
Could the Urease of the Gut Bacterium Proteus mirabilis Play a Role in the Altered Gut–Brain Talk Associated with Parkinson’s Disease?

 ,
,  , , and
, , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Purifications
2.1.1. Holoenzyme–PMU
2.1.2. PMU B Subunit–PmUreβ
2.1.3. HPU
2.2. Protein Determination and Sterilization
2.3. In Vivo Assays
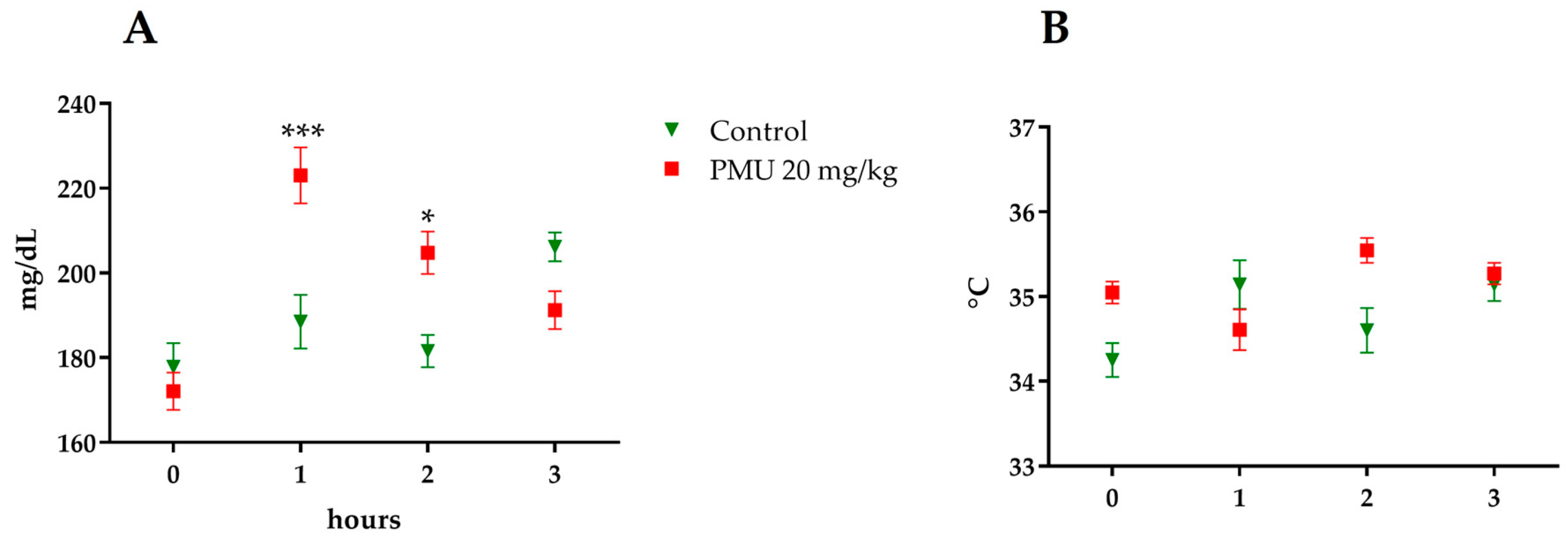
2.3.1. Acute Effects and Toxicity Monitoring
2.3.2. Treatments and Behavioral Analysis
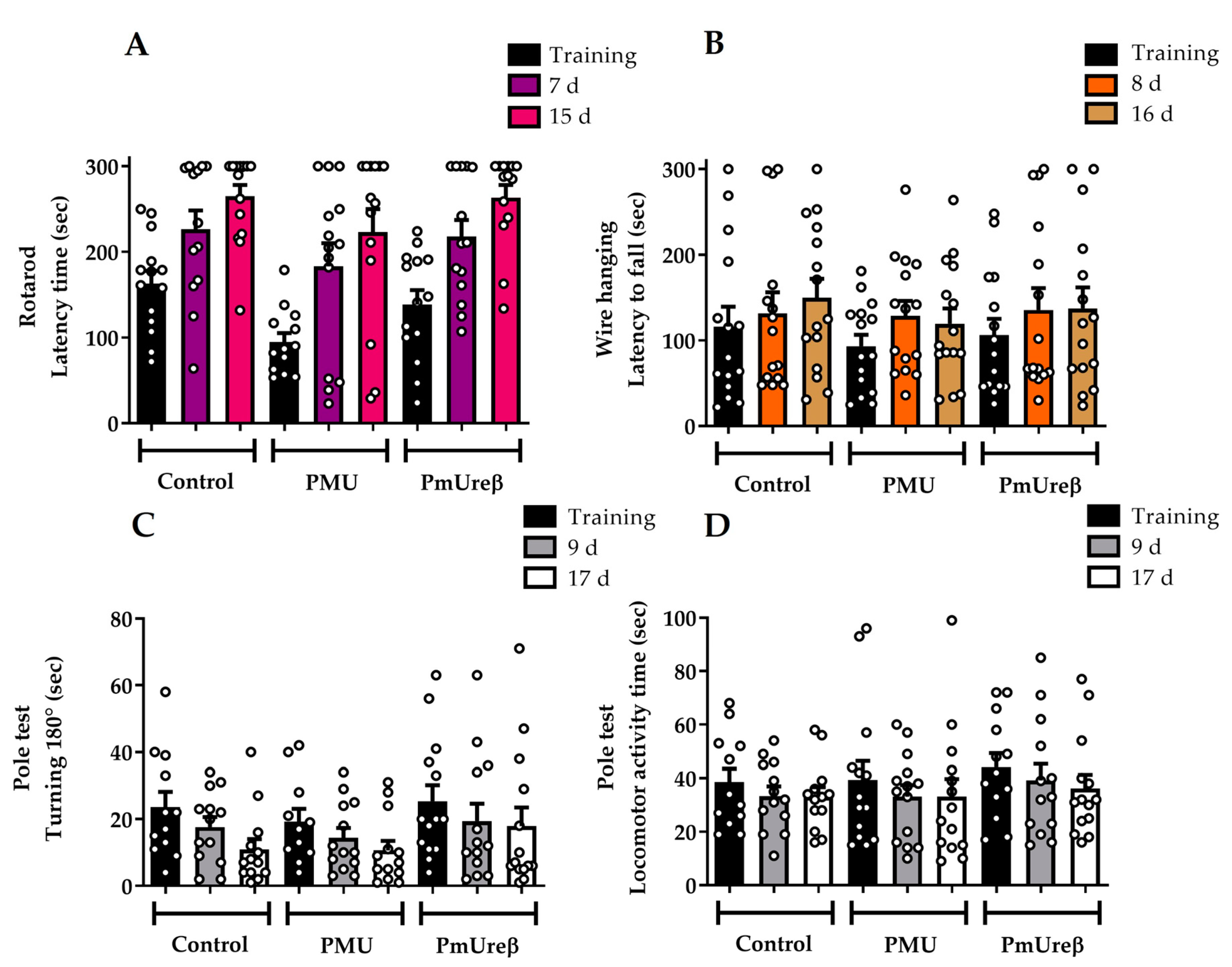
2.3.3. Motor Tests
Rotarod
Wire Hanging Box
Pole Test
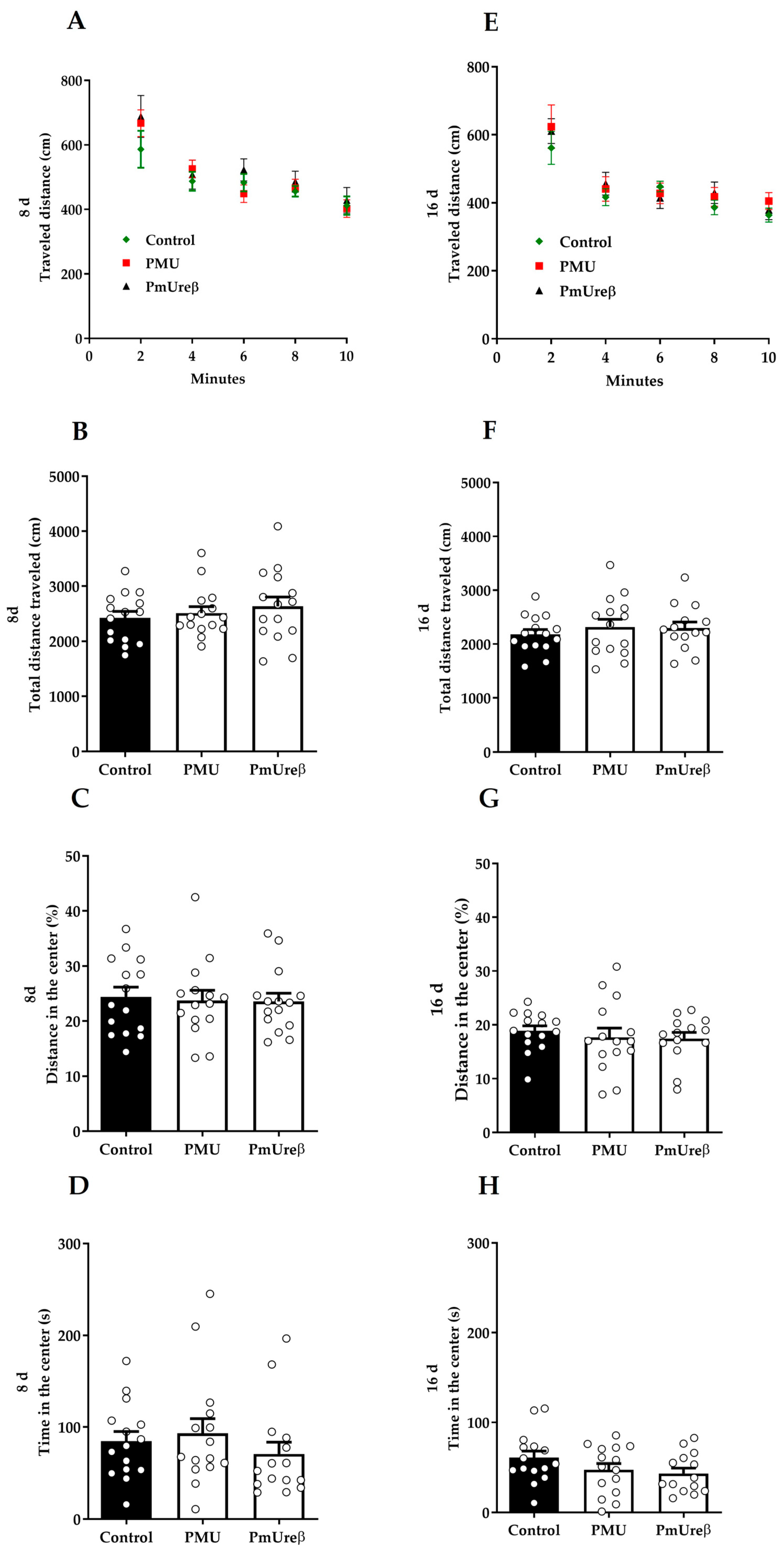
2.3.4. Open Field
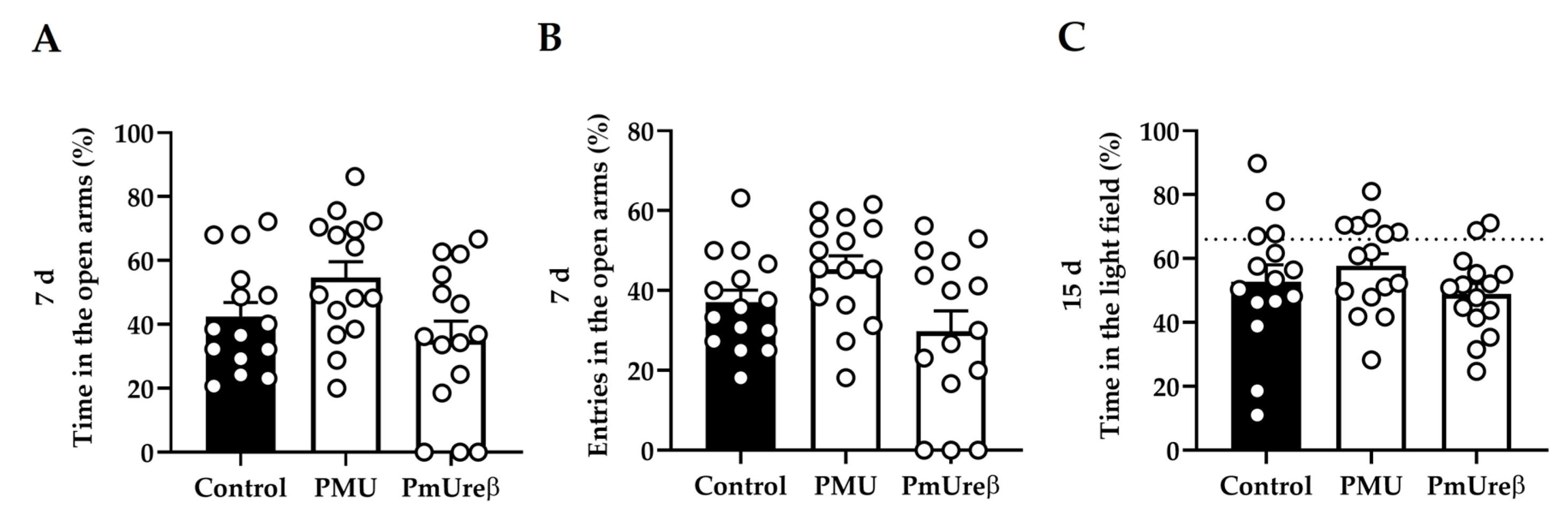
2.3.5. Anxiety Tests
Elevated Plus Maze
Dark–Light Box
2.3.6. Tail Suspension Test
2.4. Integrity of the Blood–Brain Barrier
2.5. Western Blot
2.5.1. Preparation of Brain Homogenates
2.5.2. Western Blot Analyses
2.6. Cytokine Detection
2.7. Permeability of Cell Junction
2.8. Fibrillation of α-Synuclein
2.9. Statistical Analysis
3. Results
3.1. Acute In Vivo Effects of PMU
3.2. Effects in Mice of 7-Day Treatment with Purified PMU or PmUreβ
3.2.1. Motor Tests
3.2.2. Open Field Test
3.2.3. Anxiety Tests: Elevated Plus Maze and Dark–Light Box
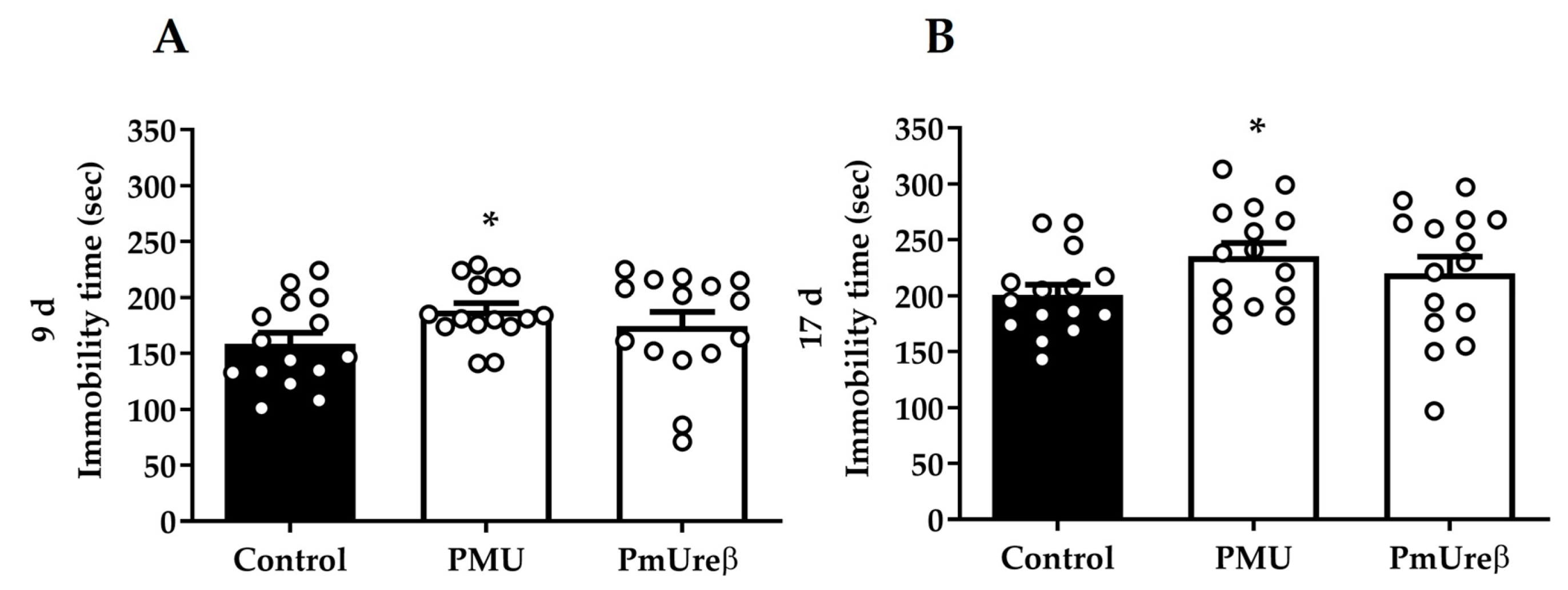
3.2.4. Tail Suspension Test
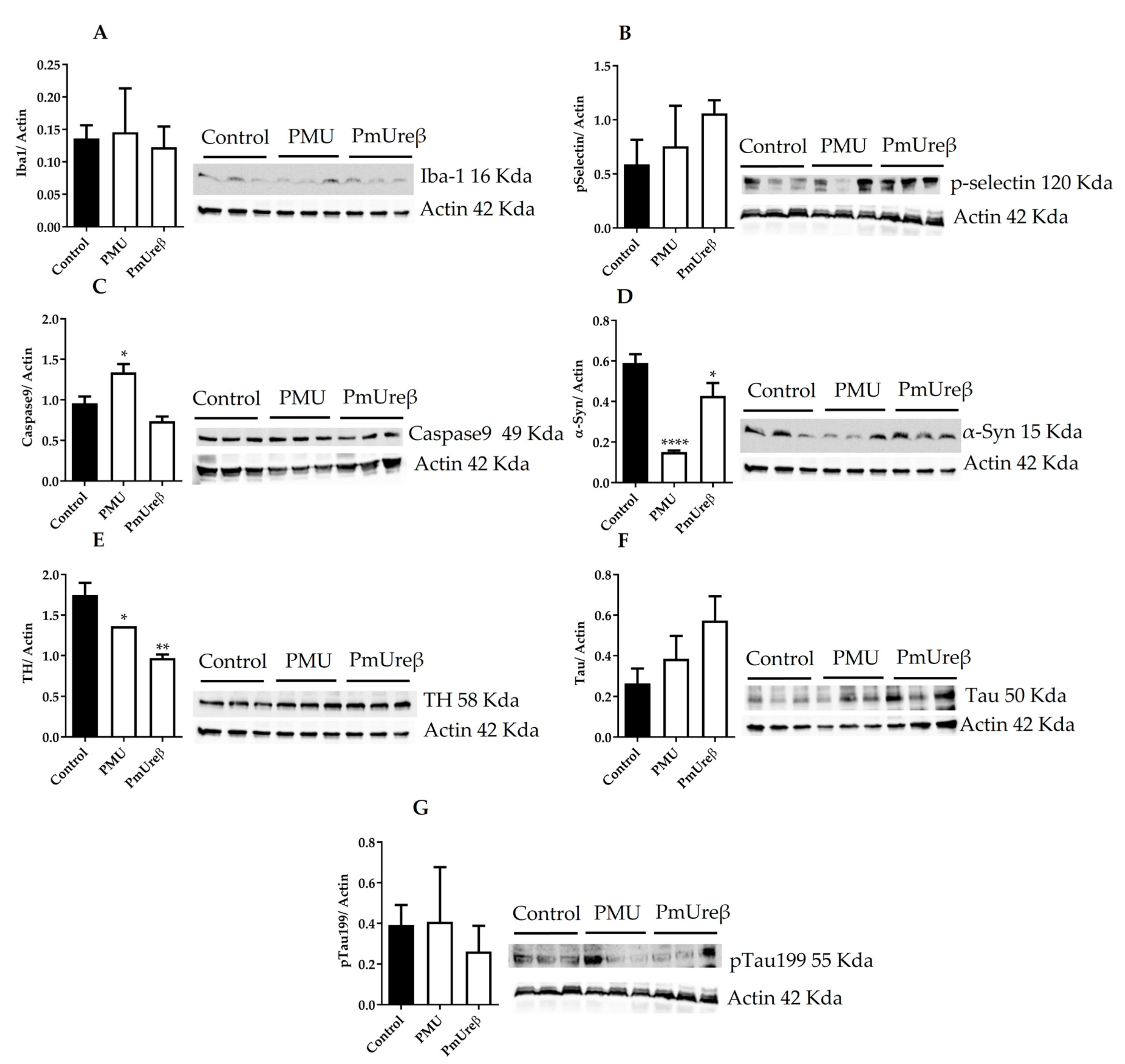
3.3. Western Blot Analyses
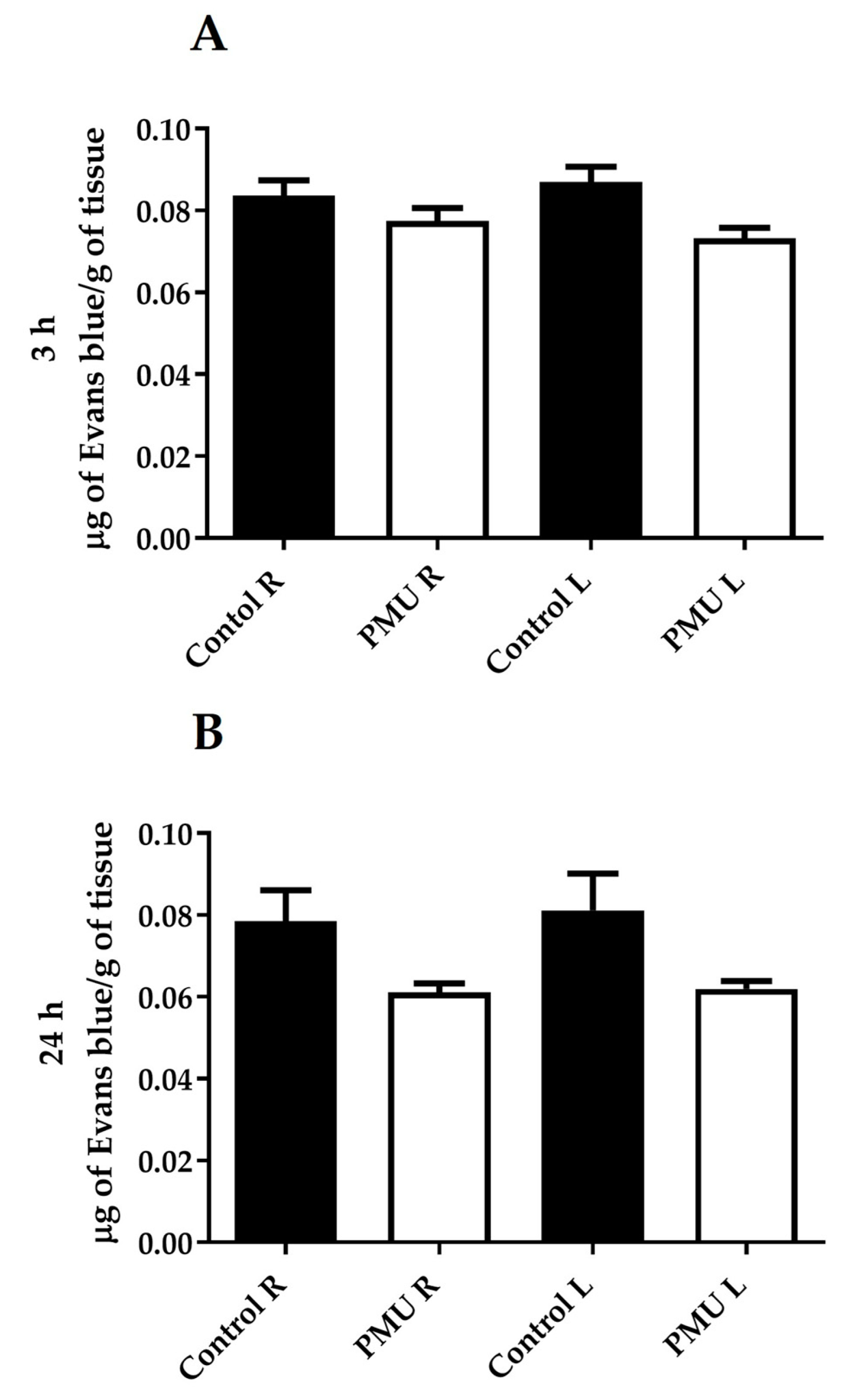
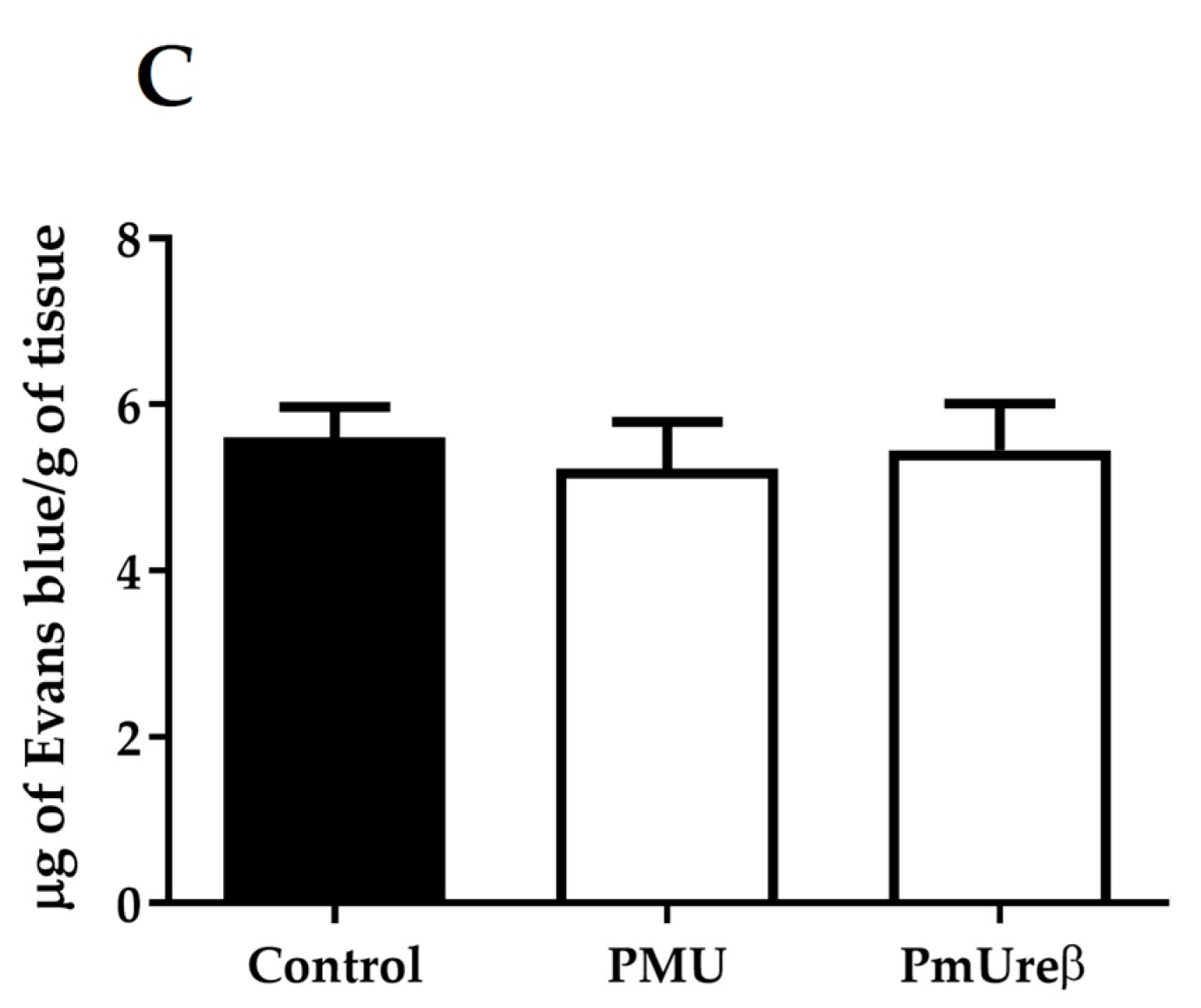
3.4. Integrity of the Blood–Brain Barrier
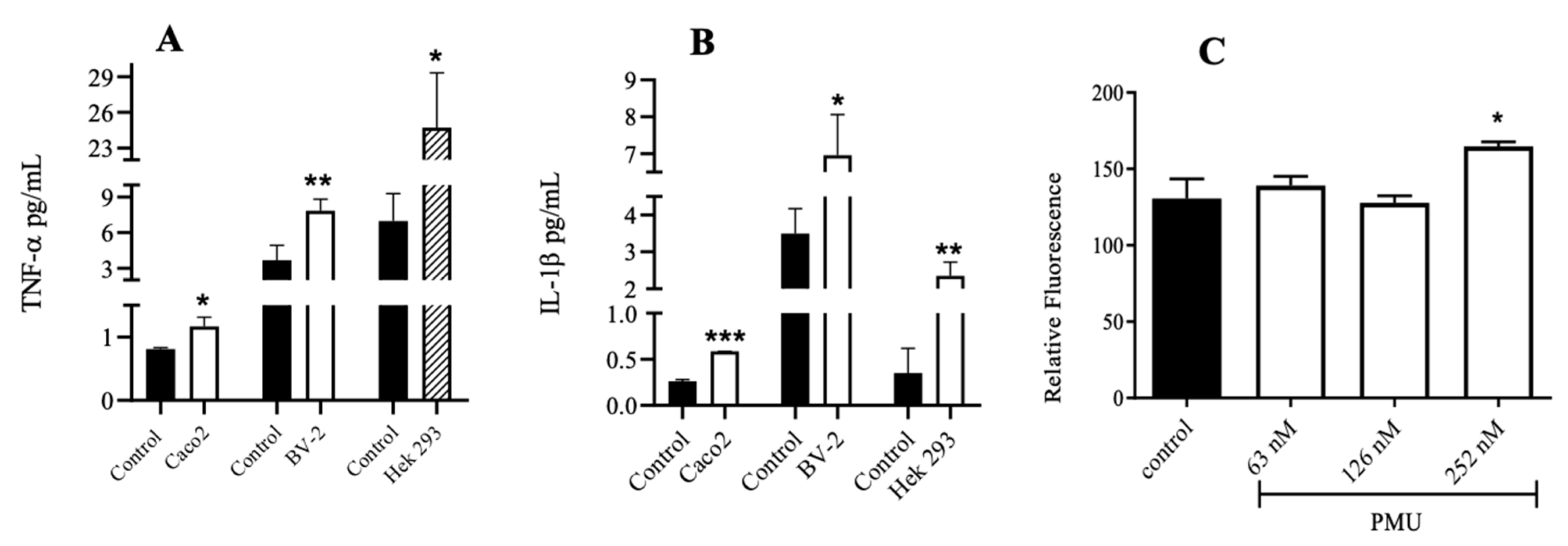
3.5. Effects of PMU in Cell Cultures
3.6. Paracellular Permeability of Cell Monolayers
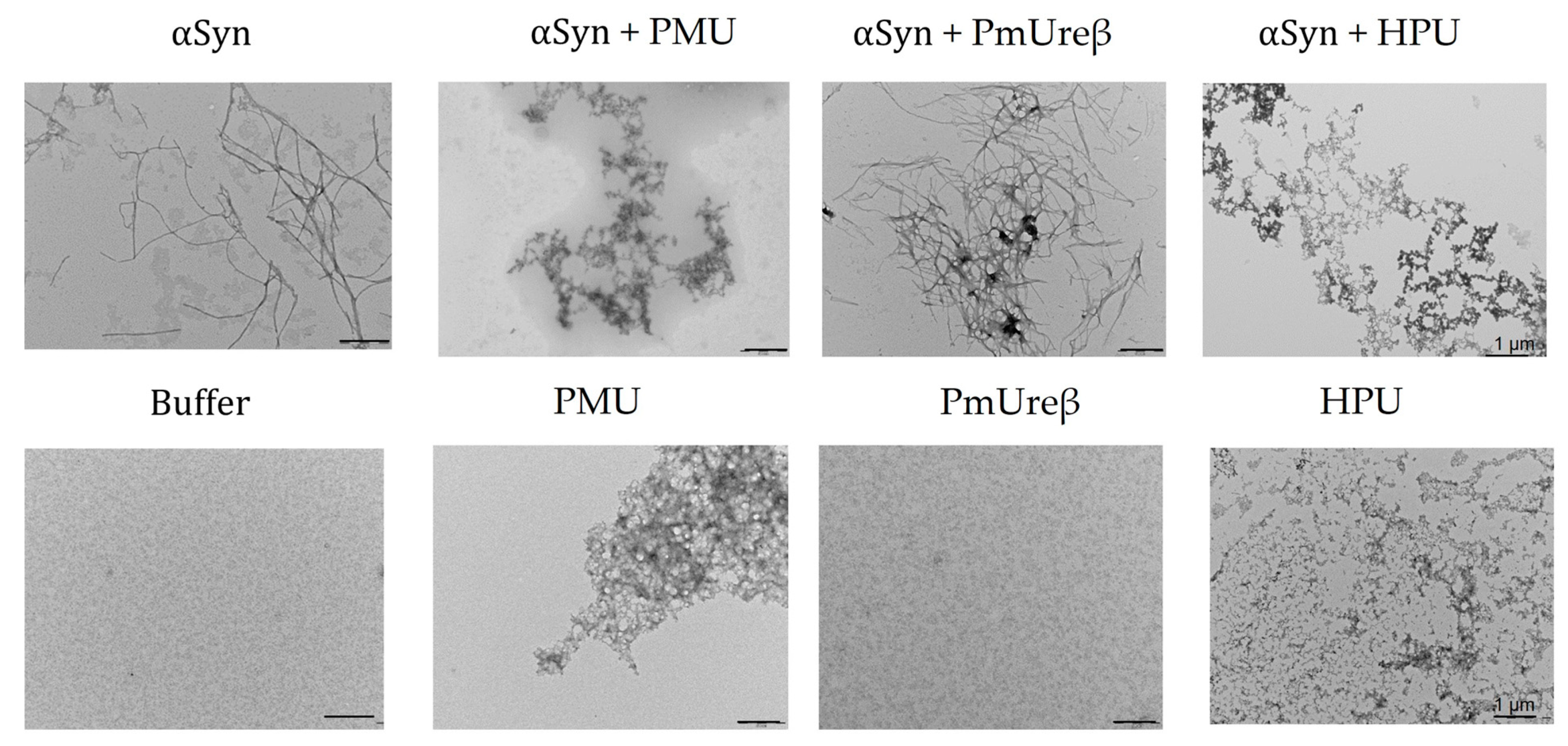
3.7. Morphology of α-Synuclein Aggregates Formed in the Presence of Ureases
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Carlini, C.R.; Ligabue-Braun, R. Ureases as Multifunctional Toxic Proteins: A Review. Toxicon 2016, 110, 90–109. [Google Scholar] [CrossRef] [PubMed]
- Kappaun, K.; Piovesan, A.R.; Carlini, C.R.; Ligabue-Braun, R. Ureases: Historical Aspects, Catalytic, and Non-Catalytic Properties—A Review. J. Adv. Res. 2018, 13, 3–17. [Google Scholar] [CrossRef] [PubMed]
- Drzewiecka, D. Significance and Roles of Proteus Spp. Bacteria in Natural Environments. Microb. Ecol. 2016, 72, 741–758. [Google Scholar] [CrossRef]
- Jones, B.D.; Mobley, H.L. Proteus Mirabilis Urease: Genetic Organization, Regulation, and Expression of Structural Genes. J. Bacteriol. 1988, 170, 3342–3349. [Google Scholar] [CrossRef]
- Schaffer, J.N.; Pearson, M.M. Proteus Mirabilis and Urinary Tract Infections. Microbiol. Spectr. 2015, 3, 383–433. [Google Scholar] [CrossRef] [PubMed]
- Jacobsen, S.M.; Shirtliff, M.E. Proteus Mirabilis Biofilms and Catheter-Associated Urinary Tract Infections. Virulence 2011, 2, 460–465. [Google Scholar] [CrossRef] [PubMed]
- Mo, S.; Durrani, A.F.; Safiullah, Z.; Kowalski, R.P.; Jhanji, V. Proteus Mirabilis Keratitis: Risk Factors, Clinical Features, Treatment Outcomes, and Microbiological Characteristics. Cornea 2021, 40, 704–709. [Google Scholar] [CrossRef] [PubMed]
- Snorf, L.D.; Shepanek, L.; Foltz, E.E.; Harding, H. Pneumonia Due to Proteus Mirabilis Treated with Penicillin and Streptomycin. JAMA 1947, 135, 222. [Google Scholar] [CrossRef]
- Earasi, K.; Welch, C.; Zelickson, A.; Westover, C.; Ramani, C.; Sumner, C.; Davis, E.M. Proteus Empyema as a Rare Complication from an Infected Renal Cyst, a Case Report. BMC Pulm. Med. 2020, 20, 314. [Google Scholar] [CrossRef]
- Chiang, M.-H.; Lee, M.-H.; Liu, Y.-C.; Lee, C.-H. Proteus Mirabilis Thoracic Vertebral Osteomyelitis: A Case Report. J. Med. Case Rep. 2021, 15, 274. [Google Scholar] [CrossRef]
- Şah İpek, M. Neonatal Bacterial Meningitis. In Neonatal Medicine; IntechOpen: London, UK, 2019. [Google Scholar]
- Phan, H.; Lehman, D. Cerebral Abscess Complicating Proteus Mirabilis Meningitis in a Newborn Infant. J. Child. Neurol. 2012, 27, 405–407. [Google Scholar] [CrossRef] [PubMed]
- Grahl, M.V.C.; Uberti, A.F.; Broll, V.; Bacaicoa-Caruso, P.; Meirelles, E.F.; Carlini, C.R. Proteus Mirabilis Urease: Unsuspected Non-Enzymatic Properties Relevant to Pathogenicity. Int. J. Mol. Sci. 2021, 22, 7205. [Google Scholar] [CrossRef] [PubMed]
- Dugger, B.N.; Dickson, D.W. Pathology of Neurodegenerative Diseases. Cold Spring Harb. Perspect. Biol. 2017, 9, a028035. [Google Scholar] [CrossRef] [PubMed]
- Peng, C.; Trojanowski, J.Q.; Lee, V.M.-Y. Protein Transmission in Neurodegenerative Disease. Nat. Rev. Neurol. 2020, 16, 199–212. [Google Scholar] [CrossRef]
- Reith, W. Neurodegenerative Erkrankungen. Radiologe 2018, 58, 241–258. [Google Scholar] [CrossRef]
- Cabreira, V.; Massano, J. Parkinson’s Disease: Clinical Review and Update. Acta Med. Port. 2019, 32, 661–670. [Google Scholar] [CrossRef]
- Choi, J.G.; Kim, N.; Ju, I.G.; Eo, H.; Lim, S.-M.; Jang, S.-E.; Kim, D.-H.; Oh, M.S. Oral Administration of Proteus Mirabilis Damages Dopaminergic Neurons and Motor Functions in Mice. Sci. Rep. 2018, 8, 1275. [Google Scholar] [CrossRef]
- Caputi, V.; Giron, M. Microbiome-Gut-Brain Axis and Toll-Like Receptors in Parkinson’s Disease. Int. J. Mol. Sci. 2018, 19, 1689. [Google Scholar] [CrossRef]
- Holmqvist, S.; Chutna, O.; Bousset, L.; Aldrin-Kirk, P.; Li, W.; Björklund, T.; Wang, Z.-Y.; Roybon, L.; Melki, R.; Li, J.-Y. Direct Evidence of Parkinson Pathology Spread from the Gastrointestinal Tract to the Brain in Rats. Acta Neuropathol. 2014, 128, 805–820. [Google Scholar] [CrossRef]
- Uemura, N.; Yagi, H.; Uemura, M.T.; Hatanaka, Y.; Yamakado, H.; Takahashi, R. Inoculation of α-Synuclein Preformed Fibrils into the Mouse Gastrointestinal Tract Induces Lewy Body-like Aggregates in the Brainstem via the Vagus Nerve. Mol. Neurodegener. 2018, 13, 21. [Google Scholar] [CrossRef]
- Van Den Berge, N.; Ferreira, N.; Gram, H.; Mikkelsen, T.W.; Alstrup, A.K.O.; Casadei, N.; Tsung-Pin, P.; Riess, O.; Nyengaard, J.R.; Tamgüney, G.; et al. Evidence for Bidirectional and Trans-Synaptic Parasympathetic and Sympathetic Propagation of Alpha-Synuclein in Rats. Acta Neuropathol. 2019, 138, 535–550. [Google Scholar] [CrossRef]
- Takahashi, H.; Wakabayashi, K. The Cellular Pathology of Parkinson’s Disease. Neuropathology 2001, 21, 315–322. [Google Scholar] [CrossRef]
- Braak, H.; Ghebremedhin, E.; Rüb, U.; Bratzke, H.; Del Tredici, K. Stages in the Development of Parkinson’s Disease-Related Pathology. Cell Tissue Res. 2004, 318, 121–134. [Google Scholar] [CrossRef]
- Braak, H.; de Vos, R.A.I.; Bohl, J.; Del Tredici, K. Gastric α-Synuclein Immunoreactive Inclusions in Meissner’s and Auerbach’s Plexuses in Cases Staged for Parkinson’s Disease-Related Brain Pathology. Neurosci. Lett. 2006, 396, 67–72. [Google Scholar] [CrossRef]
- Li, W.; Wu, X.; Hu, X.; Wang, T.; Liang, S.; Duan, Y.; Jin, F.; Qin, B. Structural Changes of Gut Microbiota in Parkinson’s Disease and Its Correlation with Clinical Features. Sci. China Life Sci. 2017, 60, 1223–1233. [Google Scholar] [CrossRef]
- Tan, A.H.; Lim, S.Y.; Lang, A.E. The Microbiome–Gut–Brain Axis in Parkinson Disease—from Basic Research to the Clinic. Nat. Rev. Neurol. 2022, 18, 476–495. [Google Scholar] [CrossRef]
- Marizzoni, M.; Provasi, S.; Cattaneo, A.; Frisoni, G.B. Microbiota and Neurodegenerative Diseases. Curr. Opin. Neurol. 2017, 30, 630–638. [Google Scholar] [CrossRef] [PubMed]
- Quigley, E.M.M. Microbiota-Brain-Gut Axis and Neurodegenerative Diseases. Curr. Neurol. Neurosci. Rep. 2017, 17, 94. [Google Scholar] [CrossRef] [PubMed]
- Svensson, E.; Horváth-Puhó, E.; Thomsen, R.W.; Djurhuus, J.C.; Pedersen, L.; Borghammer, P.; Sørensen, H.T. Vagotomy and Subsequent Risk of Parkinson’s Disease. Ann. Neurol. 2015, 78, 522–529. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Fang, F.; Pedersen, N.L.; Tillander, A.; Ludvigsson, J.F.; Ekbom, A.; Svenningsson, P.; Chen, H.; Wirdefeldt, K. Vagotomy and Parkinson Disease. Neurology 2017, 88, 1996–2002. [Google Scholar] [CrossRef]
- Killinger, B.A.; Madaj, Z.; Sikora, J.W.; Rey, N.; Haas, A.J.; Vepa, Y.; Lindqvist, D.; Chen, H.; Thomas, P.M.; Brundin, P.; et al. The Vermiform Appendix Impacts the Risk of Developing Parkinson’s Disease. Sci. Transl. Med. 2018, 10, eaar5280. [Google Scholar] [CrossRef]
- Armbruster, C.E.; Mobley, H.L.T.; Pearson, M.M. Pathogenesis of Proteus Mirabilis Infection. EcoSal Plus 2018, 8, 8. [Google Scholar] [CrossRef]
- Lamb, A.; Chen, J.; Blanke, S.R.; Chen, L.-F. Helicobacter Pylori Activates NF-ΚB by Inducing Ubc13-Mediated Ubiquitination of Lysine 158 of TAK1. J. Cell. Biochem. 2013, 114, 2284–2292. [Google Scholar] [CrossRef] [PubMed]
- Grau, A.J.; Buggle, F.; Lichy, C.; Brandt, T.; Becher, H.; Rudi, J. Helicobacter pylori Infection as an Independent Risk Factor for Cerebral Ischemia of Atherothrombotic Origin. J. Neurol. Sci. 2001, 186, 1–5. [Google Scholar] [CrossRef]
- Torres, A.M.; Gaensly, M.M. Helicobacter pylori: A New Cardiovascular Risk Factor? Rev. Esp. Cardiol. 2002, 55, 652–656. [Google Scholar] [CrossRef] [PubMed]
- Kountouras, J.; Tsolaki, M.; Boziki, M.; Gavalas, E.; Zavos, C.; Stergiopoulos, C.; Kapetanakis, N.; Chatzopoulos, D.; Venizelos, I. Association between Helicobacter pylori Infection and Mild Cognitive Impairment. Eur. J. Neurol. 2007, 14, 976–982. [Google Scholar] [CrossRef] [PubMed]
- Kountouras, J.; Zavos, C.; Polyzos, S.A.; Deretzi, G.; Vardaka, E.; Giartza-Taxidou, E.; Katsinelos, P.; Rapti, E.; Chatzopoulos, D.; Tzilves, D.; et al. Helicobacter pylori Infection and Parkinson’s Disease: Apoptosis as an Underlying Common Contributor. Eur. J. Neurol. 2012, 19, e56. [Google Scholar] [CrossRef]
- Doulberis, M.; Kotronis, G.; Thomann, R.; Polyzos, S.A.; Boziki, M.; Gialamprinou, D.; Deretzi, G.; Katsinelos, P.; Kountouras, J. Review: Impact of Helicobacter Pylori on Alzheimer’s Disease: What Do We Know so Far? Helicobacter 2018, 23, e12454. [Google Scholar] [CrossRef]
- Kountouras, J.; Tsolaki, M.; Gavalas, E.; Boziki, M.; Zavos, C.; Karatzoglou, P.; Chatzopoulos, D.; Venizelos, I. Relationship between Helicobacter pylori Infection and Alzheimer Disease. Neurology 2006, 66, 938–940. [Google Scholar] [CrossRef]
- Kountouras, J.; Boziki, M.; Gavalas, E.; Zavos, C.; Deretzi, G.; Grigoriadis, N.; Tsolaki, M.; Chatzopoulos, D.; Katsinelos, P.; Tzilves, D.; et al. Increased Cerebrospinal Fluid Helicobacter pylori Antibody in Alzheimer’s Disease. Int. J. Neurosci. 2009, 119, 765–777. [Google Scholar] [CrossRef]
- Burucoa, C.; Axon, A. Epidemiology of Helicobacter pylori Infection. Helicobacter 2017, 22, e12403. [Google Scholar] [CrossRef] [PubMed]
- Scopel-Guerra, A.; Olivera-Severo, D.; Staniscuaski, F.; Uberti, A.F.; Callai-Silva, N.; Jaeger, N.; Porto, B.N.; Carlini, C.R. The Impact of Helicobacter pylori Urease upon Platelets and Consequent Contributions to Inflammation. Front. Microbiol. 2017, 8, 2447. [Google Scholar] [CrossRef]
- Wassermann, G.E.; Olivera-Severo, D.; Uberti, A.F.; Carlini, C.R. Helicobacter pylori Urease Activates Blood Platelets through a Lipoxygenase-Mediated Pathway. J. Cell. Mol. Med. 2010, 14, 2025–2034. [Google Scholar] [CrossRef] [PubMed]
- Uberti, A.F.; Olivera-Severo, D.; Wassermann, G.E.; Scopel-Guerra, A.; Moraes, J.A.; Barcellos-de-Souza, P.; Barja-Fidalgo, C.; Carlini, C.R. Pro-Inflammatory Properties and Neutrophil Activation by Helicobacter pylori Urease. Toxicon 2013, 69, 240–249. [Google Scholar] [CrossRef]
- Uberti, A.F.; Callai-Silva, N.; Grahl, M.V.C.; Piovesan, A.R.; Nachtigall, E.G.; Furini, C.R.G.; Carlini, C.R. Helicobacter pylori Urease: Potential Contributions to Alzheimer’s Disease. Int. J. Mol. Sci. 2022, 23, 3091. [Google Scholar] [CrossRef] [PubMed]
- Souza, M.; Moraes, J.A.; Da Silva, V.N.; Helal-Neto, E.; Uberti, A.F.; Scopel-Guerra, A.; Olivera-Severo, D.; Carlini, C.R.; Barja-Fidalgo, C. Helicobacter pylori Urease Induces Pro-inflammatory Effects and Differentiation of Human Endothelial Cells: Cellular and Molecular Mechanism. Helicobacter 2019, 24, e12573. [Google Scholar] [CrossRef]
- Carlini, C.R.; Gomes, C.; Guimaraes, J.A.; Markus, R.P.; Sato, H.; Trolin, G. Central Nervous Effects of the Convulsant Protein Canatoxin. Copenh 1984, 54, 161–166. [Google Scholar] [CrossRef]
- Follmer, C.; Barcellos, G.B.S.; Zingali, R.B.; Machado, O.L.T.; Alves, E.W.; Barja-Fidalgo, C.; Guimarães, J.A.; Carlini, C.R. Canatoxin, a Toxic Protein from Jack Beans (Canavalia ensiformis), Is a Variant Form of Urease (EC 3.5.1.5): Biological Effects of Urease Independent of Its Ureolytic Activity. Biochem. J. 2001, 360, 217. [Google Scholar] [CrossRef]
- Broll, V.; Perin, A.P.A.; Lopes, F.C.; Martinelli, A.H.S.; Moyetta, N.R.; Fruttero, L.L.; Grahl, M.V.C.; Uberti, A.F.; Demartini, D.R.; Ligabue-Braun, R.; et al. Non-Enzymatic Properties of Proteus mirabilis Urease Subunits. Process Biochem. 2021, 110, 263–274. [Google Scholar] [CrossRef]
- Carlini, C.R.; Guimarães, J.A.; Ribeiro, J.M.C. Platelet Release Reaction and Aggregation Induced by Canatoxin, a Convulsant Protein: Evidence for the Involvement of the Platelet Lipoxygenase Pathway. Br. J. Pharmacol. 1985, 84, 551–560. [Google Scholar] [CrossRef]
- Olivera-Severo, D.; Wassermann, G.E.; Carlini, C.R. Bacillus pasteurii Urease Shares with Plant Ureases the Ability to Induce Aggregation of Blood Platelets. Arch. Biochem. Biophys. 2006, 452, 149–155. [Google Scholar] [CrossRef]
- Barja-Fidalgo, C.; Carlini, C.R.; Guimarães, J.A.; Flores, C.A.; Cunha, F.Q.; Ferreira, S.H. Role of Resident Macrophages in Canatoxin-Induced in Vivo Neutrophil Migration. Inflammation 1992, 16, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Barja-Fidalgo, C.; Guimarães, J.A.; Carlini, C.R. Lipoxygenase-Mediated Secretory Effect of Canatoxin the Toxic Protein from Canavalia ensiformis Seeds. Toxicon 1991, 29, 453–459. [Google Scholar] [CrossRef] [PubMed]
- Almeida, C.G.M.; Costa-Higuchi, K.; Piovesan, A.R.; Moro, C.F.; Venturin, G.T.; Greggio, S.; Costa-Ferro, Z.S.; Salamoni, S.D.; Peigneur, S.; Tytgat, J.; et al. Neurotoxic and Convulsant Effects Induced by Jack Bean Ureases on the Mammalian Nervous System. Toxicology 2021, 454, 152737. [Google Scholar] [CrossRef] [PubMed]
- Bradford, M.M. A Rapid and Sensitive Method for the Quantitation of Microgram Quantities of Protein Utilizing the Principle of Protein-Dye Binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Ribeiro-DaSilva, G.; Carlini, C.R.; Pires-Barbosa, R.; Guimarães, J.A. Blood Glucose Alterations Induced in Rats by Canatoxin, a Protein Isolated from Jack Bean (Canavalia ensiformis) Seeds. Toxicon 1986, 24, 775–782. [Google Scholar] [CrossRef]
- Neves, G.; Menegatti, R.; Antonio, C.B.; Grazziottin, L.R.; Vieira, R.O.; Rates, S.M.K.; Noël, F.; Barreiro, E.J.; Fraga, C.A.M. Searching for Multi-Target Antipsychotics: Discovery of Orally Active Heterocyclic N-Phenylpiperazine Ligands of D2-like and 5-HT1A Receptors. Bioorg. Med. Chem. 2010, 18, 1925–1935. [Google Scholar] [CrossRef]
- Souza, I.; Frost, P.S.; França, J.V.; Nascimento-Viana, J.B.; Neris, R.L.S.; Freitas, L.; Pinheiro, D.J.L.L.; Nogueira, C.O.; Neves, G.; Chimelli, L.; et al. Acute and Chronic Neurological Consequences of Early-Life Zika Virus Infection in Mice. Sci. Transl. Med. 2018, 10, eaar2749. [Google Scholar] [CrossRef]
- Oliván, S.; Calvo, A.C.; Rando, A.; Muñoz, M.J.; Zaragoza, P.; Osta, R. Comparative Study of Behavioural Tests in the SOD1G93A Mouse Model of Amyotrophic Lateral Sclerosis. Exp. Anim. 2015, 64, 147–153. [Google Scholar] [CrossRef]
- Ogawa, N.; Hirose, Y.; Ohara, S.; Ono, T.; Watanabe, Y. A Simple Quantitative Bradykinesia Test in MPTP-Treated Mice. Res. Commun. Chem. Pathol. Pharmacol. 1985, 50, 435–441. [Google Scholar]
- Borsoi, M.; Nunes, L.E.D.; Barbosa, A.R.; Lima, M.S.; Medeiros, I.; Pranke, M.A.; Antonio, C.B.; Rates, S.M.K.; Neves, G.A. Intermittent Repeated Stress but Not Ketamine Changes Mice Response to Antidepressants. Neurosci. Lett. 2021, 741, 135452. [Google Scholar] [CrossRef] [PubMed]
- Lister, R.G. The Use of a Plus-Maze to Measure Anxiety in the Mouse. Psychopharmacology 1987, 92, 180–185. [Google Scholar] [CrossRef] [PubMed]
- Bourin, M.; Hascoët, M. The Mouse Light/Dark Box Test. Eur. J. Pharmacol. 2003, 463, 55–65. [Google Scholar] [CrossRef] [PubMed]
- Steru, L.; Chermat, R.; Thierry, B.; Simon, P. The Tail Suspension Test: A New Method for Screening Antidepressants in Mice. Psychopharmacology 1985, 85, 367–370. [Google Scholar] [CrossRef] [PubMed]
- Manaenko, A.; Chen, H.; Kammer, J.; Zhang, J.H.; Tang, J. Comparison Evans Blue Injection Routes: Intravenous versus Intraperitoneal, for Measurement of Blood–Brain Barrier in a Mice Hemorrhage Model. J. Neurosci. Methods 2011, 195, 206–210. [Google Scholar] [CrossRef]
- Coelho-Cerqueira, E.; Carmo-Gonçalves, P.; Sá Pinheiro, A.; Cortines, J.; Follmer, C. α-Synuclein as an Intrinsically Disordered Monomer—Fact or Artefact? FEBS J. 2013, 280, 4915–4927. [Google Scholar] [CrossRef]
- Carmo-Gonçalves, P.; Romão, L.; Follmer, C. In Vitro Protective Action of Monomeric and Fibrillar α-Synuclein on Neuronal Cells Exposed to the Dopaminergic Toxins Salsolinol and DOPAL. ACS Chem. 2020, 11, 3541–3548. [Google Scholar] [CrossRef]
- Hoffman, E.; Winder, S.J. A Modified Wire Hanging Apparatus for Small Animal Muscle Function Testing. PLoS Curr. 2016, 8, ecurrents.md.1e2bec4e78697b7b0ff80ea25a1d38be. [Google Scholar] [CrossRef]
- Pinto, W.B.V.d.R.; Ko, G.M. The Rotarod Teste: Contributions to the Study of Neuromuscular Diseases, Extrapyramidal Syndromes and Cerebellar Ataxia. R. Soc. Bras. Ci. Anim. Lab. 2012, 1, 202–212. [Google Scholar]
- Denenberg, V.H. Open-Field Behavior in the Rat: What Does It Mean? Ann. N. Y. Acad. Sci. 1969, 159, 852–859. [Google Scholar] [CrossRef]
- Stanford, S.C. The Open Field Test: Reinventing the Wheel. J. Psychopharmacol. 2007, 21, 134–135. [Google Scholar] [CrossRef]
- Can, A.; Dao, D.T.; Terrillion, C.E.; Piantadosi, S.C.; Bhat, S.; Gould, T.D. The Tail Suspension Test. J. Vis. Exp. 2011, 59, e3769. [Google Scholar] [CrossRef]
- Yeini, E.; Ofek, P.; Pozzi, S.; Albeck, N.; Ben-Shushan, D.; Tiram, G.; Golan, S.; Kleiner, R.; Sheinin, R.; Israeli Dangoor, S.; et al. P-Selectin Axis Plays a Key Role in Microglia Immunophenotype and Glioblastoma Progression. Nat. Commun. 2021, 12, 1912. [Google Scholar] [CrossRef]
- Metcalfe, M.J.; Figueiredo-Pereira, M.E. Relationship Between Tau Pathology and Neuroinflammation in Alzheimer’s Disease. Mt. Sinai J. Med: J. Transl. Med. 2010, 77, 50–58. [Google Scholar] [CrossRef]
- Thakur, P.; Breger, L.S.; Lundblad, M.; Wan, O.W.; Mattsson, B.; Luk, K.C.; Lee, V.M.Y.; Trojanowski, J.Q.; Björklund, A. Modeling Parkinson’s Disease Pathology by Combination of Fibril Seeds and α-Synuclein Overexpression in the Rat Brain. Proc. Natl. Acad. Sci. USA 2017, 114, E8284–E8293. [Google Scholar] [CrossRef]
- Gibb, W.R.; Lees, A.J. The Relevance of the Lewy Body to the Pathogenesis of Idiopathic Parkinson’s Disease. J. Neurol. Neurosurg. Psychiatry 1988, 51, 745–752. [Google Scholar] [CrossRef]
- Adler, C.H.; Connor, D.J.; Hentz, J.G.; Sabbagh, M.N.; Caviness, J.N.; Shill, H.A.; Noble, B.; Beach, T.G. Incidental Lewy Body Disease: Clinical Comparison to a Control Cohort. Mov. Disord. 2010, 25, 642–646. [Google Scholar] [CrossRef]
- Evidente, V.G.H.; Adler, C.H.; Sabbagh, M.N.; Connor, D.J.; Hentz, J.G.; Caviness, J.N.; Sue, L.I.; Beach, T.G. Neuropathological Findings of PSP in the Elderly without Clinical PSP: Possible Incidental PSP? Park. Relat. Disord. 2011, 17, 365–371. [Google Scholar] [CrossRef]
- Frigerio, R.; Fujishiro, H.; Ahn, T.-B.; Josephs, K.A.; Maraganore, D.M.; DelleDonne, A.; Parisi, J.E.; Klos, K.J.; Boeve, B.F.; Dickson, D.W.; et al. Incidental Lewy Body Disease: Do Some Cases Represent a Preclinical Stage of Dementia with Lewy Bodies? Neurobiol. Aging 2011, 32, 857–863. [Google Scholar] [CrossRef]
- Vaquer-Alicea, J.; Diamond, M.I. Propagation of Protein Aggregation in Neurodegenerative Diseases. Annu. Rev. Biochem. 2019, 88, 785–810. [Google Scholar] [CrossRef]
- Jucker, M.; Walker, L.C. Self-Propagation of Pathogenic Protein Aggregates in Neurodegenerative Diseases. Nature 2013, 501, 45–51. [Google Scholar] [CrossRef]
- Brettschneider, J.; Del Tredici, K.; Lee, V.M.-Y.; Trojanowski, J.Q. Spreading of Pathology in Neurodegenerative Diseases: A Focus on Human Studies. Nat. Rev. Neurosci. 2015, 16, 109–120. [Google Scholar] [CrossRef]
- Bousset, L.; Pieri, L.; Ruiz-Arlandis, G.; Gath, J.; Jensen, P.H.; Habenstein, B.; Madiona, K.; Olieric, V.; Böckmann, A.; Meier, B.H.; et al. Structural and Functional Characterization of Two Alpha-Synuclein Strains. Nat. Commun. 2013, 4, 2575. [Google Scholar] [CrossRef] [PubMed]
- Peelaerts, W.; Bousset, L.; van der Perren, A.; Moskalyuk, A.; Pulizzi, R.; Giugliano, M.; van den Haute, C.; Melki, R.; Baekelandt, V. α-Synuclein Strains Cause Distinct Synucleinopathies after Local and Systemic Administration. Nature 2015, 522, 340–344. [Google Scholar] [CrossRef]
- Prusiner, S.B.; Woerman, A.L.; Mordes, D.A.; Watts, J.C.; Rampersaud, R.; Berry, D.B.; Patel, S.; Oehler, A.; Lowe, J.K.; Kravitz, S.N.; et al. Evidence for α-Synuclein Prions Causing Multiple System Atrophy in Humans with Parkinsonism. Proc. Natl. Acad. Sci. USA 2015, 112, E5308–E5317. [Google Scholar] [CrossRef] [PubMed]
- Woerman, A.L.; Stöhr, J.; Aoyagi, A.; Rampersaud, R.; Krejciova, Z.; Watts, J.C.; Ohyama, T.; Patel, S.; Widjaja, K.; Oehler, A.; et al. Propagation of Prions Causing Synucleinopathies in Cultured Cells. Proc. Natl. Acad. Sci. USA 2015, 112, E4949–E4958. [Google Scholar] [CrossRef]
- Peng, C.; Gathagan, R.J.; Covell, D.J.; Medellin, C.; Stieber, A.; Robinson, J.L.; Zhang, B.; Pitkin, R.M.; Olufemi, M.F.; Luk, K.C.; et al. Cellular Milieu Imparts Distinct Pathological α-Synuclein Strains in α-Synucleinopathies. Nature 2018, 557, 558–563. [Google Scholar] [CrossRef] [PubMed]
- Yamasaki, T.R.; Holmes, B.B.; Furman, J.L.; Dhavale, D.D.; Su, B.W.; Song, E.-S.; Cairns, N.J.; Kotzbauer, P.T.; Diamond, M.I. Parkinson’s Disease and Multiple System Atrophy Have Distinct α-Synuclein Seed Characteristics. J. Biol. Chem. 2019, 294, 1045–1058. [Google Scholar] [CrossRef]
- Werner, T.; Kumar, R.; Horvath, I.; Scheers, N.; Wittung-Stafshede, P. Abundant Fish Protein Inhibits α-Synuclein Amyloid Formation. Sci. Rep. 2018, 8, 5465. [Google Scholar] [CrossRef]
- Poewe, W.; Seppi, K.; Tanner, C.M.; Halliday, G.M.; Brundin, P.; Volkmann, J.; Schrag, A.-E.; Lang, A.E. Parkinson Disease. Nat. Rev. Dis. Primers 2017, 3, 17013. [Google Scholar] [CrossRef]
- Foster, J.A.; McVey Neufeld, K.-A. Gut–Brain Axis: How the Microbiome Influences Anxiety and Depression. Trends Neurosci. 2013, 36, 305–312. [Google Scholar] [CrossRef]
- Jiang, H.; Ling, Z.; Zhang, Y.; Mao, H.; Ma, Z.; Yin, Y.; Wang, W.; Tang, W.; Tan, Z.; Shi, J.; et al. Altered Fecal Microbiota Composition in Patients with Major Depressive Disorder. Brain Behav. Immun. 2015, 48, 186–194. [Google Scholar] [CrossRef]
- Mayer, E.A.; Padua, D.; Tillisch, K. Altered Brain-Gut Axis in Autism: Comorbidity or Causative Mechanisms? BioEssays 2014, 36, 933–939. [Google Scholar] [CrossRef]
- Escobar, Y.-N.H.; O’Piela, D.; Wold, L.E.; Mackos, A.R. Influence of the Microbiota-Gut-Brain Axis on Cognition in Alzheimer’s Disease. J. Alzheimer’s Dis. 2022, 87, 17–31. [Google Scholar] [CrossRef]
- Scheperjans, F.; Aho, V.; Pereira, P.A.B.; Koskinen, K.; Paulin, L.; Pekkonen, E.; Haapaniemi, E.; Kaakkola, S.; Eerola-Rautio, J.; Pohja, M.; et al. Gut Microbiota Are Related to Parkinson’s Disease and Clinical Phenotype. Mov. Disord. 2015, 30, 350–358. [Google Scholar] [CrossRef]
- Álvarez-Arellano, L. Helicobacter pylori and Neurological Diseases: Married by the Laws of Inflammation. World J. Gastrointest. Pathophy. 2014, 5, 400. [Google Scholar] [CrossRef]
- McGee, D.J.; Lu, X.-H.; Disbrow, E.A. Stomaching the Possibility of a Pathogenic Role for Helicobacter pylori in Parkinson’s Disease. J. Park. Dis. 2018, 8, 367–374. [Google Scholar] [CrossRef]
- Tan, A.H.; Mahadeva, S.; Marras, C.; Thalha, A.M.; Kiew, C.K.; Yeat, C.M.; Ng, S.W.; Ang, S.P.; Chow, S.K.; Loke, M.F.; et al. Helicobacter pylori Infection Is Associated with Worse Severity of Parkinson’s Disease. Park. Relat. Disord. 2015, 21, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Zhong, R.; Chen, Q.; Zhang, X.; Li, M.; Lin, W. Helicobacter pylori Infection Is Associated with a Poor Response to Levodopa in Patients with Parkinson’s Disease: A Systematic Review and Meta-Analysis. J. Neurol. 2022, 269, 703–711. [Google Scholar] [CrossRef]
- Keshavarzian, A.; Green, S.J.; Engen, P.A.; Voigt, R.M.; Naqib, A.; Forsyth, C.B.; Mutlu, E.; Shannon, K.M. Colonic Bacterial Composition in Parkinson’s Disease. Mov. Disord. 2015, 30, 1351–1360. [Google Scholar] [CrossRef]
- Heintz-Buschart, A.; Pandey, U.; Wicke, T.; Sixel-Döring, F.; Janzen, A.; Sittig-Wiegand, E.; Trenkwalder, C.; Oertel, W.H.; Mollenhauer, B.; Wilmes, P. The Nasal and Gut Microbiome in Parkinson’s Disease and Idiopathic Rapid Eye Movement Sleep Behavior Disorder. Mov. Disord. 2018, 33, 88–98. [Google Scholar] [CrossRef] [PubMed]
- Dong, S.; Sun, M.; He, C.; Cheng, H. Brain-Gut-Microbiota Axis in Parkinson’s Disease: A Historical Review and Future Perspective. Brain Res. Bull. 2022, 183, 84–93. [Google Scholar] [CrossRef]
- Verbaan, D.; Marinus, J.; Visser, M.; van Rooden, S.M.; Stiggelbout, A.M.; van Hilten, J.J. Patient-Reported Autonomic Symptoms in Parkinson Disease. Neurology 2007, 69, 333–341. [Google Scholar] [CrossRef] [PubMed]
- Mertsalmi, T.H.; Aho, V.T.E.; Pereira, P.A.B.; Paulin, L.; Pekkonen, E.; Auvinen, P.; Scheperjans, F. More than Constipation—Bowel Symptoms in Parkinson’s Disease and Their Connection to Gut Microbiota. Eur. J. Neurol. 2017, 24, 1375–1383. [Google Scholar] [CrossRef]
- Sakakibara, R.; Doi, H.; Fukudo, S. Lewy Body Constipation. J. Anus Rectum Colon. 2019, 3, 10–17. [Google Scholar] [CrossRef]
- Erny, D.; Hrabě de Angelis, A.L.; Jaitin, D.; Wieghofer, P.; Staszewski, O.; David, E.; Keren-Shaul, H.; Mahlakoiv, T.; Jakobshagen, K.; Buch, T.; et al. Host Microbiota Constantly Control Maturation and Function of Microglia in the CNS. Nat. Neurosci. 2015, 18, 965–977. [Google Scholar] [CrossRef]
- Klingelhoefer, L.; Reichmann, H. Pathogenesis of Parkinson Disease—The Gut–Brain Axis and Environmental Factors. Nat. Rev. Neurol. 2015, 11, 625–636. [Google Scholar] [CrossRef] [PubMed]
- Sampson, T.R.; Debelius, J.W.; Thron, T.; Janssen, S.; Shastri, G.G.; Ilhan, Z.E.; Challis, C.; Schretter, C.E.; Rocha, S.; Gradinaru, V.; et al. Gut Microbiota Regulate Motor Deficits and Neuroinflammation in a Model of Parkinson’s Disease. Cell 2016, 167, 1469–1480.e12. [Google Scholar] [CrossRef]
- Zhou, X.; Lu, J.; Wei, K.; Wei, J.; Tian, P.; Yue, M.; Wang, Y.; Hong, D.; Li, F.; Wang, B.; et al. Neuroprotective Effect of Ceftriaxone on MPTP-Induced Parkinson’s Disease Mouse Model by Regulating Inflammation and Intestinal Microbiota. Oxid. Med. Cell. Longev. 2021, 2021, 9424582. [Google Scholar] [CrossRef]
- Wang, X.-L.; Zeng, J.; Yang, Y.; Xiong, Y.; Zhang, Z.-H.; Qiu, M.; Yan, X.; Sun, X.-Y.; Tuo, Q.-Z.; Liu, R.; et al. Helicobacter pylori Filtrate Induces Alzheimer-Like Tau Hyperphosphorylation by Activating Glycogen Synthase Kinase-3β. J. Alzheimer’s Dis. 2015, 43, 153–165. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.-L.; Zeng, J.; Feng, J.; Tian, Y.-T.; Liu, Y.-J.; Qiu, M.; Yan, X.; Yang, Y.; Xiong, Y.; Zhang, Z.-H.; et al. Helicobacter pylori Filtrate Impairs Spatial Learning and Memory in Rats and Increases Î2-Amyloid by Enhancing Expression of Presenilin-2. Front. Aging Neurosci. 2014, 6, 66. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro-DaSilva, G.; Prado, J.F. Increased Insulin Circulating Levels Induced by Canatoxin in Rats. Toxicon 1993, 31, 1131–1136. [Google Scholar] [CrossRef] [PubMed]
- Micheletto, Y.M.S.; Moro, C.F.; Lopes, F.C.; Ligabue-Braun, R.; Martinelli, A.H.S.; Marques, C.M.; Schroder, A.P.; Carlini, C.R.; da Silveira, N.P. Interaction of Jack Bean (Canavalia ensiformis) Urease and a Derived Peptide with Lipid Vesicles. Colloids Surf. B 2016, 145, 576–585. [Google Scholar] [CrossRef]
- Piovesan, A.R.; Martinelli, A.H.S.; Ligabue-Braun, R.; Schwartz, J.-L.; Carlini, C.R. Canavalia ensiformis Urease, Jaburetox and Derived Peptides Form Ion Channels in Planar Lipid Bilayers. Arch. Biochem. Biophys. 2014, 547, 6–17. [Google Scholar] [CrossRef]
- Baik, S.-C.; Kang, H.-L.; Seo, J.-H.; Park, E.-S.; Rhee, K.-H.; Cho, M.-J. Helicobacter pylori Urease Induces Mouse Death. J. Bacteriol. Virol. 2005, 3, 175–181. [Google Scholar]
- Becker-Ritt, A.B.; Martinelli, A.H.S.; Mitidieri, S.; Feder, V.; Wassermann, G.E.; Santi, L.; Vainstein, M.H.; Oliveira, J.T.A.; Fiuza, L.M.; Pasquali, G.; et al. Antifungal Activity of Plant and Bacterial Ureases. Toxicon 2007, 50, 971–983. [Google Scholar] [CrossRef]
- Mulinari, F.; Becker-Ritt, A.B.; Demartini, D.R.; Ligabue-Braun, R.; Stanisçuaski, F.; Verli, H.; Fragoso, R.R.; Schroeder, E.K.; Carlini, C.R.; Grossi-de-Sá, M.F. Characterization of JBURE-IIb Isoform of Canavalia ensiformis (L.) DC Urease. Biochim. Biophys. Acta—Proteins Proteom. 2011, 1814, 1758–1768. [Google Scholar] [CrossRef]
- Ferreira-DaSilva, C.T.; Gombarovits, M.E.; Masuda, H.; Oliveira, C.M.; Carlini, C.R. Proteolytic Activation of Canatoxin, a Plant Toxic Protein, by Insect Cathepsin-like Enzymes. Arch. Biochem. Biophys. 2000, 44, 162–171. [Google Scholar] [CrossRef]
- Mulinari, F.; Stanisçuaski, F.; Bertholdo-Vargas, L.R.; Postal, M.; Oliveira-Neto, O.B.; Rigden, D.J.; Grossi-de-Sá, M.F.; Carlini, C.R. Jaburetox-2Ec: An Insecticidal Peptide Derived from an Isoform of Urease from the Plant Canavalia ensiformis. Peptides 2007, 28, 2042–2050. [Google Scholar] [CrossRef]
- Moro, C.F.; Nogueira, F.C.S.; Almeida, C.G.M.; Real-Guerra, R.; Dalberto, P.F.; Bizarro, C.V.; Ligabue-Braun, R.; Carlini, C.R. One Enzyme, Many Faces: Urease Is Also Canatoxin. J. Biomol. Struct. Dyn. 2022, 1–12. [Google Scholar] [CrossRef]
- Krajewska, B.; Ureases, I. Functional, Catalytic and Kinetic Properties: A Review. J. Mol. Catal. B Enzym. 2009, 59, 9–21. [Google Scholar] [CrossRef]
- Mendez, M.F. The Relationship Between Anxiety and Alzheimer’s Disease. J. Alzheimers Dis. Rep. 2021, 5, 171–177. [Google Scholar] [CrossRef]
- Kalia, L.V.; Lang, A.E. Parkinson’s Disease. Lancet 2015, 386, 896–912. [Google Scholar] [CrossRef] [PubMed]
- O’Mahony, S.M.; Clarke, G.; Borre, Y.E.; Dinan, T.G.; Cryan, J.F. Serotonin, Tryptophan Metabolism and the Brain-Gut-Microbiome Axis. Behav. Brain Res. 2015, 277, 32–48. [Google Scholar] [CrossRef] [PubMed]
- Yan, T.; Li, F.; Xiong, W.; Wu, B.; Xiao, F.; He, B.; Jia, Y. Nootkatone Improves Anxiety- and Depression-like Behavior by Targeting Hyperammonemia-induced Oxidative Stress in D-galactosamine Model of Liver Injury. Environ. Toxicol. 2021, 36, 694–706. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Shal, B.; Naveed, M.; Shah, F.A.; Atiq, A.; Khan, N.U.; Kim, Y.S.; Khan, S. Matrine Ameliorates Anxiety and Depression-like Behaviour by Targeting Hyperammonemia-Induced Neuroinflammation and Oxidative Stress in CCl4 Model of Liver Injury. Neurotoxicology 2019, 72, 38–50. [Google Scholar] [CrossRef]
- Agrawal, A.; Gupta, A.; Chandra, M.; Koowar, S. Role of Helicobacter pylori Infection in the Pathogenesis of Minimal Hepatic Encephalopathy and Effect of Its Eradication. Indian. J. Gastroenterol. 2011, 30, 29–32. [Google Scholar] [CrossRef]
- Albersen, M.; Joniau, S.; Van Poppel, H.; Cuyle, P.-J.; Knockaert, D.C.; Meersseman, W. Urea-Splitting Urinary Tract Infection Contributing to Hyperammonemic Encephalopathy. Nat. Clin. Pr. Pract. Urol. 2007, 4, 455–458. [Google Scholar] [CrossRef]
- Gorantla, A.; Kishore, A.; Zaman, A.; Ramirez, M.; Taluru, H.; Horton, N.; Sivakumar, S.; Geraghty, P.; McFarlane, S.I. Hyperammonemic Encephalopathy Secondary to Urinary Tract Infection. Cureus 2022, 14, e31754. [Google Scholar] [CrossRef]
- Federico, P.; Zochodne, D.W. Reversible Parkinsonism and Hyperammonemia Associated with Portal Vein Thrombosis. Acta Neurol. Scand. 2001, 103, 198–200. [Google Scholar] [CrossRef]
- Bosoi, C.R.; Rose, C.F. Identifying the Direct Effects of Ammonia on the Brain. Metab. Brain Dis. 2009, 24, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Lopes, F.F.; Sitta, A.; de Moura Coelho, D.; Ribas, G.S.; Faverzani, J.L.; dos Reis, B.G.; Wajner, M.; Vargas, C.R. Clinical Findings of Patients with Hyperammonemia Affected by Urea Cycle Disorders with Hepatic Encephalopathy. Int. J. Dev. Neurosci. 2022, 82, 771–787. [Google Scholar] [CrossRef] [PubMed]
- Woolley, J.D.; Khan, B.K.; Murthy, N.K.; Miller, B.L.; Rankin, K.P. The Diagnostic Challenge of Psychiatric Symptoms in Neurodegenerative Disease. J. Clin. Psychiatry 2011, 72, 126–133. [Google Scholar] [CrossRef]
- Lyketsos, C.G.; Carrillo, M.C.; Ryan, J.M.; Khachaturian, A.S.; Trzepacz, P.; Amatniek, J.; Cedarbaum, J.; Brashear, R.; Miller, D.S. Neuropsychiatric Symptoms in Alzheimer’s Disease. Alzheimer’s Dement. 2011, 7, 532–539. [Google Scholar] [CrossRef] [PubMed]
- Santabárbara, J.; Villagrasa, B.; López-Antón, R.; Olaya, B.; Bueno-Notivol, J.; de la Cámara, C.; Gracia-García, P.; Lobo, E.; Lobo, A. Clinically Relevant Anxiety and Risk of Alzheimer’s Disease in an Elderly Community Sample: 4.5 Years of Follow-Up. J. Affect. Disord. 2019, 250, 16–20. [Google Scholar] [CrossRef]
- Lin, C.-H.; Lin, J.-W.; Liu, Y.-C.; Chang, C.-H.; Wu, R.-M. Risk of Parkinson’s Disease Following Anxiety Disorders: A Nationwide Population-Based Cohort Study. Eur. J. Neurol. 2015, 22, 1280–1287. [Google Scholar] [CrossRef]
- Shen, C.-C.; Tsai, S.-J.; Perng, C.-L.; Kuo, B.I.-T.; Yang, A.C. Risk of Parkinson Disease after Depression: A Nationwide Population-Based Study. Neurology 2013, 81, 1538–1544. [Google Scholar] [CrossRef]
- Fang, F.; Xu, Q.; Park, Y.; Huang, X.; Hollenbeck, A.; Blair, A.; Schatzkin, A.; Kamel, F.; Chen, H. Depression and the Subsequent Risk of Parkinson’s Disease in the NIH-AARP Diet and Health Study. Mov. Disord. 2010, 25, 1157–1162. [Google Scholar] [CrossRef]
- Benjamin, C.F.; Carlini, C.R.; Barja-Fidalgo, C. Pharmacological Characterization of Rat Paw Edema Induced by Canatoxin, the Toxic Protein from Canavalia ensiformis (Jack Bean) Seeds. Toxicon 1992, 30, 879–885. [Google Scholar] [CrossRef]
- Sprague, A.H.; Khalil, R.A. Inflammatory Cytokines in Vascular Dysfunction and Vascular Disease. Biochem. Pharmacol. 2009, 78, 539–552. [Google Scholar] [CrossRef]
- Ahmad, A.; Patel, V.; Xiao, J.; Khan, M.M. The Role of Neurovascular System in Neurodegenerative Diseases. Mol. Neurobiol. 2020, 57, 4373–4393. [Google Scholar] [CrossRef]
- Wroblewski, L.E.; Shen, L.; Ogden, S.; Romero–Gallo, J.; Lapierre, L.A.; Israel, D.A.; Turner, J.R.; Peek, R.M. Helicobacter Pylori Dysregulation of Gastric Epithelial Tight Junctions by Urease-Mediated Myosin II Activation. Gastroenterol. 2009, 136, 236–246. [Google Scholar] [CrossRef]
- Caron, T.J. Tight Junction Disruption: Helicobacter pylori and Dysregulation of the Gastric Mucosal Barrier. World J. Gastroenterol. 2015, 21, 11411. [Google Scholar] [CrossRef]
- Wroblewski, L.E.; Peek, R.M. Targeted Disruption of the Epithelial-Barrier by Helicobacter pylori. Cell Commun. Signal 2011, 9, 29. [Google Scholar] [CrossRef] [PubMed]
- Chow, B.W.; Gu, C. The Molecular Constituents of the Blood–Brain Barrier. Trends Neurosci. 2015, 38, 598–608. [Google Scholar] [CrossRef]
- Skowrońska, M.; Albrecht, J. Alterations of Blood Brain Barrier Function in Hyperammonemia: An Overview. Neurotox. Res. 2012, 21, 236–244. [Google Scholar] [CrossRef] [PubMed]
- Skowrońska, M.; Zielińska, M.; Wójcik-Stanaszek, L.; Ruszkiewicz, J.; Milatovic, D.; Aschner, M.; Albrecht, J. Ammonia Increases Paracellular Permeability of Rat Brain Endothelial Cells by a Mechanism Encompassing Oxidative/Nitrosative Stress and Activation of Matrix Metalloproteinases. J. Neurochem. 2012, 121, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Adlimoghaddam, A.; Sabbir, M.G.; Albensi, B.C. Ammonia as a Potential Neurotoxic Factor in Alzheimer’s Disease. Front. Mol. Neurosci. 2016, 9, 57. [Google Scholar] [CrossRef]
- Yasunishi, M.; Koumura, A.; Hayashi, Y.; Nishida, S.; Inuzuka, T. A Case of Hyperammonemia Resulting from Urinary Tract Infection Caused by Urease-Producing Bacteria in a Parkinson’s Disease Patient with Drug-Induced Urinary Retention. Jpn. J. Geriatr. 2017, 54, 560–566. [Google Scholar] [CrossRef][Green Version]
- Saunders, N.R.; Dziegielewska, K.M.; Møllgård, K.; Habgood, M.D. Markers for Blood-Brain Barrier Integrity: How Appropriate Is Evans Blue in the Twenty-First Century and What Are the Alternatives? Front. Neurosci. 2015, 9, 385. [Google Scholar] [CrossRef]
- Yen, L.F.; Wei, V.C.; Kuo, E.Y.; Lai, T.W. Distinct Patterns of Cerebral Extravasation by Evans Blue and Sodium Fluorescein in Rats. PLoS ONE 2013, 8, e68595. [Google Scholar] [CrossRef]
- Bentivoglio, M.; Kristensson, K.; Rottenberg, M.E. Circumventricular Organs and Parasite Neurotropism: Neglected Gates to the Brain? Front. Immunol. 2018, 9, 2877. [Google Scholar] [CrossRef]
- Wilhelm, I.; Nyúl-Tóth, Á.; Suciu, M.; Hermenean, A.; Krizbai, I.A. Heterogeneity of the Blood-Brain Barrier. Tissue Barriers 2016, 4, e1143544. [Google Scholar] [CrossRef]
- Olofsson, A.; Vallström, A.; Petzold, K.; Tegtmeyer, N.; Schleucher, J.; Carlsson, S.; Haas, R.; Backert, S.; Wai, S.N.; Gröbner, G.; et al. Biochemical and Functional Characterization of Helicobacter pylori Vesicles. Mol. Microbiol. 2010, 77, 1539–1555. [Google Scholar] [CrossRef]
- Wei, S.; Li, X.; Wang, J.; Wang, Y.; Zhang, C.; Dai, S.; Wang, X.; Deng, X.; Zhao, L.; Shan, B. Outer Membrane Vesicles Secreted by Helicobacter pylori Transmitting Gastric Pathogenic Virulence Factors. ACS Omega 2022, 7, 240–258. [Google Scholar] [CrossRef]
- Zingl, F.G.; Thapa, H.B.; Scharf, M.; Kohl, P.; Müller, A.M.; Schild, S. Outer Membrane Vesicles of Vibrio Cholerae Protect and Deliver Active Cholera Toxin to Host Cells via Porin-Dependent Uptake. mBio 2021, 12, e0053421. [Google Scholar] [CrossRef]
- Alves, N.J.; Turner, K.B.; Medintz, I.L.; Walper, S.A. Protecting Enzymatic Function through Directed Packaging into Bacterial Outer Membrane Vesicles. Sci. Rep. 2016, 6, 24866. [Google Scholar] [CrossRef] [PubMed]
- Banks, W.A.; Sharma, P.; Bullock, K.M.; Hansen, K.M.; Ludwig, N.; Whiteside, T.L. Transport of Extracellular Vesicles across the Blood-Brain Barrier: Brain Pharmacokinetics and Effects of Inflammation. Int. J. Mol. Sci. 2020, 21, 4407. [Google Scholar] [CrossRef] [PubMed]
- Cuesta, C.M.; Guerri, C.; Ureña, J.; Pascual, M. Role of Microbiota-Derived Extracellular Vesicles in Gut-Brain Communication. Int. J. Mol. Sci. 2021, 22, 4235. [Google Scholar] [CrossRef]
- McGeer, P.L.; Itagaki, S.; Boyes, B.E.; McGeer, E.G. Reactive Microglia Are Positive for HLA-DR in the Substantia Nigra of Parkinson’s and Alzheimer’s Disease Brains. Neurology 1988, 38, 1285. [Google Scholar] [CrossRef] [PubMed]
- Dickson, D.W.; Lee, S.C.; Mattiace, L.A.; Yen, S.C.; Brosnan, C. Microglia and Cytokines in Neurological Disease, with Special Reference to AIDS and Alzheimer’s Disease. Glia 1993, 7, 75–83. [Google Scholar] [CrossRef]
- Bö, L.; Mörk, S.; Kong, P.A.; Nyland, H.; Pardo, C.A.; Trapp, B.D. Detection of MHC Class II-Antigens on Macrophages and Microglia, but Not on Astrocytes and Endothelia in Active Multiple Sclerosis Lesions. J. Neuroimmunol. 1994, 51, 135–146. [Google Scholar] [CrossRef]
- Kouli, A.; Camacho, M.; Allinson, K.; Williams-Gray, C.H. Neuroinflammation and Protein Pathology in Parkinson’s Disease Dementia. Acta Neuropathol. Commun. 2020, 8, 211. [Google Scholar] [CrossRef]
- Chaudhry, Z.L.; Ahmed, B.Y. The Role of Caspases in Parkinson’s Disease Pathogenesis: A Brief Look at the Mitochondrial Pathway. Austin Alzheimers Park. Dis. 2014, 1, 5. [Google Scholar]
- Li, Q.-X.; Mok, S.S.; Laughton, K.M.; McLean, C.A.; Cappai, R.; Masters, C.L.; Culvenor, J.G.; Horne, M.K. Plasma α-Synuclein Is Decreased in Subjects with Parkinson’s Disease. Exp. Neurol. 2007, 204, 583–588. [Google Scholar] [CrossRef]
- Tokuda, T.; Salem, S.A.; Allsop, D.; Mizuno, T.; Nakagawa, M.; Qureshi, M.M.; Locascio, J.J.; Schlossmacher, M.G.; El-Agnaf, O.M.A. Decreased α-Synuclein in Cerebrospinal Fluid of Aged Individuals and Subjects with Parkinson’s Disease. Biochem. Biophys. Res. Commun. 2006, 349, 162–166. [Google Scholar] [CrossRef]
- Guivernau, B.; Bonet, J.; Valls-Comamala, V.; Bosch-Morató, M.; Godoy, J.A.; Inestrosa, N.C.; Perálvarez-Marín, A.; Fernández-Busquets, X.; Andreu, D.; Oliva, B.; et al. Amyloid-β Peptide Nitrotyrosination Stabilizes Oligomers and Enhances NMDAR-Mediated Toxicity. J. Neurosci. 2016, 36, 11693–11703. [Google Scholar] [CrossRef]
- Yuan, B.; Sierks, M.R. Intracellular Targeting and Clearance of Oligomeric Alpha-Synuclein Alleviates Toxicity in Mammalian Cells. Neurosci. Lett. 2009, 459, 16–18. [Google Scholar] [CrossRef] [PubMed]
- Bigi, A.; Ermini, E.; Chen, S.W.; Cascella, R.; Cecchi, C. Exploring the Release of Toxic Oligomers from α-Synuclein Fibrils with Antibodies and STED Microscopy. Life 2021, 11, 431. [Google Scholar] [CrossRef] [PubMed]
- Yoo, G.; Yeou, S.; Son, J.B.; Shin, Y.-K.; Lee, N.K. Cooperative Inhibition of SNARE-Mediated Vesicle Fusion by α-Synuclein Monomers and Oligomers. Sci. Rep. 2021, 11, 10955. [Google Scholar] [CrossRef]










Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grahl, M.V.C.; Andrade, B.d.S.; Perin, A.P.A.; Neves, G.A.; Duarte, L.d.S.; Uberti, A.F.; Hohl, K.S.; Follmer, C.; Carlini, C.R. Could the Urease of the Gut Bacterium Proteus mirabilis Play a Role in the Altered Gut–Brain Talk Associated with Parkinson’s Disease? Microorganisms 2023, 11, 2042. https://doi.org/10.3390/microorganisms11082042
Grahl MVC, Andrade BdS, Perin APA, Neves GA, Duarte LdS, Uberti AF, Hohl KS, Follmer C, Carlini CR. Could the Urease of the Gut Bacterium Proteus mirabilis Play a Role in the Altered Gut–Brain Talk Associated with Parkinson’s Disease? Microorganisms. 2023; 11(8):2042. https://doi.org/10.3390/microorganisms11082042
Chicago/Turabian StyleGrahl, Matheus V. C., Brenda da Silva Andrade, Ana Paula A. Perin, Gilda A. Neves, Laura de Souza Duarte, Augusto Frantz Uberti, Kelvin Siqueira Hohl, Cristian Follmer, and Celia Regina Carlini. 2023. "Could the Urease of the Gut Bacterium Proteus mirabilis Play a Role in the Altered Gut–Brain Talk Associated with Parkinson’s Disease?" Microorganisms 11, no. 8: 2042. https://doi.org/10.3390/microorganisms11082042
APA StyleGrahl, M. V. C., Andrade, B. d. S., Perin, A. P. A., Neves, G. A., Duarte, L. d. S., Uberti, A. F., Hohl, K. S., Follmer, C., & Carlini, C. R. (2023). Could the Urease of the Gut Bacterium Proteus mirabilis Play a Role in the Altered Gut–Brain Talk Associated with Parkinson’s Disease? Microorganisms, 11(8), 2042. https://doi.org/10.3390/microorganisms11082042