure to environmental variables. Mar. Ecol. Prog. Ser. 1993, 92, 205–219. [Google Scholar] [CrossRef]
- Zanne, A.E.; Tank, D.C.; Cornwell, W.K.; Eastman, J.M.; Smith, S.A.; FitzJohn, R.G.; McGlinn, D.J.; O’Meara, B.C.; Moles, A.T.; Reich, P.B.; et al. Data from: Three Keys to the Radiation of Angiosperms into Freezing Environments. Nature 2014, 506, 89–92. [Google Scholar] [CrossRef] [PubMed]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing mothur: Open-Source, Platform-Independent, Community-Supported Software for Describing and Comparing Microbial Communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef]
- Schloss, P.D. Evaluating different approaches that test whether microbial communities have the same structure. ISME J. 2008, 2, 265–275. [Google Scholar] [CrossRef]
- Parks, D.H.; Tyson, G.W.; Hugenholtz, P.; Beiko, R.G. STAMP User’s Guide v2.0.0. Bioinformatics 2014, 30, 3123–3124. [Google Scholar] [CrossRef] [PubMed]
- Grice, E.A.; Segre, J.A. The skin microbiome. Nat. Rev. Microbiol. 2011, 9, 244–253. [Google Scholar] [CrossRef]
- Nematian, J.; Matini, E.; Shayeghi, F.; Vaghar, M.; Hosseini, S.S.; Mojri, N.; Taherabadi, N.T.; Hakimi, R.; Ahmadi, N.; Badkoubeh, N.; et al. A survey of public restrooms microbial contamination in Tehran city, capital of Iran, during 2019. J. Fam. Med. Prim. Care 2020, 9, 3131–3135. [Google Scholar] [CrossRef] [PubMed]
- Gizaw, Z.; Yalew, A.W.; Bitew, B.D.; Lee, J.; Bisesi, M. Effects of local handwashing agents on microbial contamination of the hands in a rural setting in Northwest Ethiopia: A cluster randomised controlled trial. BMJ Open 2022, 12, e056411. [Google Scholar] [CrossRef] [PubMed]
- Henschel, A.; Anwar, M.Z.; Manohar, V. Comprehensive Meta-analysis of Ontology Annotated 16S rRNA Profiles Identifies Beta Diversity Clusters of Environmental Bacterial Communities. PLoS Comput. Biol. 2015, 11, e1004468. [Google Scholar] [CrossRef]
- Flores, G.E.; Bates, S.T.; Knights, D.; Lauber, C.L.; Stombaugh, J.; Knight, R.; Fierer, N. Microbial Biogeography of Public Restroom Surfaces. PLoS ONE 2011, 6, e28132. [Google Scholar] [CrossRef]
- Pausan, M.-R.; Blohs, M.; Mahnert, A.; Moissl-Eichinger, C. The sanitary indoor environment—A potential source for intact human-associated anaerobes. NPJ Biofilms Microbiomes 2022, 8, 44. [Google Scholar] [CrossRef]
- Dobbler, P.C.T.; Laureano, M.; Sarzi, D.S.; Cañón, E.R.P.; Metz, G.F.; de Freitas, A.S.; Takagaki, B.M.; D’oliveira, C.B.; Pylro, V.S.; Copetti, A.C.; et al. Differences in bacterial composition between men’s and women’s restrooms and other common areas within a public building. Antonie Leeuwenhoek Int. J. Gen. Mol. Microbiol. 2018, 111, 551–561. [Google Scholar] [CrossRef]
- Roy, M.; Ahmad, S. Rare case of Corynebacterium striatum septic arthritis. BMJ Case Rep. 2016, 2016, bcr2016216914. [Google Scholar] [CrossRef]
- Concha, C.-D.; Maestre, F.T.; Eldridge, D.J.; Singh, B.K.; Bardgett, R.D.; Fierer, N.; Delgado-Baquerizo, M. Ecological Niche Differentiation in Soil Cyanobacterial Communities across the Globe. BioRxiv 2019, 15, 13–23. [Google Scholar] [CrossRef]
- Gibbons, S.M.; Schwartz, T.; Fouquier, J.; Mitchell, M.; Sangwan, N.; Gilbert, J.A.; Kelley, S.T. Ecological Succession and Viability of Human-Associated Microbiota on Restroom Surfaces. Appl. Environ. Microbiol. 2015, 81, 765–773. [Google Scholar] [CrossRef]
- Boone, S.A.; Gerba, C.P. The Prevalence of Human Parainfluenza Virus 1 on Indoor Office Fomites. Food Environ. Virol. 2010, 2, 41–46. [Google Scholar] [CrossRef]
- Bifulco, J.M.; Shirey, J.J.; Bissonnette, G.K. Detection of Acinetobacter spp. in rural drinking water supplies. Appl. Environ. Microbiol. 1989, 55, 2214–2219. [Google Scholar] [CrossRef] [PubMed]
- Hitzfeld, B.C.; Höger, S.J.; Dietrich, D.R. Cyanobacterial toxins: Removal during drinking water treatment, and human risk assessment. Environ. Healrh Perspect. J. 2000, 108, 113–122. [Google Scholar] [CrossRef]
- Roberts, M. Characterization and Isolation of Fecal Indicator Bacteria, Staphylococcus aureus, and Methicillin-resistant Staphylococcus aureus from Pacific Northwest Marine Beach Samples. Environ. Health 2012, 78, 50–56. [Google Scholar] [CrossRef]
- Drees, M.; Snydman, D.R.; Schmid, C.H.; Barefoot, L.; Hansjosten, K.; Vue, P.M.; Cronin, M.; Nasraway, S.A.; Golan, Y. Prior Environmental Contamination Increases the Risk of Acquisition of Vancomycin-Resistant Enterococci. Clin. Infect. Dis. 2008, 46, 678–685. [Google Scholar] [CrossRef] [PubMed]
- O’neill, A.M.; Gallo, R.L. Host-microbiome interactions and recent progress into understanding the biology of acne vulgaris. Microbiome 2018, 6, 177. [Google Scholar] [CrossRef] [PubMed]
- Fouts, D.E.; Pieper, R.; Szpakowski, S.; Pohl, H.; Knoblach, S.; Suh, M.-J.; Huang, S.-T.; Ljungberg, I.; Sprague, B.M.; Lucas, S.K.; et al. Integrated next-generation sequencing of 16S rDNA and metaproteomics differentiate the healthy urine microbiome from asymptomatic bacteriuria in neuropathic bladder associated with spinal cord injury. J. Transl. Med. 2012, 10, 174. [Google Scholar] [CrossRef]
- Mueller, S.; Saunier, K.; Hanisch, C.; Norin, E.; Alm, L.; Midtvedt, T.; Cresci, A.; Silvi, S.; Orpianesi, C.; Verdenelli, M.C.; et al. Differences in Fecal Microbiota in Different European Study Populations in Relation to Age, Gender, and Country: A Cross-Sectional Study. Appl. Environ. Microbiol. 2006, 72, 1027–1033. [Google Scholar] [CrossRef]
- Li, M.; Wang, B.; Zhang, M.; Rantalainen, M.; Wang, S.; Zhou, H.; Zhang, Y.; Shen, J.; Pang, X.; Zhang, M.; et al. Symbiotic gut microbes modulate human metabolic phenotypes. Proc. Natl. Acad. Sci. USA 2008, 105, 2117–2122. [Google Scholar] [CrossRef]
- Dominianni, C.; Sinha, R.; Goedert, J.J.; Pei, Z.; Yang, L.; Hayes, R.B.; Ahn, J. Sex, Body Mass Index, and Dietary Fiber Intake Influence the Human Gut Microbiome. PLoS ONE 2015, 10, e0124599. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.; Manning, S.D. Impact of age and sex on the composition and abundance of the intestinal microbiota in individuals with and without enteric infections. Ann. Epidemiol. 2016, 26, 380–385. [Google Scholar] [CrossRef] [PubMed]
- Haro, C.; Rangel-Zúñiga, O.A.; Alcalá-Díaz, J.F.; Gómez-Delgado, F.; Pérez-Martínez, P.; Delgado-Lista, J.; Quintana-Navarro, G.M.; Landa, B.B.; Navas-Cortés, J.A.; Tena-Sempere, M.; et al. Intestinal Microbiota Is Influenced by Gender and Body Mass Index. PLoS ONE 2016, 11, e0154090. [Google Scholar] [CrossRef] [PubMed]
- Borgo, F.; Garbossa, S.; Riva, A.; Severgnini, M.; Luigiano, C.; Benetti, A.; Pontiroli, A.E.; Morace, G.; Borghi, E. Body Mass Index and Sex Affect Diverse Microbial Niches within the Gut. Front. Microbiol. 2018, 9, 213. [Google Scholar] [CrossRef]
- Gao, X.; Zhang, M.; Xue, J.; Huang, J.; Zhuang, R.; Zhou, X.; Zhang, H.; Fu, Q.; Hao, Y. Body Mass Index Differences in the Gut Microbiota Are Gender Specific. Front. Microbiol. 2018, 9, 1250. [Google Scholar] [CrossRef] [PubMed]
- Takagi, T.; Naito, Y.; Inoue, R.; Kashiwagi, S.; Uchiyama, K.; Mizushima, K.; Tsuchiya, S.; Dohi, O.; Yoshida, N.; Kamada, K.; et al. Differences in gut microbiota associated with age, sex, and stool consistency in healthy Japanese subjects. J. Gastroenterol. 2019, 54, 53–63. [Google Scholar] [CrossRef]
- Lepargneur, J.-P. Lactobacillus crispatus as biomarker of the healthy vaginal tract. Ann. Biol. Clin. 2016, 74, 421–427. [Google Scholar] [CrossRef]
- You, Y.; Kwon, E.J.; Choi, S.; Hwang, H.; Choi, S.; Lee, S.M.; Kim, Y.J. Vaginal microbiome profiles of pregnant women in Korea using a 16S metagenomics approach. Am. J. Reprod. Immunol. 2019, 82, e13124. [Google Scholar] [CrossRef]
- Josephs-Spaulding, J.; Krogh, T.J.; Rettig, H.C.; Lyng, M.; Chkonia, M.; Waschina, S.; Graspeuntner, S.; Rupp, J.; Møller-Jensen, J.; Kaleta, C. Recurrent Urinary Tract Infections: Unraveling the Complicated Environment of Uncomplicated rUTIs. Front. Cell. Infect. Microbiol. 2021, 11, 562525. [Google Scholar] [CrossRef]
- Claassen-Weitz, S.; Shittu, A.O.; Ngwarai, M.R.; Thabane, L.; Nicol, M.P.; Kaba, M. Fecal Carriage of Staphylococcus aureus in the Hospital and Community Setting: A Systematic Review. Front. Microbiol. 2016, 7, 449. [Google Scholar] [CrossRef]
- Ogba, O.M.; Obio, O.M. Microbial Spectrum on Public Toilet Seats. Ann. Microbiol. Infect. Dis. 2018, 1, 58–62. [Google Scholar]
- Sherman, H.M.H.J.M.; Eqzuinus, S. Streptococcus equinus. J. Bacteriol. 1936, 1910, 283–289. [Google Scholar] [CrossRef]
- O’Sullivan, T.; Friendship, R.; Blackwell, T.; Pearl, D.; McEwen, B.; Carman, S.; Slavić, D.; Dewey, C. Microbiological identification and analysis of swine tonsils collected from carcasses at slaughter. Can. J. Vet. Res. 2011, 75, 106–111. [Google Scholar]
- Lebreton, F.; Willems, R.J.L.; Gilmore, M.S. Enterococcus Diversity, Origins in Nature, and Gut Colonization. In Enterococci: From Commensals to Leading Causes of Drug Resistant Infection; Gilmore, M.S., Clewell, D.B., Ike, Y., Eds.; Massachusetts Eye and Ear Infirmary: Boston, MA, USA, 2014. Available online: https://www.ncbi.nlm.nih.gov/books/NBK190427 (accessed on 2 February 2014).
- Deusch, S.; Camarinha-Silva, A.; Conrad, J.; Beifuss, U.; Rodehutscord, M.; Seifert, J. A Structural and Functional Elucidation of the Rumen Microbiome Influenced by Various Diets and Microenvironments. Front. Microbiol. 2017, 8, 1605. [Google Scholar] [CrossRef]
- Moriconi, M.; Acke, E.; Petrelli, D.; Preziuso, S. Multiplex PCR-based identification of Streptococcus canis, Streptococcus zooepidemicus and Streptococcus dysgalactiae subspecies from dogs. Comp. Immunol. Microbiol. Infect. Dis. 2017, 50, 48–53. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | Gender 1 | Surface Type 2 | Cleaning Status 3 |
|---|---|---|---|
| F_Handle_in_Clean | F (Female) | Handle_in | Clean |
| F_Handle_out_Clean | F (Female) | Handle_out | Clean |
| F_Handle_Cabin_in_Clean | F (Female) | Handle_Cabin_in | Clean |
| F_Handle_Cabin_out_Clean | F (Female) | Handle_Cabin_out | Clean |
| F_Tap_Clean | F (Female) | Tap | Clean |
| M_Handle_in_Clean | M (Male) | Handle_in | Clean |
| M_Handle_out_Clean | M (Male) | Hanlde_out | Clean |
| M_Handle_Cabin_in_Clean | M (Male) | Handle_Cabin_in | Clean |
| M_Handle_Cabin_out_Clean | M (Male) | Handle_Cabin_out | Clean |
| M_Tap_Clean | M (Male) | Tap | Clean |
| F_Flush_Clean | F (Female) | Flush | Clean |
| M_Flush_Clean | M (Male) | Flush | Clean |
| M_Flush_M_Clean | M (Male) | Urinal_Flush | Clean |
| F_Seat_Clean | F (Female) | Seat | Clean |
| M_Seat_Clean | M (Male) | Seat | Clean |
| F_Handle_in_Dirty | F (Female) | Handle_in | Dirty |
| F_Handle_out_Dirty | F (Female) | Handle_out | Dirty |
| F_Handle_Cabin_in_Dirty | F (Female) | Handle_Cabin_in | Dirty |
| F_Handle_Cabin_out_Dirty | F (Female) | Handle_Cabin_out | Dirty |
| F_Tap_Dirty | F (Female) | Tap | Dirty |
| M_Handle_in_Dirty | M (Male) | Handle_in | Dirty |
| M_Handle_out_Dirty | M (Male) | Hanlde_out | Dirty |
| M_Handle_Cabin_in_Dirty | M (Male) | Handle_Cabin_in | Dirty |
| M_Handle_Cabin_out_Dirty | M (Male) | Handle_Cabin_out | Dirty |
| M_Tap_Dirty | M (Male) | Tap | Dirty |
| F_Flush_Dirty | F (Female) | Flush | Dirty |
| M_Flush_Dirty | M (Male) | Flush | Dirty |
| M_Flush_M_Dirty | M (Male) | Urinal_Flush | Dirty |
| F_Seat_Dirty | F (Female) | Seat | Dirty |
| M_Seat_Dirty | M (Male) | Seat | Dirty |
| Family | Species | Host-Sources in Bibliography Research (SNT) |
|---|---|---|
| Bacteroidaceae | Bacteroides dore | ANIMAL and HUMAN |
| Bacteroides pyogenes | ANIMAL | |
| Bifidobacteriaceae | Bifidobacterium merycicum | ANIMAL |
| Bifidobacterium pseudolongum | ANIMAL | |
| Clostridiaceae | Clostridium algidicarnis | ANIMAL |
| Clostridium frigidicarnis | ANIMAL | |
| Clostridium novyi | ANIMAL | |
| Clostridium ruminantium | ANIMAL | |
| Clostridium septicum | ANIMAL | |
| Enterococcaceae | Enterococcus cecorum | ANIMAL |
| Enterobacteriaceae | Escherichia coli | ANIMAL and HUMAN |
| Erysipelotrichaceae | Faecalicoccus pleomorphus | ANIMAL |
| Streptococcaceae | Streptococcus equinus | ANIMAL |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jabri, H.; Krings, S.; Fall, P.A.; Baurain, D.; Daube, G.; Taminiau, B. Microbiota Profiling on Veterinary Faculty Restroom Surfaces and Source Tracking. Microorganisms 2023, 11, 2053. https://doi.org/10.3390/microorganisms11082053
Jabri H, Krings S, Fall PA, Baurain D, Daube G, Taminiau B. Microbiota Profiling on Veterinary Faculty Restroom Surfaces and Source Tracking. Microorganisms. 2023; 11(8):2053. https://doi.org/10.3390/microorganisms11082053
Chicago/Turabian StyleJabri, Hiba, Simone Krings, Papa Abdoulaye Fall, Denis Baurain, Georges Daube, and Bernard Taminiau. 2023. "Microbiota Profiling on Veterinary Faculty Restroom Surfaces and Source Tracking" Microorganisms 11, no. 8: 2053. https://doi.org/10.3390/microorganisms11082053
APA StyleJabri, H., Krings, S., Fall, P. A., Baurain, D., Daube, G., & Taminiau, B. (2023). Microbiota Profiling on Veterinary Faculty Restroom Surfaces and Source Tracking. Microorganisms, 11(8), 2053. https://doi.org/10.3390/microorganisms11082053