

Metatranscriptome Analysis of Nasopharyngeal Swabs across the Varying Severity of COVID-19 Disease Demonstrated Unprecedented Species Diversity
 , ,
, ,
Abstract
1. Introduction
2. Material and Methods
2.1. Patients and Samples
2.2. RNA Isolation, RT-PCR and Library Preparation
2.3. Sequencing and Bioinformatic Analysis
2.4. Statistical Analysis
3. Results
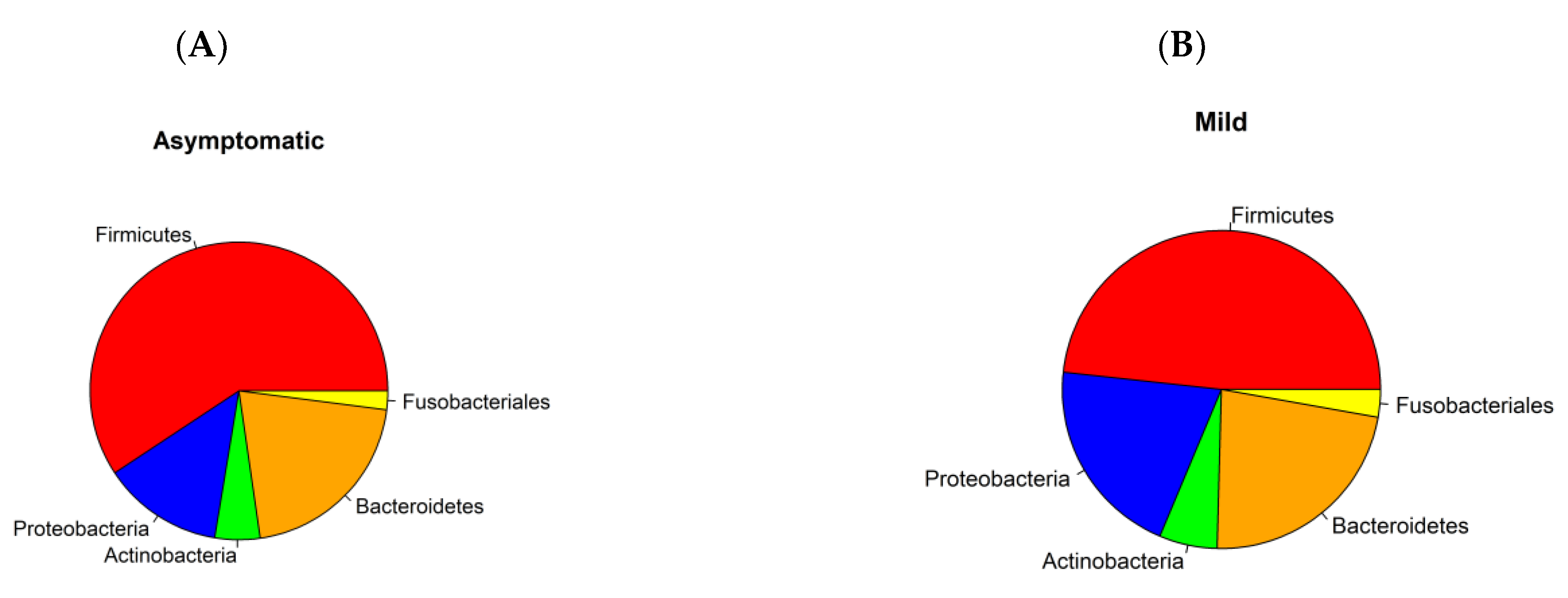
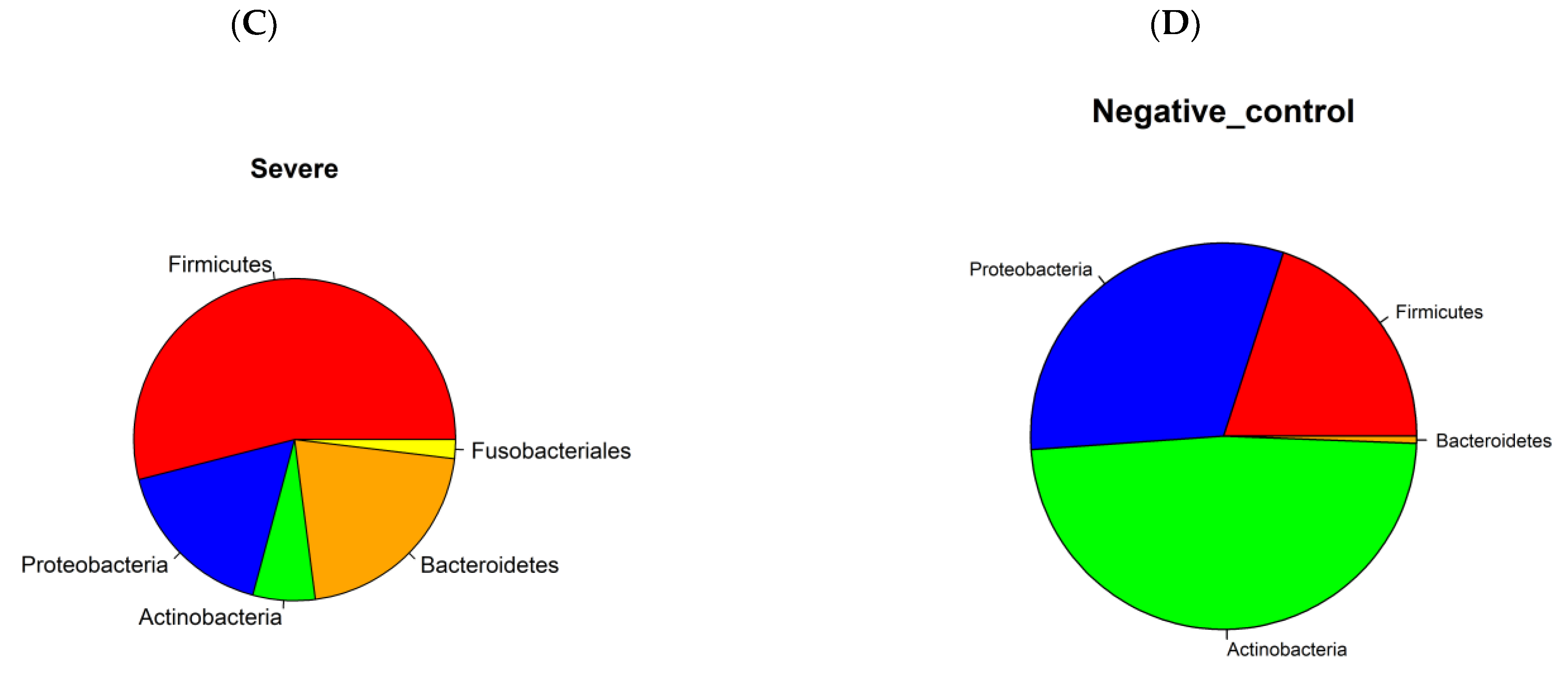
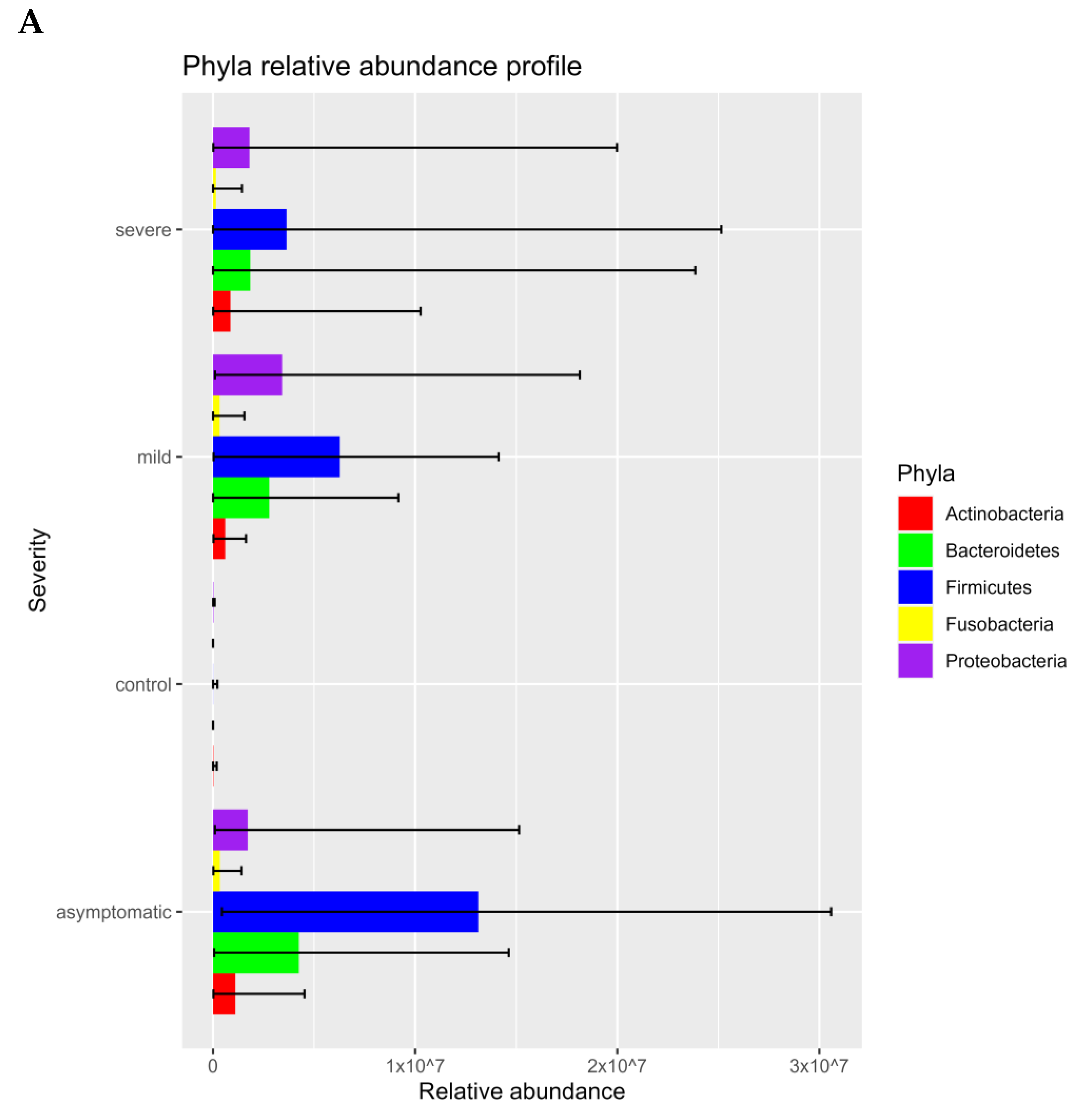
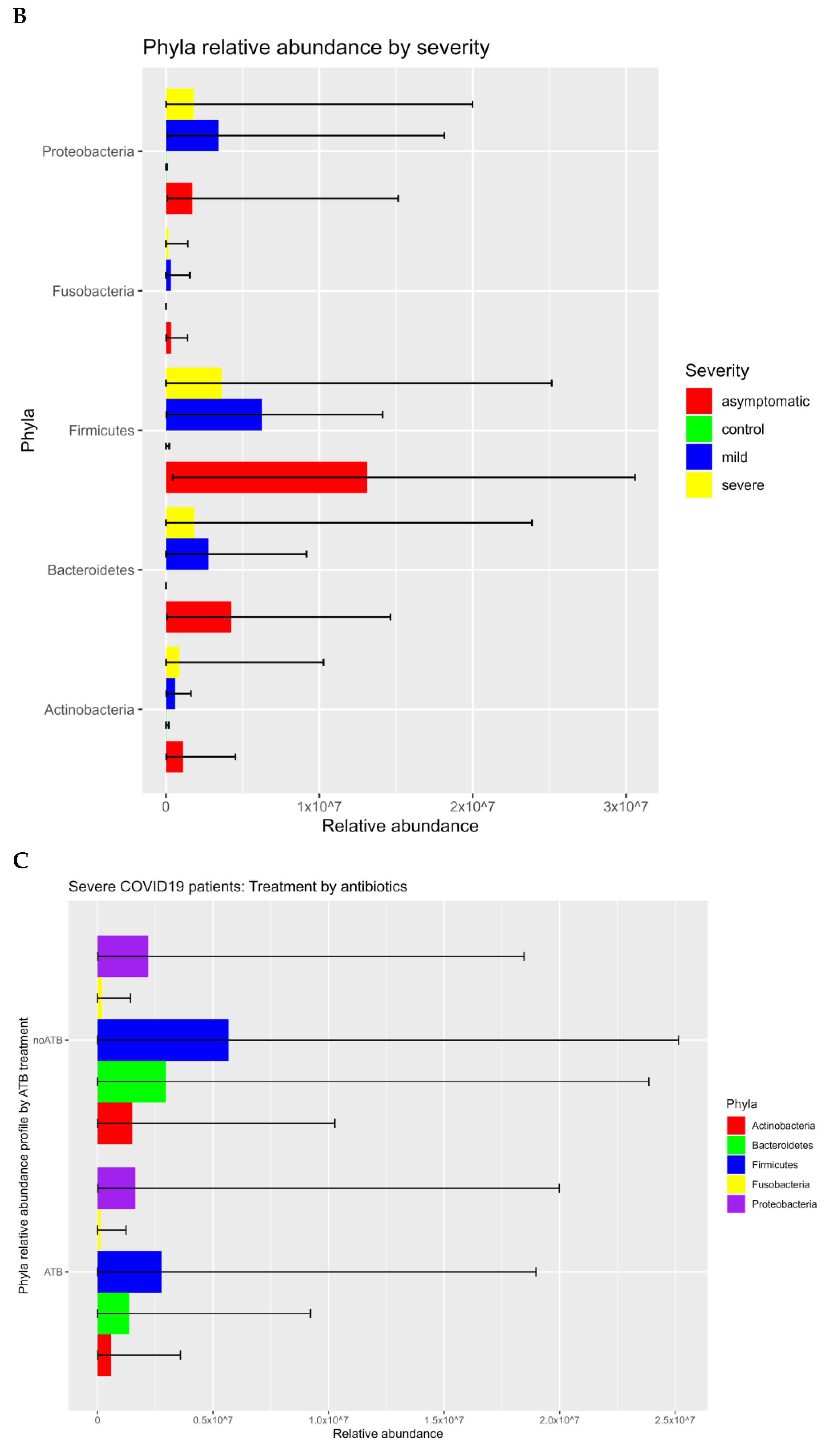
3.1. Prokaryotic Microbiome
3.2. Virome Analyses
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Burbank, A.J.; Sood, A.K.; Kesic, M.J.; Peden, D.B.; Hernandez, M.L. Environmental determinants of allergy and asthma in early life. J. Allergy Clin. Immunol. 2017, 140, 1–12. [Google Scholar] [CrossRef]
- Bai, X.; Narayanan, A.; Skagerberg, M.; Ceña-Diez, R.; Giske, C.G.; Strålin, K.; Sönnerborg, A. Characterization of the Upper Respiratory Bacterial Microbiome in Critically Ill COVID-19 Patients. Biomedicines 2022, 10, 982. [Google Scholar] [CrossRef]
- Zhu, T.; Jin, J.; Chen, M.; Chen, Y. The impact of infection with COVID-19 on the respiratory microbiome: A narrative review. Virulence 2022, 13, 1076–1087. [Google Scholar] [CrossRef] [PubMed]
- Candel, S.; Tyrkalska, S.D.; Álvarez-Santacruz, C.; Mulero, V. The nasopharyngeal microbiome in COVID-19. Emerg. Microbes Infect. 2023, 12, e2165970. [Google Scholar] [CrossRef] [PubMed]
- Kumpitsch, C.; Koskinen, K.; Schöpf, V.; Moissl-Eichinger, C. The microbiome of the upper respiratory tract in health and disease. BMC Biol. 2019, 17, 87. [Google Scholar] [CrossRef]
- Di Simone, S.K.; Rudloff, I.; Nold-Petry, C.A.; Forster, S.C.; Nold, M.F. Understanding respiratory microbiome–immune system interactions in health and disease. Sci. Transl. Med. 2023, 15, eabq5126. [Google Scholar] [CrossRef] [PubMed]
- Bassis, C.M.; Tang, A.L.; Young, V.B.; Pynnonen, M.A. The nasal cavity microbiota of healthy adults. Microbiome 2014, 2, 27. [Google Scholar] [CrossRef]
- Stearns, J.C.; Davidson, C.J.; McKeon, S.; Whelan, F.J.; Fontes, M.E.; Schryvers, A.B.; Bowdish, D.M.E.; Kellner, J.D.; Surette, M.G. Culture and molecular-based profiles show shifts in bacterial communities of the upper respiratory tract that occur with age. ISME J. 2015, 9, 1246–1259. [Google Scholar] [CrossRef] [PubMed]
- Baehren, C.; Buedding, E.; Bellm, A.; Schult, F.; Pembaur, A.; Wirth, S.; Ehrhardt, A.; Paulsen, F.; Postberg, J.; Aydin, M. The Relevance of the Bacterial Microbiome, Archaeome and Mycobiome in Pediatric Asthma and Respiratory Disorders. Cells 2022, 11, 1287. [Google Scholar] [CrossRef]
- Nemergut, D.R.; Schmidt, S.K.; Fukami, T.; O’Neill, S.P.; Bilinski, T.M.; Stanish, L.F.; Knelman, J.E.; Darcy, J.L.; Lynch, R.C.; Wickey, P.; et al. Patterns and Processes of Microbial Community Assembly. Microbiol. Mol. Biol. Rev. 2013, 77, 342–356. [Google Scholar] [CrossRef]
- Minot, S.; Bryson, A.; Chehoud, C.; Wu, G.D.; Lewis, J.D.; Bushman, F.D. Rapid evolution of the human gut virome. Proc. Natl. Acad. Sci. USA 2013, 110, 12450–12455. [Google Scholar] [CrossRef]
- Rajagopala, S.V.; Bakhoum, N.G.; Pakala, S.B.; Shilts, M.H.; Rosas-Salazar, C.; Mai, A.; Boone, H.H.; McHenry, R.; Yooseph, S.; Halasa, N.; et al. Metatranscriptomics to characterize respiratory virome, microbiome, and host response directly from clinical samples. Cell Rep. Methods 2021, 1, 100091. [Google Scholar] [CrossRef]
- Rubió, J.R.; Megremis, S.; Pasioti, M.; Lakoumentas, J.; Constantinides, B.; Xepapadaki, P.; Bachert, C.; Finotto, S.; Jartti, T.; Andreakos, E.; et al. Respiratory virome profiles reflect antiviral immune responses. Allergy 2023, 78, 1258–1268. [Google Scholar] [CrossRef]
- Jankauskaitė, L.; Misevičienė, V.; Vaidelienė, L.; Kėvalas, R. Lower Airway Virology in Health and Disease—From Invaders to Symbionts. Medicina 2018, 54, 72. [Google Scholar] [CrossRef]
- Porto, B.N. Insights Into the Role of the Lung Virome During Respiratory Viral Infections. Front. Immunol. 2022, 13, 885341. [Google Scholar] [CrossRef] [PubMed]
- Tessler, M.; Neumann, J.S.; Afshinnekoo, E.; Pineda, M.; Hersch, R.; Velho, L.F.M.; Segovia, B.T.; Lansac-Toha, F.A.; Lemke, M.; DeSalle, R.; et al. Large-scale differences in microbial biodiversity discovery between 16S amplicon and shotgun sequencing. Sci. Rep. 2017, 7, 6589. [Google Scholar] [CrossRef]
- Brumfield, K.D.; Huq, A.; Colwell, R.R.; Olds, J.; Leddy, M.B. Microbial resolution of whole genome shotgun and 16S amplicon metagenomic sequencing using publicly available NEON data. PLoS ONE 2020, 15, e0228899. [Google Scholar] [CrossRef] [PubMed]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High throughput Sequence Data. 2010. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 10 April 2023).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar] [CrossRef]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve years of SAMtools and BCFtools. GigaScience 2021, 10, giab008. [Google Scholar] [CrossRef] [PubMed]
- Picard Toolkit. Broad Institute, GitHub Repository. 2019. Available online: https://broadinstitute.github.io/picard/ (accessed on 10 April 2023).
- Wood, D.E.; Salzberg, S.L. Kraken: Ultrafast metagenomic sequence classification using exact alignments. Genome Biol. 2014, 15, R46. [Google Scholar] [CrossRef]
- Hadzega, D.; Minarik, G.; Karaba, M.; Kalavska, K.; Benca, J.; Ciernikova, S.; Sedlackova, T.; Nemcova, P.; Bohac, M.; Pindak, D.; et al. Uncovering Microbial Composition in Human Breast Cancer Primary Tumour Tissue Using Transcriptomic RNA-seq. Int. J. Mol. Sci. 2021, 22, 9058. [Google Scholar] [CrossRef] [PubMed]
- Meleshko, D.; Hajirasouliha, I.; Korobeynikov, A. CoronaSPAdes: From biosynthetic gene clusters to RNA viral assemblies. BioRxiv 2021, 38, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Maier, W.; Batut, B. Mutation Calling, Viral Genome Reconstruction and Lineage/Clade Assignment from SARS-CoV-2 Sequencing Data. 2023. Available online: https://training.galaxyproject.org/training-material/topics/variant-analysis/tutorials/sars-cov-2-variant-discovery/tutorial.html (accessed on 14 April 2023).
- Lu, J.; Rincon, N.; Wood, D.E.; Breitwieser, F.P.; Pockrandt, C.; Ben Langmead, B.; Salzberg, S.L.; Steinegger, M. Metagenome analysis using the Kraken software suite. Nat. Protoc. 2022, 17, 2815–2839. [Google Scholar] [CrossRef]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef] [PubMed]
- Mallick, H.; Rahnavard, A.; McIver, L.J.; Ma, S.; Zhang, Y.; Nguyen, L.H.; Tickle, T.L.; Weingart, G.; Ren, B.; Schwager, E.H.; et al. Multivariable association discovery in population-scale meta-omics studies. PLoS Comput. Biol. 2021, 17, e1009442. [Google Scholar] [CrossRef]
- Hartmann, J.E.; Albrich, W.C.; Dmitrijeva, M.; Kahlert, C.R. The Effects of Corticosteroids on the Respiratory Microbiome: A Systematic Review. Front. Med. 2021, 8, 588584. [Google Scholar] [CrossRef]
- Shilts, M.H.; Rosas-Salazar, C.; Strickland, B.A.; Kimura, K.S.; Asad, M.; Sehanobish, E.; Freeman, M.H.; Wessinger, B.C.; Gupta, V.; Brown, H.M.; et al. Severe COVID-19 Is Associated With an Altered Upper Respiratory Tract Microbiome. Front. Cell. Infect. Microbiol. 2022, 11, 781968. [Google Scholar] [CrossRef]
- Li, J.; Jing, Q.; Li, J.; Hua, M.; Di, L.; Song, C.; Huang, Y.; Wang, J.; Chen, C.; Wu, A.R. Assessment of microbiota in the gut and upper respiratory tract associated with SARS-CoV-2 infection. Microbiome 2023, 11, 38. [Google Scholar] [CrossRef]
- Liu, J.; Liu, S.; Zhang, Z.; Lee, X.; Wu, W.; Huang, Z.; Lei, Z.; Xu, W.; Chen, D.; Wu, X.; et al. Association between the nasopharyngeal microbiome and metabolome in patients with COVID-19. Synth. Syst. Biotechnol. 2021, 6, 135–143. [Google Scholar] [CrossRef]
- Ventero, M.P.; Cuadrat, R.R.C.; Vidal, I.; Andrade, B.G.N.; Molina-Pardines, C.; Haro-Moreno, J.M.; Coutinho, F.H.; Merino, E.; Regitano, L.C.A.; Silveira, C.B.; et al. Nasopharyngeal Microbial Communities of Patients Infected With SARS-CoV-2 That Developed COVID-19. Front. Microbiol. 2021, 12, 637430. [Google Scholar] [CrossRef]
- Rattanaburi, S.; Sawaswong, V.; Chitcharoen, S.; Sivapornnukul, P.; Nimsamer, P.; Suntronwong, N.; Puenpa, J.; Poovorawan, Y.; Payungporn, S. Bacterial microbiota in upper respiratory tract of COVID-19 and influenza patients. Exp. Biol. Med. 2022, 247, 409–415. [Google Scholar] [CrossRef]
- Horn, K.J.; Schopper, M.A.; Drigot, Z.G.; Clark, S.E. Airway Prevotella promote TLR2-dependent neutrophil activation and rapid clearance of Streptococcus pneumoniae from the lung. Nat. Commun. 2022, 13, 3321. [Google Scholar] [CrossRef] [PubMed]
- Looney, W.J.; Narita, M.; Mühlemann, K. Stenotrophomonas maltophilia: An emerging opportunist human pathogen. Lancet Infect. Dis. 2009, 9, 312–323. [Google Scholar] [CrossRef]
- Bello-Morales, R.; Andreu, S.; López-Guerrero, J.A. The Role of Herpes Simplex Virus Type 1 Infection in Demyelination of the Central Nervous System. Int. J. Mol. Sci. 2020, 21, 5026. [Google Scholar] [CrossRef] [PubMed]
- Bloom, D.C. HSV LAT and neuronal survival. Int. Rev. Immunol. 2004, 23, 187–198. [Google Scholar] [CrossRef]
- Jacobs, S.E.; Lamson, D.M.; George, K.S.; Walsh, T.J. Human Rhinoviruses. Clin. Microbiol. Rev. 2013, 26, 135–162. [Google Scholar] [CrossRef]
- Lynch, J.P., 3rd; Kajon, A.E. Adenovirus: Epidemiology, Global Spread of Novel Serotypes, and Advances in Treatment and Prevention. Semin. Respir. Crit. Care Med. 2016, 37, 586–602. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
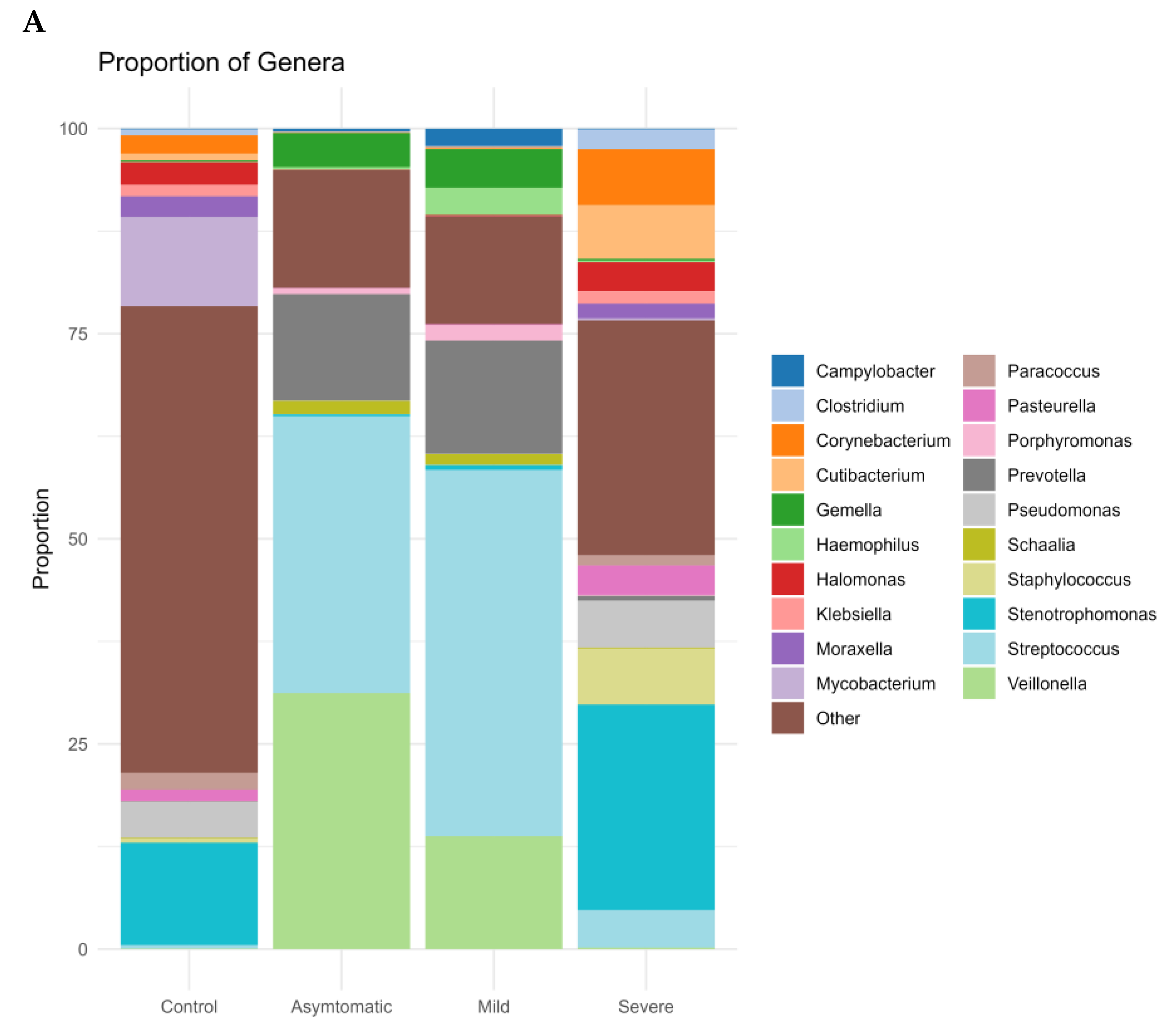
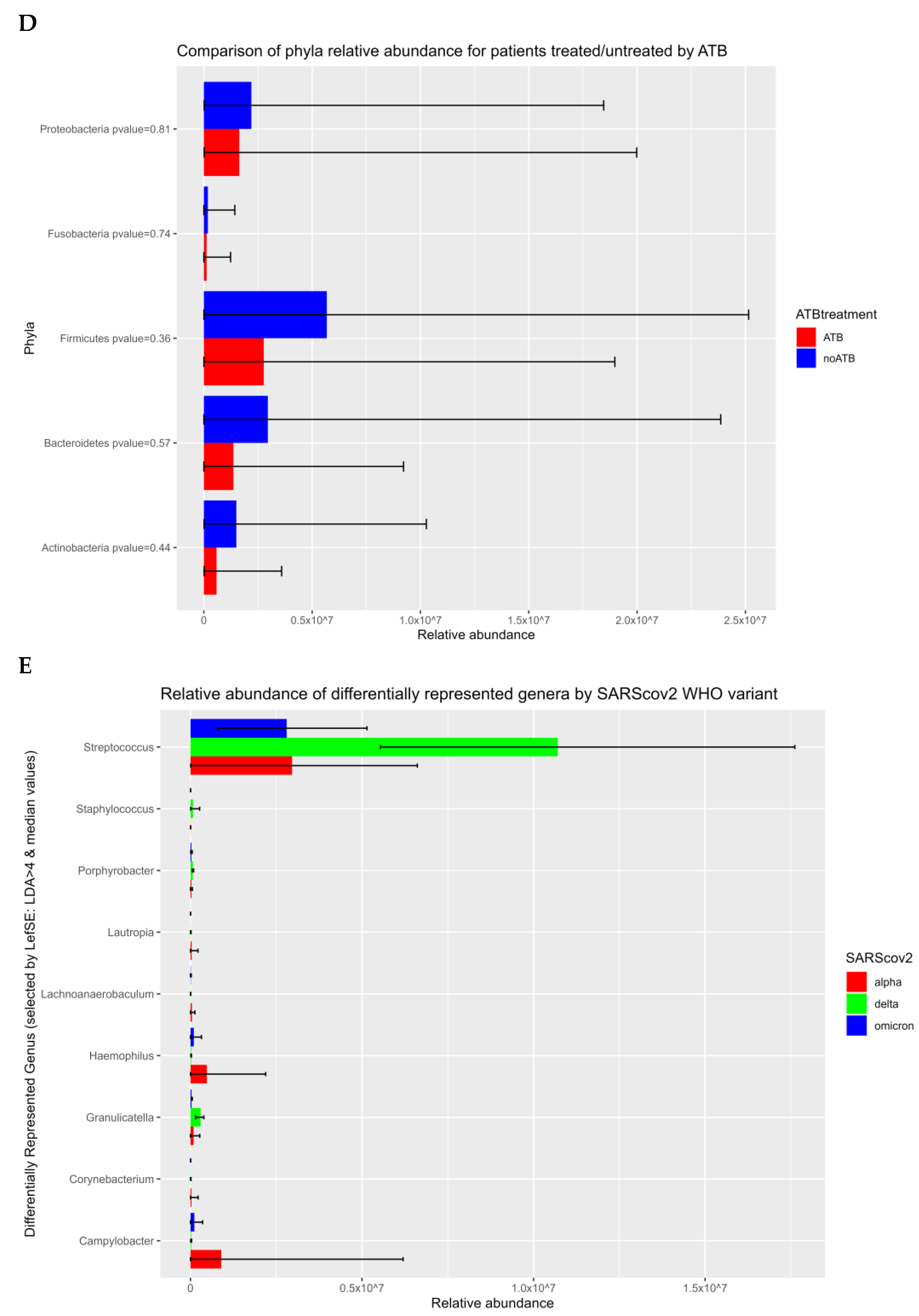{kind=link}
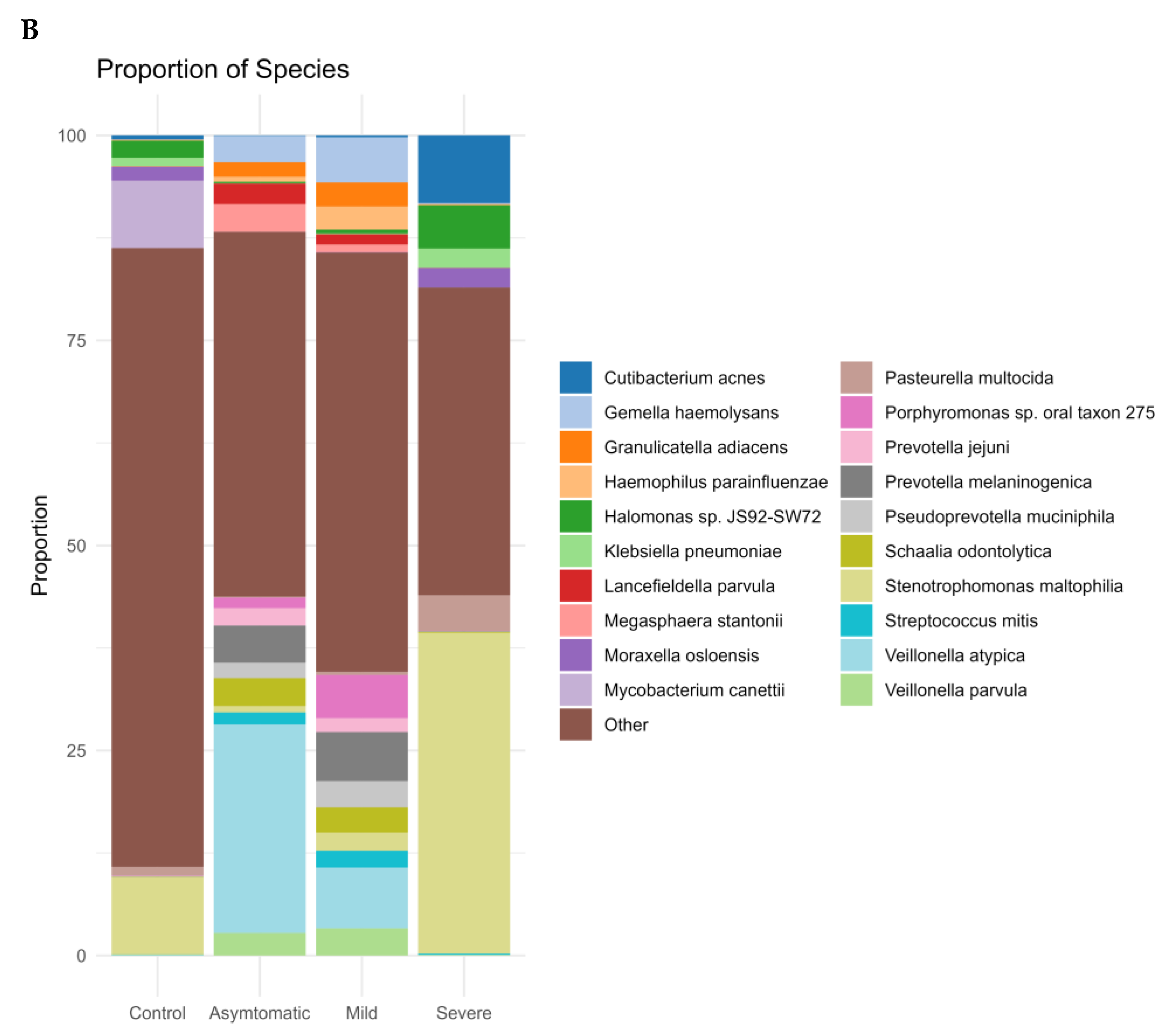
{kind=link}
{kind=link}
| Number | Median (Years) | |
|---|---|---|
| Age | 151 | 39.5 (17–90) |
| Negative controls | 72 | 37 (25–75) |
| male | 26 | 42 (25–58) |
| female | 46 | 35 (25–75) |
| Severe | 30 | 68 (32–90) |
| male | 16 | 69.5 (32–90) |
| female | 14 | 64 (41–77) |
| Mild | 25 | 37 (17–57) |
| male | 13 | 38 (17–57) |
| female | 12 | 32.5 (19–57) |
| Asymtomatic | 24 | 42 (20–49) |
| male | 14 | 39 (21–49) |
| female | 10 | 43 (36–48) |
| BMI | 132 | |
| Negative controls | 57 | 24.48 (17.2–42.4) |
| male | 25 | 28.4 (19.7–42.4) |
| female | 32 | 21 (17.2–32.6) |
| Severe | 19 | 30.39 (23.33–49.21) |
| male | 8 | 30 (23.66–34.60) |
| female | 11 | 30 (23.33–49.21) |
| Mild | 25 | 25.64 (18.62–33.46) |
| male | 13 | 28.27 (21.32–33.46) |
| female | 12 | 21.71 (18.62–29.15) |
| Asymtomatic | 24 | 25.72 (16.9–32.5) |
| male | 14 | 28.10 (23.54–32.50) |
| female | 10 | 20.73 (16.9–30.11) |
| Associated diseases | 38/151 | 25.16% |
| Diabetes | 12/151 | 7.94% |
| severe | 10 | 33.33% |
| mild | 1 | 4% |
| negative controls | 1 | 1.30% |
| Arterial hypertension | 21/151 | 13.90% |
| severe | 15 | 50% |
| asymtomatic | 3 | 12.50% |
| negative controls | 2 | 2.70% |
| mild | 1 | 4% |
| Ischaemic heart disease | 7/151 | 4.63% |
| only severe | 7 | 23.33% |
| Oncological disease | 4/151 | 2.64% |
| severe | 3 | 10% |
| negative controls | 1 | 1.30% |
| Multiple diagnosis (more than 2 ) | 11/151 | 7.28% |
| severe | 10 | 33.33% |
| mild | 1 | 4% |
| ATB (severe only) | 30 | |
| Ruzinov hospital | 18 | |
| ATB | 18 | 100% |
| no ATB | 0 | 0% |
| Kramare hospital | 12 | |
| ATB | 2 | 16.70% |
| no ATB | 10 | 83.30% |
| Median Ct value E gene | SD | |
| Severe | 28.06 (13.9-37.3) | 7.34 |
| Mild | 24.89 (15-38.77) | 5.32 |
| Asymptomatic | 28.56 (19.24-33.59) | 4.72 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hyblova, M.; Hadzega, D.; Babisova, K.; Krumpolec, P.; Gnip, A.; Sabaka, P.; Lassan, S.; Minarik, G. Metatranscriptome Analysis of Nasopharyngeal Swabs across the Varying Severity of COVID-19 Disease Demonstrated Unprecedented Species Diversity. Microorganisms 2023, 11, 1804. https://doi.org/10.3390/microorganisms11071804
Hyblova M, Hadzega D, Babisova K, Krumpolec P, Gnip A, Sabaka P, Lassan S, Minarik G. Metatranscriptome Analysis of Nasopharyngeal Swabs across the Varying Severity of COVID-19 Disease Demonstrated Unprecedented Species Diversity. Microorganisms. 2023; 11(7):1804. https://doi.org/10.3390/microorganisms11071804
Chicago/Turabian StyleHyblova, Michaela, Dominik Hadzega, Klaudia Babisova, Patrik Krumpolec, Andrej Gnip, Peter Sabaka, Stefan Lassan, and Gabriel Minarik. 2023. "Metatranscriptome Analysis of Nasopharyngeal Swabs across the Varying Severity of COVID-19 Disease Demonstrated Unprecedented Species Diversity" Microorganisms 11, no. 7: 1804. https://doi.org/10.3390/microorganisms11071804
APA StyleHyblova, M., Hadzega, D., Babisova, K., Krumpolec, P., Gnip, A., Sabaka, P., Lassan, S., & Minarik, G. (2023). Metatranscriptome Analysis of Nasopharyngeal Swabs across the Varying Severity of COVID-19 Disease Demonstrated Unprecedented Species Diversity. Microorganisms, 11(7), 1804. https://doi.org/10.3390/microorganisms11071804