
The Genome of Varunaivibrio sulfuroxidans Strain TC8T, a Metabolically Versatile Alphaproteobacterium from the Tor Caldara Gas Vents in the Tyrrhenian Sea

Abstract
1. Introduction
2. Materials and Methods
2.1. Genome Project History
2.2. Growth Conditions and Genomic DNA Preparation
2.3. Genome Sequencing and Assembly
2.4. Genome Annotation
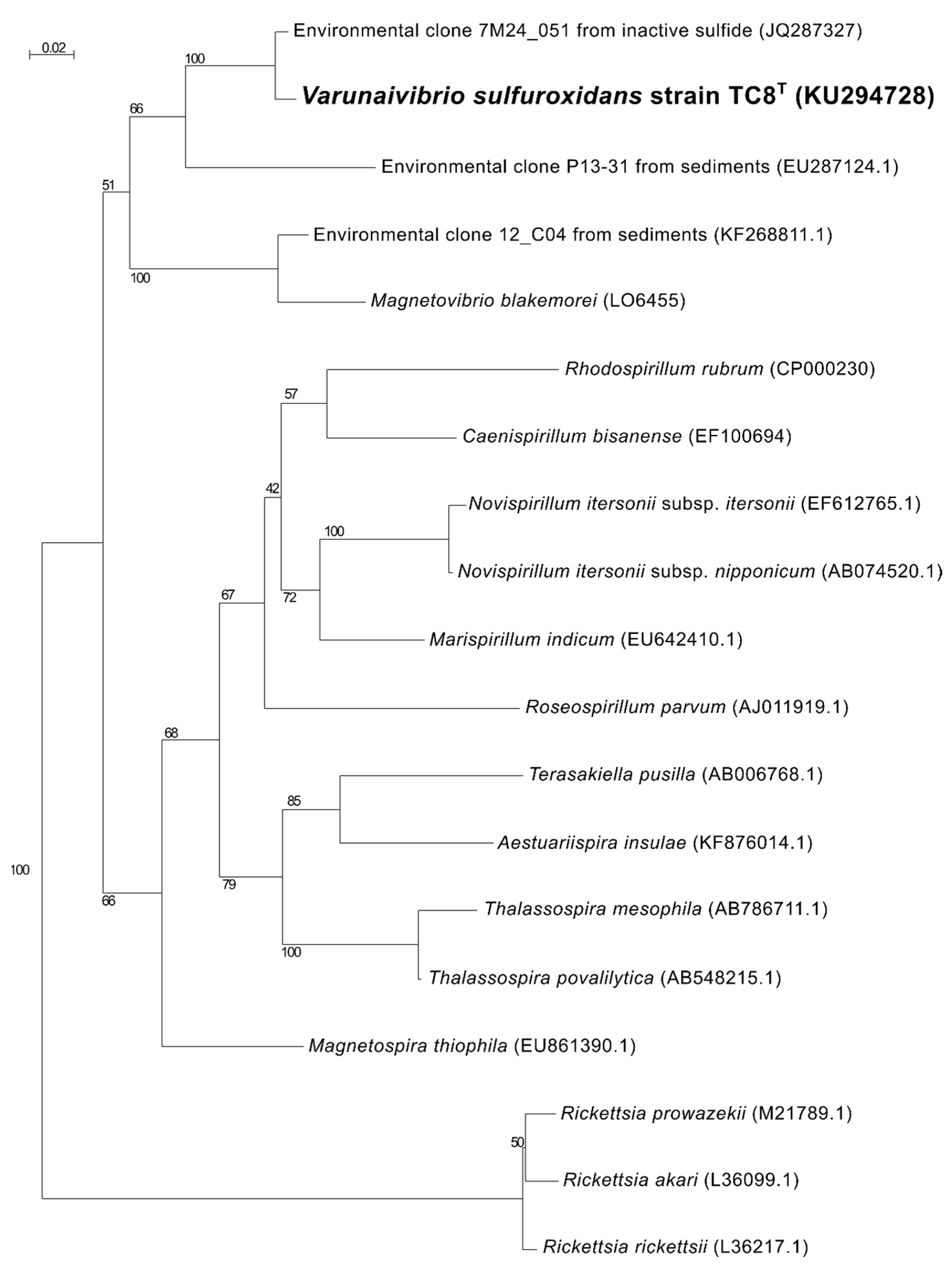
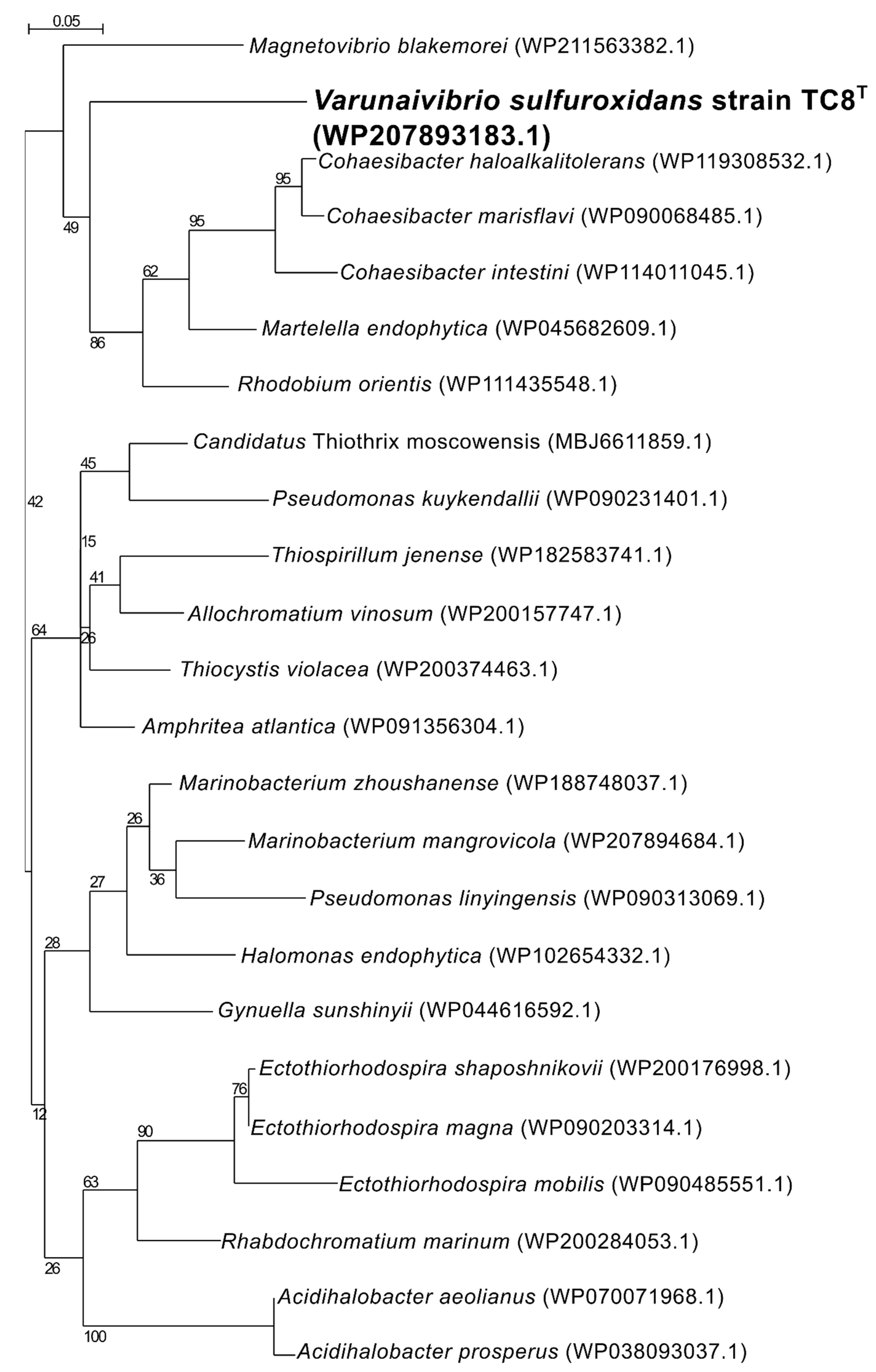
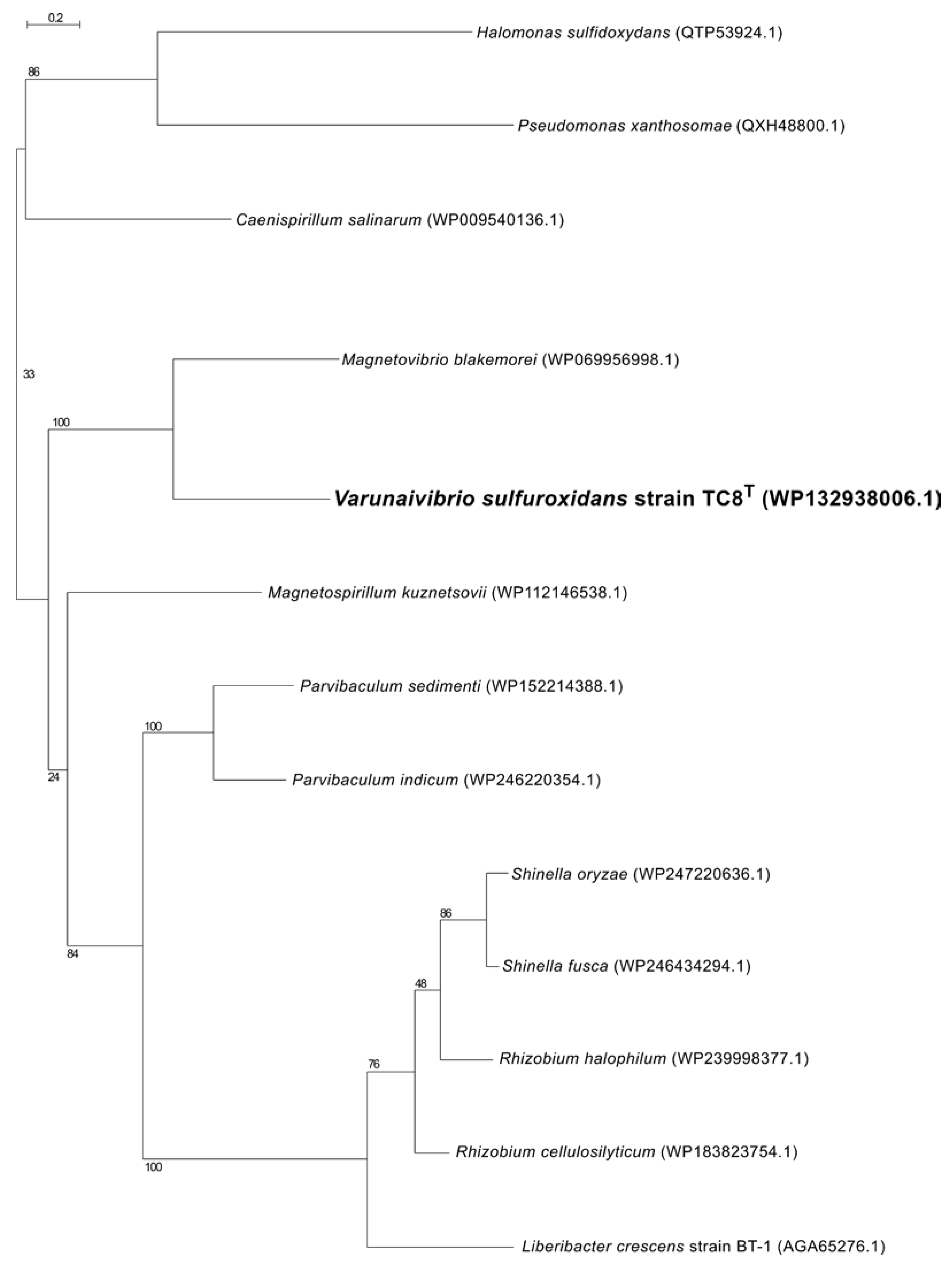
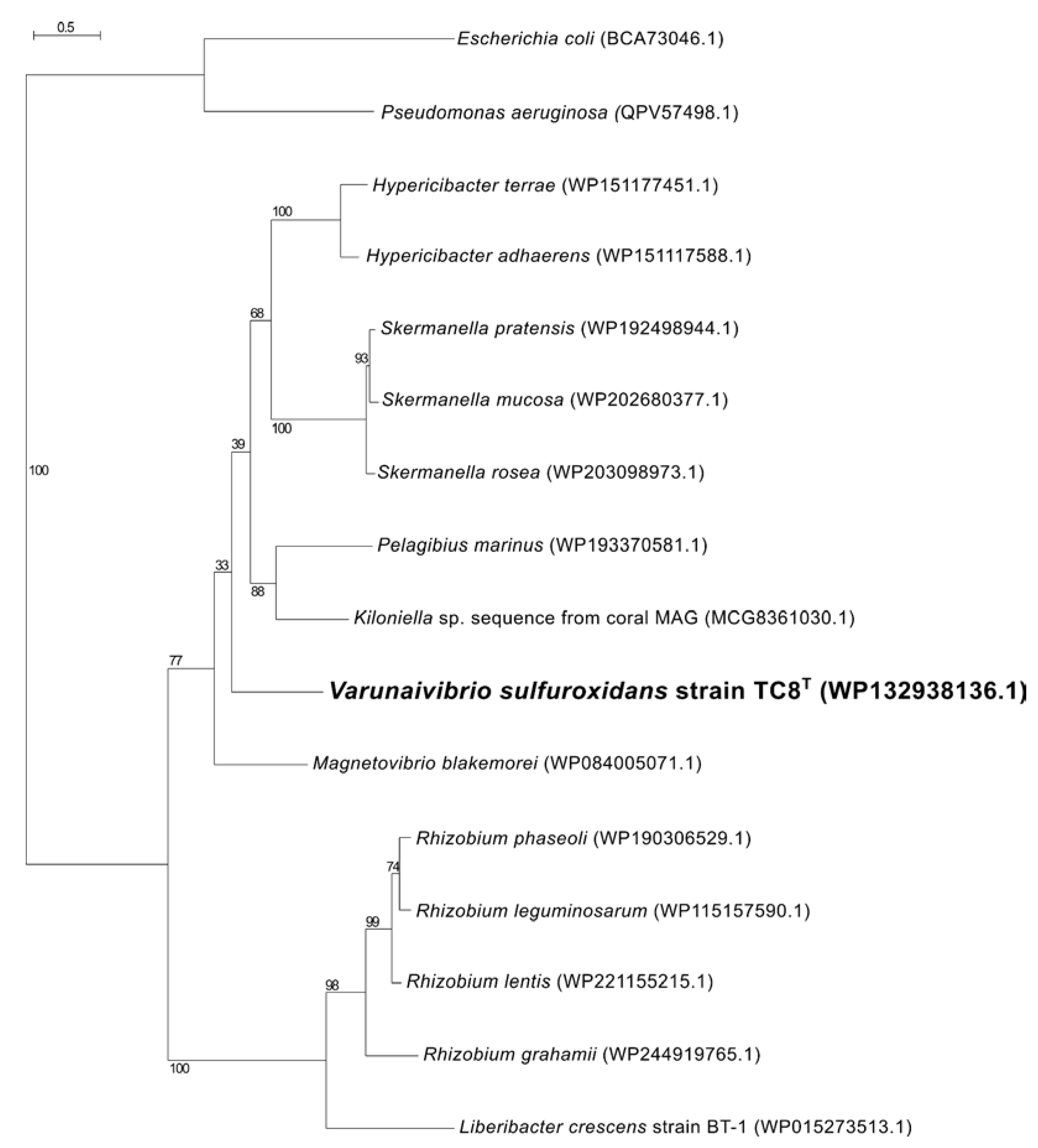
2.5. Phylogenetic, Average Nucleotide Identity and Digital DNA–DNA Hybridization Analyses
3. Results and Discussion
3.1. Overview of Varunaivibrio Sulfuroxidans Strain TC8T
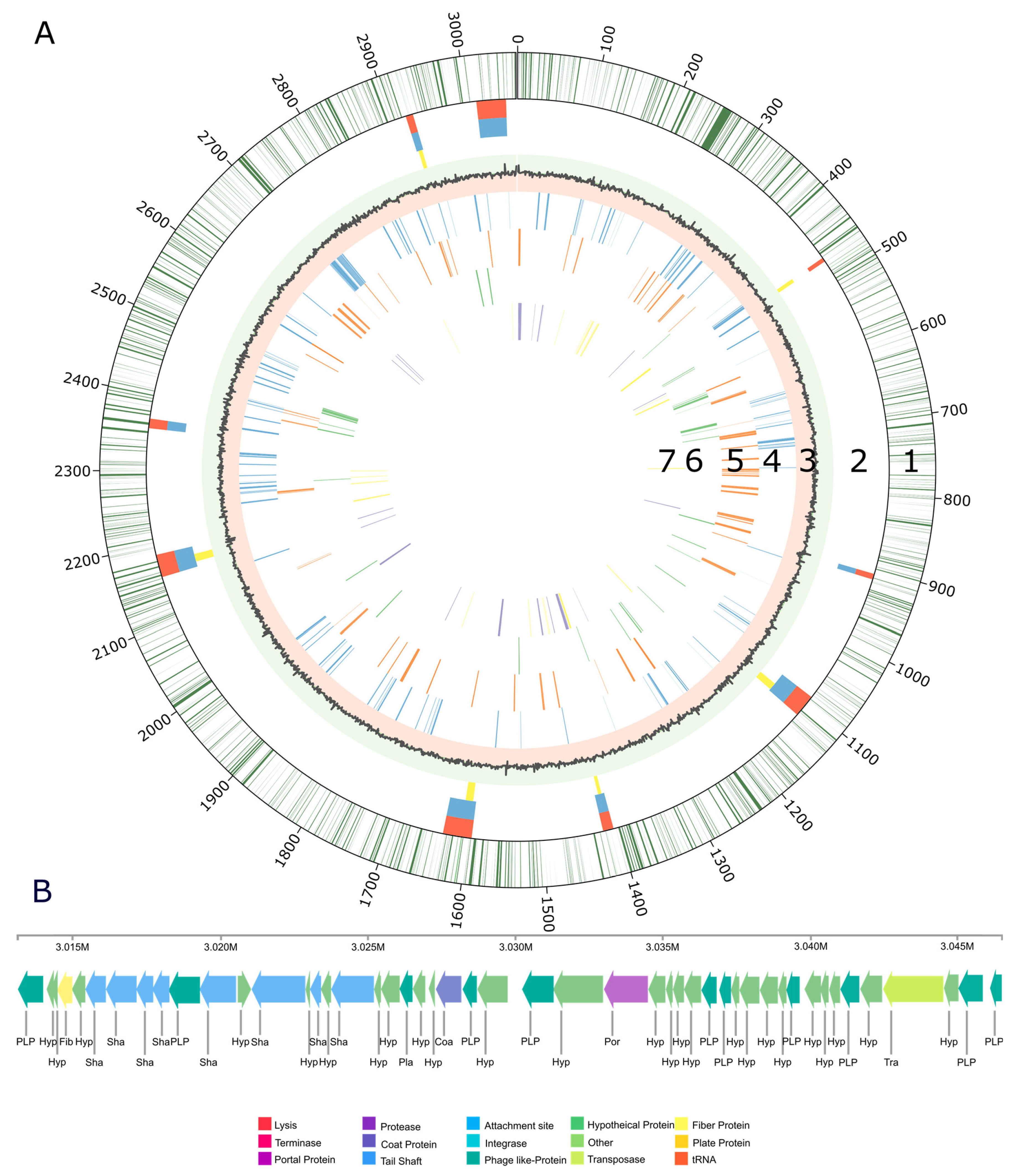
3.2. Genome Structure
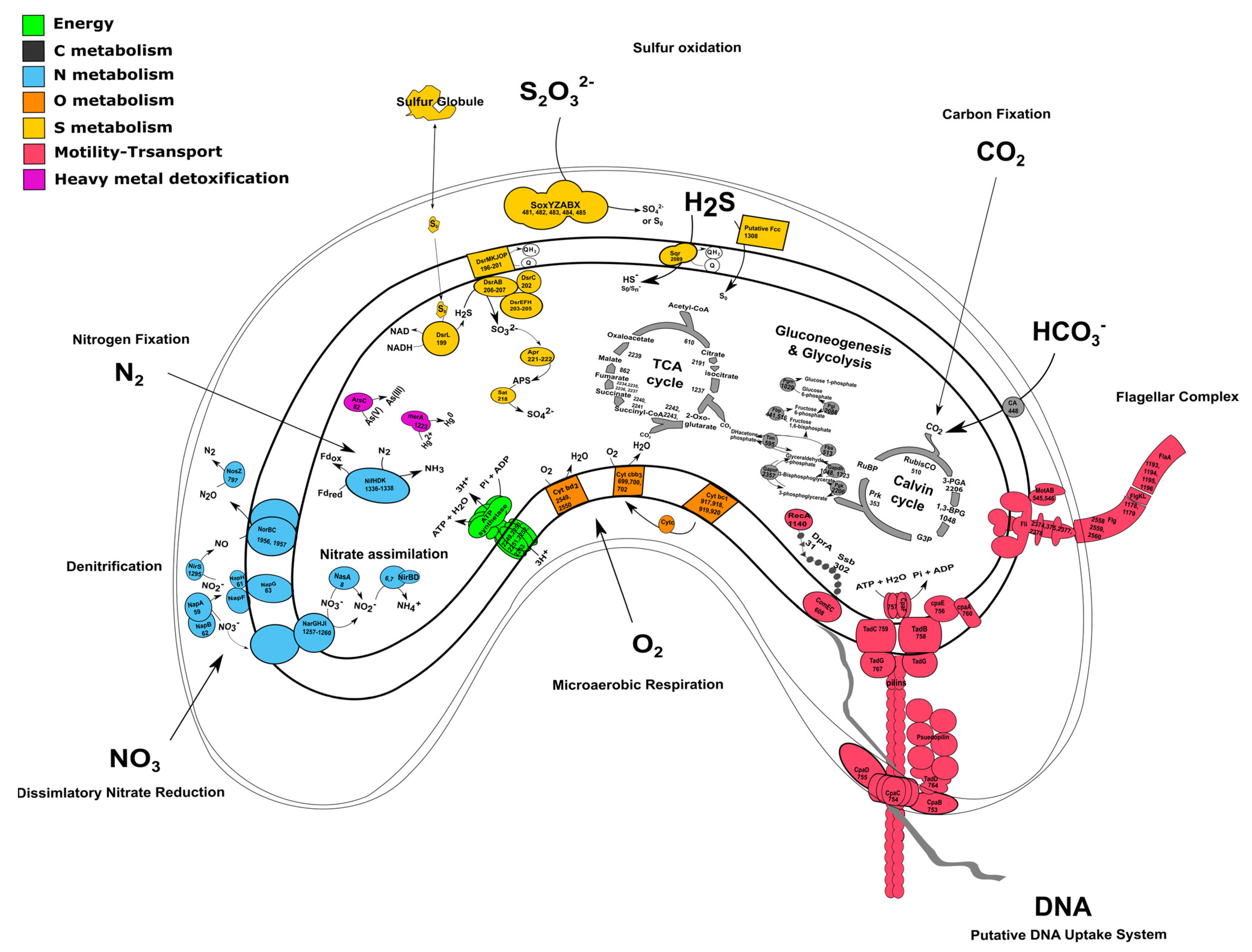
3.3. Carbon Metabolism and Microaerobic Respiration
3.4. Sulfur Metabolism
3.5. Nitrogen Metabolism
3.6. Heavy Metal Detoxification
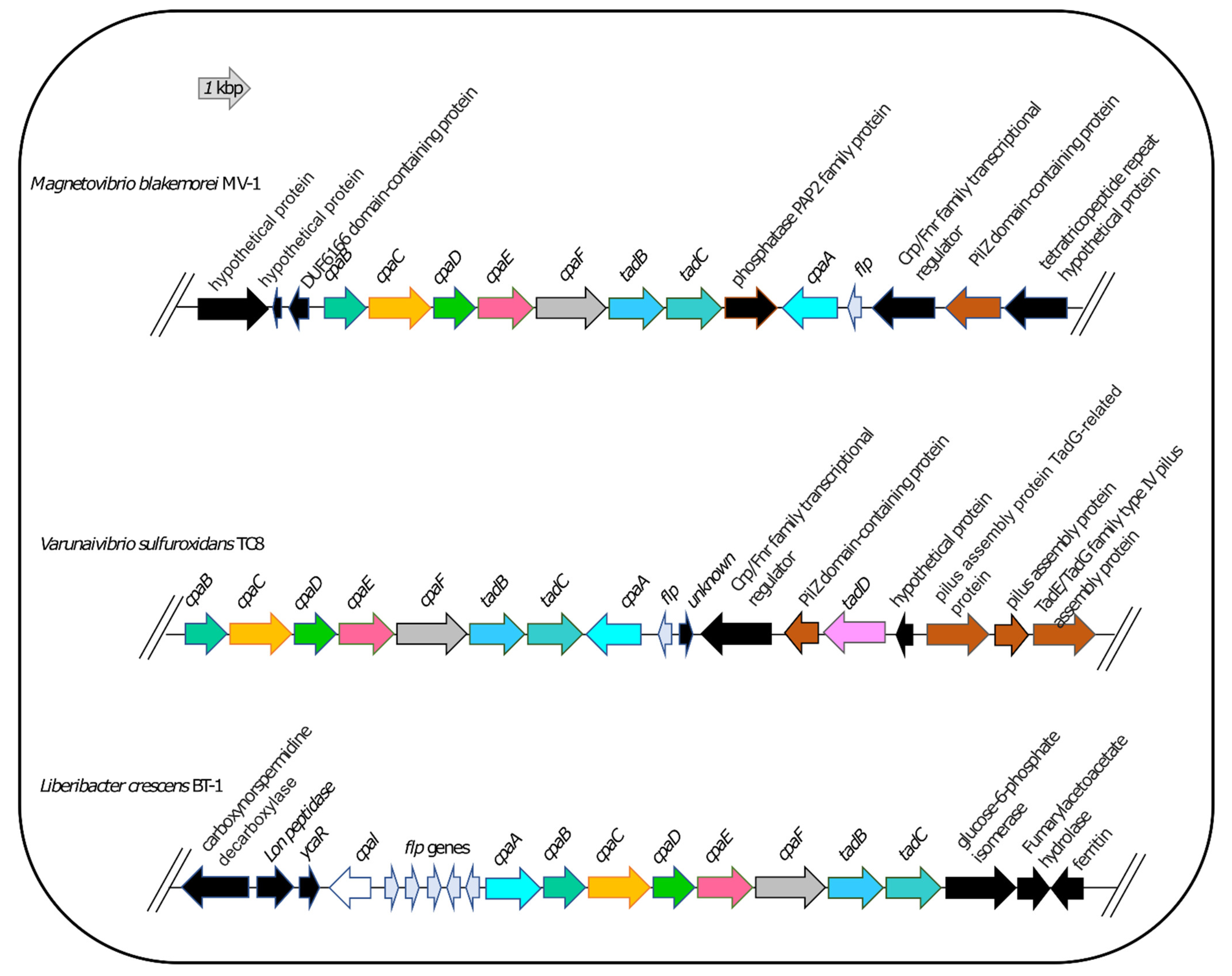
3.7. DNA Uptake System
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Patwardhan, S.; Foustoukos, D.I.; Giovannelli, D.; Yücel, M.; Vetriani, C. Ecological Succession of Sulfur-Oxidizing Epsilon- and Gammaproteobacteria During Colonization of a Shallow-Water Gas Vent. Front. Microbiol. 2018, 9, 2970. [Google Scholar] [CrossRef] [PubMed]
- Carapezza, M.L.; Barberi, F.; Ranaldi, M.; Ricci, T.; Tarchini, L.; Barrancos, J.; Fischer, C.; Granieri, D.; Lucchetti, C.; Melian, G.; et al. Hazardous gas emissions from the flanks of the quiescent Colli Albani volcano (Rome, Italy). Appl. Geochem. 2012, 27, 1767–1782. [Google Scholar] [CrossRef]
- Patwardhan, S.; Smedile, F.; Giovannelli, D.; Vetriani, C. Metaproteogenomic Profiling of Chemosynthetic Microbial Biofilms Reveals Metabolic Flexibility during Colonization of a Shallow-Water Gas Vent. Front. Microbiol. 2021, 12, 638300. [Google Scholar] [CrossRef] [PubMed]
- Patwardhan, S.S. Prokaryotic diversity, physiology and function at Tor Caldara, a shallow-water gas vent in the Tyrrhenian Sea. Doctoral Dissertation, Rutgers University-School of Graduate Studies, New Brunswick, NJ, USA, 2018. [Google Scholar] [CrossRef]
- Lopez-Garcia, P.; Duperron, S.; Philippot, P.; Foriel, J.; Susini, J.; Moreira, D. Bacterial diversity in hydrothermal sediment and epsilonproteobacterial dominance in experimental microcolonizers at the Mid-Atlantic Ridge. Environ. Microbiol. 2003, 5, 961–976. [Google Scholar] [CrossRef] [PubMed]
- Giovannelli, D.; d’Errico, G.; Manini, E.; Yakimov, M.M.; Vetriani, C. Diversity and phylogenetic analyses of bacteria from a shallow-water hydrothermal vent in Milos island (Greece). Front. Microbiol. 2013, 4, 184. [Google Scholar] [CrossRef]
- O’Brien, C.E.; Giovannelli, D.; Govenar, B.; Luther, G.W.; Lutz, R.A.; Shank, T.M.; Vetriani, C. Microbial biofilms associated with fluid chemistry and megafaunal colonization at post-eruptive deep-sea hydrothermal vents. Deep Sea Res. Part II Top. Stud. Oceanogr. 2015, 121, 31–40. [Google Scholar] [CrossRef]
- Pérez-Rodríguez, I.; Bolognini, M.; Ricci, J.; Bini, E.; Vetriani, C. From deep-sea volcanoes to human pathogens: A conserved quorum-sensing signal in Epsilonproteobacteria. ISME J. 2015, 9, 1222–1234. [Google Scholar] [CrossRef]
- Huber, J.A.; Mark Welch, D.B.; Morrison, H.G.; Huse, S.M.; Neal, P.R.; Butterfield, D.A.; Sogin, M.L. Microbial Population Structures in the Deep Marine Biosphere. Science 2007, 318, 97–100. [Google Scholar] [CrossRef]
- Miranda, P.J.; McLain, N.K.; Hatzenpichler, R.; Orphan, V.J.; Dillon, J.G. Characterization of Chemosynthetic Microbial Mats Associated with Intertidal Hydrothermal Sulfur Vents in White Point, San Pedro, CA, USA. Front. Microbiol. 2016, 7, 1163. [Google Scholar] [CrossRef]
- Kerfahi, D.; Hall-Spencer, J.M.; Tripathi, B.M.; Milazzo, M.; Lee, J.; Adams, J.M. Shallow Water Marine Sediment Bacterial Community Shifts Along a Natural CO2 Gradient in the Mediterranean Sea off Vulcano, Italy. Microb. Ecol. 2014, 67, 819–828. [Google Scholar] [CrossRef]
- Gugliandolo, C.; Lentini, V.; Bunk, B.; Overmann, J.; Italiano, F.; Maugeri, T.L. Changes in prokaryotic community composition accompanying a pronounced temperature shift of a shallow marine thermal brine pool (Panarea Island, Italy). Extremophiles 2015, 19, 547–559. [Google Scholar] [CrossRef] [PubMed]
- Lentini, V.; Gugliandolo, C.; Bunk, B.; Overmann, J.; Maugeri, T.L. Diversity of Prokaryotic Community at a Shallow Marine Hydrothermal Site Elucidated by Illumina Sequencing Technology. Curr. Microbiol. 2014, 69, 457–466. [Google Scholar] [CrossRef] [PubMed]
- Tang, K. Microbial Communities in a Shallow-Sea Hydrothermal System. In Encyclopedia of Metagenomics; Nelson, K.E., Ed.; Springer: New York, NY, USA, 2013; pp. 1–8. ISBN 978-1-4614-6418-1. [Google Scholar]
- Patwardhan, S.; Vetriani, C. Varunaivibrio sulfuroxidans gen. nov., sp. nov., a facultatively chemolithoautotrophic, mesophilic alphaproteobacterium from a shallow-water gas vent at Tor Caldara, Tyrrhenian Sea. Int. J. Syst. Evol. Microbiol. 2016, 66, 3579–3584. [Google Scholar] [CrossRef] [PubMed]
- Koziaeva, V.V.; Sorokin, D.Y.; Kolganova, T.V.; Grouzdev, D.S. Magnetospirillum sulfuroxidans sp. nov., capable of sulfur-dependent lithoautotrophy and a taxonomic reevaluation of the order Rhodospirillales. Syst. Appl. Microbiol. 2023, 46, 126406. [Google Scholar] [CrossRef] [PubMed]
- Hördt, A.; López, M.G.; Meier-Kolthoff, J.P.; Schleuning, M.; Weinhold, L.-M.; Tindall, B.J.; Gronow, S.; Kyrpides, N.C.; Woyke, T.; Göker, M. Analysis of 1000+ Type-Strain Genomes Substantially Improves Taxonomic Classification of Alphaproteobacteria. Front. Microbiol. 2020, 11, 468. [Google Scholar] [CrossRef] [PubMed]
- Williams, T.J.; Lefèvre, C.T.; Zhao, W.; Beveridge, T.J.; Bazylinski, D.A. Magnetospira thiophila gen. nov., sp. nov., a marine magnetotactic bacterium that represents a novel lineage within the Rhodospirillaceae (Alphaproteobacteria). Int. J. Syst. Evol. Microbiol. 2012, 62, 2443–2450. [Google Scholar] [CrossRef] [PubMed]
- Bazylinski, D.A.; Williams, T.J.; Lefevre, C.T.; Trubitsyn, D.; Fang, J.; Beveridge, T.J.; Moskowitz, B.M.; Ward, B.; Schubbe, S.; Dubbels, B.L.; et al. Magnetovibrio blakemorei gen. nov., sp. nov., a magnetotactic bacterium (Alphaproteobacteria: Rhodospirillaceae) isolated from a salt marsh. Int. J. Syst. Evol. Microbiol. 2013, 63, 1824–1833. [Google Scholar] [CrossRef]
- Tang, K.; Zhang, Y.; Lin, D.; Han, Y.; Chen, C.-T.A.; Wang, D.; Lin, Y.-S.; Sun, J.; Zheng, Q.; Jiao, N. Cultivation-Independent and Cultivation-Dependent Analysis of Microbes in the Shallow-Sea Hydrothermal System off Kueishantao Island, Taiwan: Unmasking Heterotrophic Bacterial Diversity and Functional Capacity. Front. Microbiol. 2018, 9, 279. [Google Scholar] [CrossRef]
- Jain, M.; Olsen, H.E.; Paten, B.; Akeson, M. The Oxford Nanopore MinION: Delivery of nanopore sequencing to the genomics community. Genome Biol. 2016, 17, 239. [Google Scholar] [CrossRef]
- Inagaki, F.; Takai, K.; Nealson, K.H.; Horikoshi, K. Sulfurovum lithotrophicum gen. nov., sp. nov., a novel sulfur-oxidizing chemolithoautotroph within the ε-Proteobacteria isolated from Okinawa trough hydrothermal sediments. Int. J. Syst. Evol. Microbiol. 2004, 54, 1477–1482. [Google Scholar] [CrossRef]
- Wick, R.R.; Judd, L.M.; Gorrie, C.L.; Holt, K.E. Unicycler: Resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput. Biol. 2017, 13, e1005595. [Google Scholar] [CrossRef] [PubMed]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed]
- Chen, I.-M.A.; Markowitz, V.M.; Chu, K.; Palaniappan, K.; Szeto, E.; Pillay, M.; Ratner, A.; Huang, J.; Andersen, E.; Huntemann, M.; et al. IMG/M: Integrated genome and metagenome comparative data analysis system. Nucleic Acids Res. 2017, 45, D507–D516. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Sato, Y.; Morishima, K. BlastKOALA and GhostKOALA: KEGG Tools for Functional Characterization of Genome and Metagenome Sequences. J. Mol. Biol. 2016, 428, 726–731. [Google Scholar] [CrossRef] [PubMed]
- Grissa, I.; Vergnaud, G.; Pourcel, C. CRISPRFinder: A web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic Acids Res. 2007, 35, W52–W57. [Google Scholar] [CrossRef]
- Arndt, D.; Grant, J.R.; Marcu, A.; Sajed, T.; Pon, A.; Liang, Y.; Wishart, D.S. PHASTER: A better, faster version of the PHAST phage search tool. Nucleic Acids Res. 2016, 44, W16–W21. [Google Scholar] [CrossRef]
- Bertelli, C.; Laird, M.R.; Williams, K.P.; Simon Fraser University Research Computing Group; Lau, B.Y.; Hoad, G.; Winsor, G.L.; Brinkman , F.S. IslandViewer 4: Expanded prediction of genomic islands for larger-scale datasets. Nucleic Acids Res. 2017, 45, W30–W35. [Google Scholar] [CrossRef]
- Rancurel, C.; Legrand, L.; Danchin, E. Alienness: Rapid Detection of Candidate Horizontal Gene Transfers across the Tree of Life. Genes 2017, 8, 248. [Google Scholar] [CrossRef]
- Reed, A.J.; Dorn, R.; Van Dover, C.L.; Lutz, R.A.; Vetriani, C. Phylogenetic diversity of methanogenic, sulfate-reducing and methanotrophic prokaryotes from deep-sea hydrothermal vents and cold seeps. Deep Sea Res. Part II Top. Stud. Oceanogr. 2009, 56, 1665–1674. [Google Scholar] [CrossRef]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef]
- Gouy, M.; Guindon, S.; Gascuel, O. SeaView Version 4: A Multiplatform Graphical User Interface for Sequence Alignment and Phylogenetic Tree Building. Mol. Biol. Evol. 2010, 27, 221–224. [Google Scholar] [CrossRef] [PubMed]
- Guindon, S.; Dufayard, J.-F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New Algorithms and Methods to Estimate Maximum-Likelihood Phylogenies: Assessing the Performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Holmquist, R.; Cantor, C.; Jukes, T. Improved procedures for comparing homologous sequences in molecules of proteins and nucleic acids. J. Mol. Biol. 1972, 64, 145–161. [Google Scholar] [CrossRef]
- Le, S.Q.; Gascuel, O. An Improved General Amino Acid Replacement Matrix. Mol. Biol. Evol. 2008, 25, 1307–1320. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.-H.; Ha, S.; Lim, J.; Kwon, S.; Chun, J. A large-scale evaluation of algorithms to calculate average nucleotide identity. Antonie Van Leeuwenhoek 2017, 110, 1281–1286. [Google Scholar] [CrossRef]
- Meier-Kolthoff, J.P.; Carbasse, J.S.; Peinado-Olarte, R.L.; Göker, M. TYGS and LPSN: A database tandem for fast and reliable genome-based classification and nomenclature of prokaryotes. Nucleic Acids Res. 2022, 50, D801–D807. [Google Scholar] [CrossRef]
- Sylvan, J.B.; Toner, B.M.; Edwards, K.J. Life and Death of Deep-Sea Vents: Bacterial Diversity and Ecosystem Succession on Inactive Hydrothermal Sulfides. MBio 2012, 3, e00279-11. [Google Scholar] [CrossRef]
- Lin, D.; Tang, K.; Han, Y.; Li, C.; Chen, X. Genome sequence of an inducible phage in Rhodovulum sp. P5 isolated from the shallow-sea hydrothermal system. Mar. Genom. 2016, 30, 93–95. [Google Scholar] [CrossRef]
- Tabita, F.R.; Hanson, T.E.; Li, H.; Satagopan, S.; Singh, J.; Chan, S. Function, Structure, and Evolution of the RubisCO-Like Proteins and Their RubisCO Homologs. Microbiol. Mol. Biol. Rev. 2007, 71, 576–599. [Google Scholar] [CrossRef]
- Friedrich, C.G.; Rother, D.; Bardischewsky, F.; Quentmeier, A.; Fischer, J. Oxidation of Reduced Inorganic Sulfur Compounds by Bacteria: Emergence of a Common Mechanism? Appl. Environ. Microbiol. 2001, 67, 2873–2882. [Google Scholar] [CrossRef]
- Hensen, D.; Sperling, D.; Trüper, H.G.; Brune, D.C.; Dahl, C. Thiosulphate oxidation in the phototrophic sulphur bacterium Allochromatium vinosum. Mol. Microbiol. 2006, 62, 794–810. [Google Scholar] [CrossRef] [PubMed]
- Dahl, C.; Friedrich, C.; Kletzin, A. Sulfur Oxidation in Prokaryotes. eLS 2008. [Google Scholar] [CrossRef]
- Griesbeck, C.; Hauska, G.; Schütz, M. Biological sulfide oxidation: Sulfide-quinone reductase (SQR), the primary reaction. In Recent Research Developments in Microbiology; Research Signpost: Trivadrum, India, 2000; Volume 4, pp. 179–203. [Google Scholar]
- Brune, D.C. Sulfur Compounds as Photosynthetic Electron Donors. In Anoxygenic Photosynthetic Bacteria; Advances in Photosynthesis and Respiration; Springer: Dordrecht, The Netherlands, 1995; pp. 847–870. ISBN 978-0-7923-3681-5. [Google Scholar]
- Stockdreher, Y.; Venceslau, S.S.; Josten, M.; Sahl, H.-G.; Pereira, I.A.C.; Dahl, C. Cytoplasmic Sulfurtransferases in the Purple Sulfur Bacterium Allochromatium vinosum: Evidence for Sulfur Transfer from DsrEFH to DsrC. PLoS ONE 2012, 7, e40785. [Google Scholar] [CrossRef]
- Vetriani, C.; Voordeckers, J.W.; Crespo-Medina, M.; O’Brien, C.E.; Giovannelli, D.; Lutz, R.A. Deep-sea hydrothermal vent Epsilonproteobacteria encode a conserved and widespread nitrate reduction pathway (Nap). ISME J. 2014, 8, 1510–1521. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Rodríguez, I.; Bohnert, K.A.; Cuebas, M.; Keddis, R.; Vetriani, C. Detection and phylogenetic analysis of the membrane-bound nitrate reductase (Nar) in pure cultures and microbial communities from deep-sea hydrothermal vents. FEMS Microbiol. Ecol. 2013, 86, 256–267. [Google Scholar] [CrossRef]
- Potter, L.C.; Millington, P.; Griffiths, L.; Thomas, G.H.; Cole, J.A. Competition between Escherichia coli strains expressing either a periplasmic or a membrane-bound nitrate reductase: Does Nap confer a selective advantage during nitrate-limited growth? Biochem. J. 1999, 344, 77–84. [Google Scholar] [CrossRef]
- Heylen, K.; Keltjens, J. Redundancy and modularity in membrane-associated dissimilatory nitrate reduction in Bacillus. Front. Microbiol. 2012, 3, 371. [Google Scholar] [CrossRef]
- Moreno-Vivian, C.; Cabello, P.; Martinez-Luque, M.; Blasco, R.; Castillo, F. Prokaryotic nitrate reduction: Molecular properties and functional distinction among bacterial nitrate reductases. J. Bacteriol. 1999, 181, 6573–6584. [Google Scholar] [CrossRef]
- Malm, S.; Tiffert, Y.; Micklinghoff, J.; Schultze, S.; Joost, I.; Weber, I.; Horst, S.; Ackermann, B.; Schmidt, M.; Wohlleben, W.; et al. The roles of the nitrate reductase NarGHJI, the nitrite reductase NirBD and the response regulator GlnR in nitrate assimilation of Mycobacterium tuberculosis. Microbiology 2009, 155, 1332–1339. [Google Scholar] [CrossRef]
- Sohm, J.A.; Webb, E.A.; Capone, D.G. Emerging patterns of marine nitrogen fixation. Nat. Rev. Microbiol. 2011, 9, 499–508. [Google Scholar] [CrossRef]
- Jouanneau, Y.; Meyer, C.; Naud, I.; Klipp, W. Characterization of an fdxN mutant of Rhodobacter capsulatus indicates that ferredoxin I serves as electron donor to nitrogenase. Biochim. Biophys. Acta (BBA)-Bioenerg. 1995, 1232, 33–42. [Google Scholar] [CrossRef]
- Price, R.E.; Lesniewski, R.; Nitzsche, K.; Meyerdierks, A.; Saltikov, C.; Pichler, T.; Amend, J. Archaeal and bacterial diversity in an arsenic-rich shallow-sea hydrothermal system undergoing phase separation. Front. Microbiol. 2013, 4, 158. [Google Scholar] [CrossRef] [PubMed]
- Rathgeber, C.; Yurkova, N.; Stackebrandt, E.; Beatty, J.T.; Yurkov, V. Isolation of Tellurite- and Selenite-Resistant Bacteria from Hydrothermal Vents of the Juan de Fuca Ridge in the Pacific Ocean. Appl. Environ. Microbiol. 2002, 68, 4613–4622. [Google Scholar] [CrossRef]
- Vetriani, C.; Chew, Y.S.; Miller, S.M.; Yagi, J.; Coombs, J.; Lutz, R.A.; Barkay, T. Mercury Adaptation among Bacteria from a Deep-Sea Hydrothermal Vent. Appl. Environ. Microbiol. 2005, 71, 220–226. [Google Scholar] [CrossRef] [PubMed]
- Crepo-Medina, M.; Chatziefthimiou, A.D.; Bloom, N.S.; Luther, G.W.I.; Wright, D.D.; Reinfelder, J.R.; Vetriani, C.; Barkay, T. Adaptation of chemosynthetic microorganisms to elevated mercury concentrations in deep-sea hydrothermal vents. Limnol. Oceanogr. 2009, 54, 41–49. [Google Scholar] [CrossRef]
- Xu, C.; Zhou, T.; Kuroda, M.; Rosen, B.P. Metalloid Resistance Mechanisms in Prokaryotes. J. Biochem. 1998, 123, 16–23. [Google Scholar] [CrossRef]
- Dopson, M.; Baker-Austin, C.; Koppineedi, P.R.; Bond, P.L. Growth in sulfidic mineral environments: Metal resistance mechanisms in acidophilic micro-organisms. Microbiology 2003, 149, 1959–1970. [Google Scholar] [CrossRef]
- Barkay, T.; Miller, S.M.; Summers, A.O. Bacterial mercury resistance from atoms to ecosystems. FEMS Microbiol. Rev. 2003, 27, 355–384. [Google Scholar] [CrossRef]
- Mathema, V.B.; Thakuri, B.C.; Sillanpää, M. Bacterial mer operon-mediated detoxification of mercurial compounds: A short review. Arch. Microbiol. 2011, 193, 837–844. [Google Scholar] [CrossRef]
- Sabaty, M.; Avazeri, C.; Pignol, D.; Vermeglio, A. Characterization of the Reduction of Selenate and Tellurite by Nitrate Reductases. Appl. Environ. Microbiol. 2001, 67, 5122–5126. [Google Scholar] [CrossRef]
- Oremland, R.S.; Blum, J.S.; Bindi, A.B.; Dowdle, P.R.; Herbel, M.; Stolz, J.F. Simultaneous Reduction of Nitrate and Selenate by Cell Suspensions of Selenium-Respiring Bacteria. Appl. Environ. Microbiol. 1999, 65, 4385–4392. [Google Scholar] [CrossRef] [PubMed]
- Ledgham, F.; Quest, B.; Vallaeys, T.; Mergeay, M.; Covès, J. A probable link between the DedA protein and resistance to selenite. Res. Microbiol. 2005, 156, 367–374. [Google Scholar] [CrossRef] [PubMed]
- Cai, L.; Jain, M.; Sena-Vélez, M.; Jones, K.M.; Fleites, L.A.; Heck, M.; Gabriel, D.W. Tad pilus-mediated twitching motility is essential for DNA uptake and survival of Liberibacters. PLoS ONE 2021, 16, e0258583. [Google Scholar] [CrossRef] [PubMed]
- Andrade, M.; Wang, N. The Tad Pilus Apparatus of ‘Candidatus Liberibacter asiaticus’ and Its Regulation by VisNR. MPMI 2019, 32, 1175–1187. [Google Scholar] [CrossRef]
- Jain, M.; Cai, L.; Fleites, L.A.; Munoz-Bodnar, A.; Davis, M.J.; Gabriel, D.W. Liberibacter crescens Is a Cultured Surrogate for Functional Genomics of Uncultured Pathogenic ‘Candidatus Liberibacter’ spp. and Is Naturally Competent for Transformation. Phytopathology 2019, 109, 1811–1819. [Google Scholar] [CrossRef]
- Markowitz, V.M.; Chen, I.-M.A.; Palaniappan, K.; Chu, K.; Szeto, E.; Grechkin, Y.; Ratner, A.; Jacob, B.; Huang, J.; Williams, P.; et al. IMG: The Integrated Microbial Genomes Database and Comparative Analysis System. Nucleic Acids Res. 2012, 40, D115–D122. [Google Scholar] [CrossRef]
- Aziz, R.K.; Bartels, D.; Best, A.A.; DeJongh, M.; Disz, T.; Edwards, R.A.; Formsma, K.; Gerdes, S.; Glass, E.M.; Kubal, M.; et al. The RAST Server: Rapid Annotations Using Subsystems Technology. BMC Genom. 2008, 9, 75. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Attribute | Genome (Total) | |
|---|---|---|
| Value | % Total 1 | |
| Size (bp) | 3,066,297 | 100.00% |
| G+C content (bp) | 1,821,569 | 59.41% |
| Coding region (bp) | 2,702,087 | 88.12% |
| Total genes | 2890 | 100.00% |
| RNA genes | 61 | 2.11% |
| Protein-coding genes | 2829 | 97.89% |
| Genes assigned to Pfam3 | 2450 | 84.78% |
| Genes assigned to COGs | 2105 | 72.84% |
| Genes assigned in KEGG Orthology (KO) | 1641 | 56.78% |
| Genes assigned to Subsystems | 1568 | 54.25% |
| Genes coding signal peptides | 160 | 5.54% |
| Genes coding transmembrane proteins | 745 | 25.78% |
| CRISPRs | 1 | |
| Intact prophage | 1 | |
| Code | Value | % Total 2 | Description |
|---|---|---|---|
| E | 10.32% | 239 | Amino acid transport and metabolism |
| C | 8.93% | 207 | Energy production and conversion |
| J | 7.60% | 176 | Translation, ribosomal structure and biogenesis |
| P | 7.51% | 174 | Inorganic ion transport and metabolism |
| R | 6.78% | 157 | General function prediction only |
| H | 5.91% | 137 | Coenzyme transport and metabolism |
| T | 5.91% | 137 | Signal transduction mechanisms |
| O | 5.74% | 133 | Posttranslational modification, protein turnover, chaperones |
| M | 5.44% | 126 | Cell wall/membrane/envelope biogenesis |
| K | 4.96% | 115 | Transcription |
| G | 4.92% | 114 | Carbohydrate transport and metabolism |
| S | 4.75% | 110 | Function unknown |
| I | 3.84% | 89 | Lipid transport and metabolism |
| L | 3.63% | 84 | Replication, recombination and repair |
| F | 2.81% | 65 | Nucleotide transport and metabolism |
| N | 2.68% | 62 | Cell motility |
| Q | 1.90% | 44 | Secondary metabolites biosynthesis, transport and catabolism |
| V | 1.77% | 41 | Defense mechanisms |
| U | 1.42% | 33 | Intracellular trafficking, secretion, and vesicular transport |
| X | 1.38% | 32 | Mobilome: prophages, transposons |
| D | 1.25% | 29 | Cell cycle control, cell division, chromosome partitioning |
| W | 0.47% | 11 | Extracellular structures |
| B | 0.04% | 1 | Chromatin structure and dynamics |
| - | 27.16% | 785 | Not in COG |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Patwardhan, S.; Phan, J.; Smedile, F.; Vetriani, C. The Genome of Varunaivibrio sulfuroxidans Strain TC8T, a Metabolically Versatile Alphaproteobacterium from the Tor Caldara Gas Vents in the Tyrrhenian Sea. Microorganisms 2023, 11, 1366. https://doi.org/10.3390/microorganisms11061366
Patwardhan S, Phan J, Smedile F, Vetriani C. The Genome of Varunaivibrio sulfuroxidans Strain TC8T, a Metabolically Versatile Alphaproteobacterium from the Tor Caldara Gas Vents in the Tyrrhenian Sea. Microorganisms. 2023; 11(6):1366. https://doi.org/10.3390/microorganisms11061366
Chicago/Turabian StylePatwardhan, Sushmita, Jonathan Phan, Francesco Smedile, and Costantino Vetriani. 2023. "The Genome of Varunaivibrio sulfuroxidans Strain TC8T, a Metabolically Versatile Alphaproteobacterium from the Tor Caldara Gas Vents in the Tyrrhenian Sea" Microorganisms 11, no. 6: 1366. https://doi.org/10.3390/microorganisms11061366
APA StylePatwardhan, S., Phan, J., Smedile, F., & Vetriani, C. (2023). The Genome of Varunaivibrio sulfuroxidans Strain TC8T, a Metabolically Versatile Alphaproteobacterium from the Tor Caldara Gas Vents in the Tyrrhenian Sea. Microorganisms, 11(6), 1366. https://doi.org/10.3390/microorganisms11061366