Abstract
Alterations in the composition of the gut microbiota is thought to play a key role in causing type 2 diabetes, yet is not fully understood, especially at the strain level. Here, we used long-read DNA sequencing technology of 16S-ITS-23S rRNA genes for high-resolution characterization of gut microbiota in the development of type 2 diabetes. Gut microbiota composition was characterized from fecal DNA from 47 participants divided into 4 cohorts based on glycemic control: normal glycemic control (healthy; n = 21), reversed prediabetes (prediabetes/healthy; n = 8), prediabetes (n = 8), or type 2 diabetes (n = 10). A total of 46 taxa were found to be possibly related to progression from healthy state to type 2 diabetes. Bacteroides coprophilus DSM 18228, Bifidobacterium pseudocatenulatum DSM 20438, and Bifidobacterium adolescentis ATCC 15703 could confer resistance to glucose intolerance. On the other hand, Odoribacter laneus YIT 12061 may be pathogenic as it was found to be more abundant in type 2 diabetes participants than other cohorts. This research increases our understanding of the structural modulation of gut microbiota in the pathogenesis of type 2 diabetes and highlights gut microbiota strains, with the potential for targeted opportunistic pathogen control or consideration for probiotic prophylaxis and treatment.
1. Introduction
Gut bacterial populations are prone to perturbations driven by intrinsic host factors (e.g., genetics and lifecycle stage) and environmental factors (e.g., pollutants, diet, lifestyle, pharmaceutical use) that can alter host physiology and health status [1]. Alterations in the gut microbiota composition (dysbiosis) have been associated with the progression of insulin resistance (IR) and type 2 diabetes [2]. Among consistently reported findings, the signatures of dysbiosis associated with type 2 diabetes include, but are not limited to, a decrease in the genera of Bifidobacterium, Bacteroides, Faecalibacterium, Akkermansia, and Roseburia; and an increase in the genera of Ruminococcus, Fusobacterium, and Blautia [3]. Several studies have found a correlation between type 2 diabetes and gut microbiota alpha (α)-diversity, as well as Firmicutes/Bacteroidetes (F/B) ratios; however, other studies have found no correlation [3]. Other commonly found gut microbiome shifts include the depletion of butyrate-producing bacteria [4] and a reduction in probiotics [5]. For example, several bacterial species, such as Lactobacillus fermentum, L. plantarum and L. casei, Roseburia intestinalis, Akkermansia muciniphila, and Bacteroides fragilis, have been shown to decrease the risk of diabetes development through maintaining intestinal barrier integrity, improving glucose metabolism and insulin sensitivity, and suppressing proinflammatory cytokines [6]. While these protective species may be candidates for probiotics, it is essential that they be carefully characterized and assigned to a given strain, and not to the entire genus or species [7].
To date, strain level data have been limited in studies identifying bacterial changes accompanying metabolic disease. Fei and Zhao (2013) found that a single strain, Enterobacter cloacae B29, in combination with a high-fat diet, induced fully developed obesity phenotypes, including endotoxin-induced inflammation, adiposity, and IR in gnotobiotic mice [8]. Aside from this case, most strain level microbiome research related to metabolic disease is focused on probiotics. For example, a comparison between Bifidobacterium animalis ssp. lactis GCL2505 (BlaG) and B. longum ssp. longum JCM1217T found that BlaG reduced visceral fat accumulation and improved glucose tolerance in a mouse model [9]. Without high resolution taxonomic information, these studies would not have been able to identify strains for development of effective probiotics. Further strain-level information regarding type 2 diabetes progression could be useful for targeted opportunistic pathogen control or probiotic prophylaxis and treatment.
In this study, we aimed to identify novel strains associated with type 2 diabetes. We investigated the fecal microbiota in humans with prediabetes and type 2 diabetes, as well as those without diabetes. Using long-read DNA sequencing technology, a high-resolution ~2500-bp genetic marker (including the entire 16S gene and two additional genes), and a recently published reference database [10], we estimated microbial abundance and diversity, assessed microbiota associated with health status, and report new stains associated with type 2 diabetes progression.
2. Methods
2.1. Consent and IRB
A convenience sample of 48 adults (mean ± SD age 51.0 ± 15.9 years, 60% women) with and without type 2 diabetes, aged 18 years and older, were recruited from a university and the surrounding community to participate in this study that included two visits to the laboratory for each patient. Eligibility for the study was determined by a pre-screening questionnaire that was completed over the phone by a trained researcher. Study exclusion criteria included a history of gastric bypass surgery, inflammatory bowel disease (i.e., irritable bowel syndrome, Crohn’s or colitis), colon cancer, celiac disease, multiple sclerosis, Parkinson’s disease, Alzheimer’s disease, or current pregnancy. The University of Idaho Institutional Review Board approved the study (protocol 20-098) and all participants provided verbal and written informed consent to participate in the study. All experiments were conducted in accordance with the Declaration of Helsinki.
2.2. HbA1c Measurement
A small sample of blood (~1 µL) was obtained from the middle or ring finger of the non-dominant hand and analyzed for HbA1c under strict standardized operating procedures using the DCA Vantage analyzer (Siemens Healthcare Diagnostics, Tarrytown, NY, USA). In brief, the finger was cleaned with alcohol prior to measurement. A disposable safety lancet (SurgiLance, MediPurpose, Duluth, GA, USA) was used to puncture the skin and the first drop of blood was wiped clean. The sample was drawn into the capillary tube, inserted into the reagent cartridge, and analyzed immediately. Participants were divided into four groups, based on measured hemoglobin A1c (HbA1c) during the first study visit: healthy (n = 21, HbA1c < 5.7%), healthy range but previously diagnosed with prediabetes by a registered physician (prediabetes/healthy (reversed prediabetes), n = 9; HbA1c < 5.7%), prediabetes (n = 8, HbA1c 5.7–6.4%), and type 2 diabetes (n = 10, HbA1c ≥ 6.5%).
2.3. Stool Collection
After collection of the blood sample, participants were provided a stool sample kit and detailed instructions to collect and store the sample. All samples were stored on ice in a Styrofoam cooler, brought to the laboratory within 24 h of collection, and frozen at −80 °C prior to extraction. The average number of days between the blood sample collection and fecal sample collection was 4.8 ± 2.1 days (range 1–8 days).
2.4. DNA Isolation, Sequencing, and Taxonomic Identification
Total genomic DNA from 250 mg of human fecal samples was extracted using the QIAamp PowerFecal Pro DNA kits (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. The DNA concentration and purity were monitored on 1.5% agarose gels. The Intus Biosciences Wave StrainID Kits (Farmington, CT, USA), SetA and SetZ, were then used to produce amplicons that span the full-length 16S, ITS, and partial 23S rRNA genes. Sequencing libraries were generated from these amplicons using PacBio SMRTbell Express Template Prep Kit v.3.0 (Pacific Biosciences, Menlo Park, CA 94025, USA) according to the manufacturer’s instructions. We included two negative controls (elution buffer and DNA extraction with water). The library was sequenced on 1 SMRT Cell 8M on a PacBio Sequel II System using the circular consensus sequencing (CCS) mode at the University of Idaho Institute for Interdisciplinary Data Sciences Genomics and Bioinformatics Resources Core. The CCS reads were determined with a minimum predicted accuracy of 0.9 and the minimum number of passes set to three in the SMRT Link software package v.10.2.1.143962 (Pacific Biosciences).
SBanalyzer v.3.0 (Intus Biosciences) was used to demultiplex and assign taxonomic identification to all reads by mapping to the Athena database [10]. In this analysis, sequences are clustered by similarity with a threshold of 97% and put into bins called ‘Operational Taxonomic Units’ (OTUs). Setting the sequence similarity threshold to 97% to delineate species is only a rough approximation and, in some cases, may not be biologically accurate to distinguish between some taxa [11]. A single sequence is selected as a representative sequence for each of the OTU bins. If a read does not match any strains or if it matches equally to multiple strains in the database, the taxonomic classification will be reported at the highest level where an unambiguous call can be made, resulting in an “unclassified” taxon. The output reports abundance for each OTU for each patient. Patients with less than 500 reads were removed from all subsequent analyses. Potential reagent contaminants were identified using the decontam package v.1.14.0 with a threshold frequency of 0.5 of the OTU in the negative control [12,13]. The decontam package has been shown to remove upward of 90% of contaminants even when the source of contamination was not well defined [14]. To determine whether sequencing depth was sufficient to capture a nearly complete microbiome profile for samples, rarefaction curves were plotted using the R-package vegan (v.2.6.2; [15]).
2.5. Alpha and Beta Diversity Analyses
Subsequent analysis of the filtered OTU table was conducted in R [16] using the phyloseq package v.1.16.2 [17]. To calculate and plot α-diversity indices (Shannon diversity, Simpson index, and Chao1 index) of the microbiome communities, we used phyloseq. To analyze whether the α-diversity was significantly different between the four health status groups (healthy, prediabetes/healthy, prediabetes, diabetes), we first tested for normal distribution of each of the α-diversity indices using the Shapiro–Wilk test of normality. For nonparametric data, we tested for significant differences using the Kruskal–Wallis test. For normally distributed data, we used an ANOVA test and subsequent Tukey’s honest significance test of the ANOVA to test for statistical differences.
We calculated beta (β) diversity using non-metric multidimensional scaling (NMDS) from Bray-Curtis dissimilarity in phyloseq. To test whether microbial communities differ by donor health status, we used the adonis2 function in R-package vegan (v 2.6.2) to run a permutational multivariate analysis of variance (PERMANOVA) after testing for multivariate homogeneity of group dispersion (beta dispersion).
2.6. Identification of Differentially Abundant Taxonomic Groups
Community composition barplots were generated using the fantaxtic v.0.2.0 package in R. To identify taxa that were significantly different between health status groups, we used the DESeq2 package as described for microbiome applications [18,19]. After controlling for donor sex in the design matrix, DESeq2 was run under the Wald significance tests and parametric fitting of dispersions to the mean intensity settings, and false discovery rate adjusted p-values were calculated with the Benjamini-Hochberg procedure using a significance threshold of p < 0.05. Differentially abundant (or “discriminatory”) taxa were assessed at the phylum, genus, and strain levels. To further investigate the level of differentiation across groups for discriminatory strains, we generated NMDS plots using Bray–Curtis dissimilarity for strain-level OTUs that were differentially abundant for one or more pairwise comparisons.
3. Results
3.1. DNA Isolation, Sequencing, and Taxonomic Identification
After removing one individual due to a low number of sequencing reads (SG11), the number of fecal microbial DNA samples sequenced was 21 healthy, 8 prediabetes/healthy, 8 prediabetes, and 10 type 2 diabetes individuals (Supplementary Table S1). The mean number of CCS reads per sample was 15,836.76. After all filtering steps, 641,305 reads were successfully classified with a mean of 13,644.79 (±5311.93) per sample.
Before removing contaminants, 884 OTUs were identified. Eight OTUs were removed at a prevalence threshold of 0.5. After removing contaminants, ten phyla were identified, including Actinobacteria, Bacteroidetes, Cyanobacteria, Elusimicrobia, Firmicutes, Fusobacteria, Proteobacteria, Synergistetes, Tenericutes (Mycoplasmatota), and Verrucomicrobia. Four phyla, namely Firmicutes, Bacteroidetes, Actinobacteria, and Proteobacteria were the most abundant across all samples (Figure 1), with a mean percent total reads across samples for these phyla of 58.80% (±16.85), 23.13% (±15.90), 10.72% (±10.37), and 5.15% (±5.76), respectively (Supplementary Table S2). These phyla were represented by 29 classes, 62 orders, 114 families, 312 genera, 602 species, and 876 strains. Of the 876 strains, 314 were known, previously published strains and 562 were unclassified (de novo) strains (Supplementary Table S3).
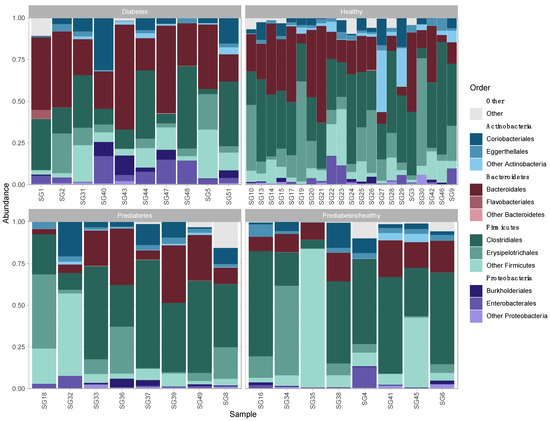
Figure 1.
Relative abundance of the three most abundant bacterial orders within each of the four most abundant phyla in fecal microbiota of patients by health status.
3.2. Alpha and Beta Diversity
Rarefaction curves of the gut microbiomes of all individuals approached, but did not reach, a plateau, indicating that our dataset likely captured the dominant patterns of the microbial communities; however, that increased sequencing depth may have captured additional information regarding low-frequency taxa (Supplementary Figure S1). We used the Chao1 index as an indicator of microbial community richness, and Shannon and Simpson indexes as indicators of diversity (Figure 2a). These alpha diversity indices were not significantly different between the bacterial gut microbiomes of individuals from any health status comparisons. The Shapiro–Wilk normality test indicated that the Shannon and Simpson diversity values were not normally distributed (Shannon: W = 0.92, p = 0.003; Simpson: W = 0.79, p = 1.13 × 10−6), and the Kruskal–Wallis test showed no evidence of differences between groups (Shannon: chi-squared = 2.65, df = 3, p = 0.45; Simpson: chi-squared = 2.02, df = 3, p = 0.57). Chao1 richness was normally distributed (W = 0.99, p = 0.91) and was not significantly different between groups (p = 0.31).
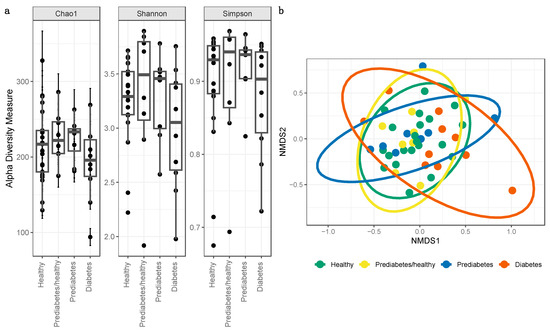
Figure 2.
(a) Microbial alpha diversity as measured by the Chao1, Shannon, and Simpson index of fecal samples, which are colored according to four health status categories. All alpha diversity metrics were not statistically different between health status categories. (b) Microbial beta diversity. Dimensional reduction of the Bray-Curtis distance between microbiome samples, using NMDS ordination method, for fecal samples in healthy and controls. Data points are colored according to healthy status.
When testing for β-diversity, the NMDS plot did not indicate distinct clustering between health status cohorts, as evidenced by overlapping ellipses (Figure 2b). However, the PERMANOVA test did identify significant differences (p < 0.05) between health status categories (F = 1.72; R2 = 0.11; p = 0.02).
3.3. Differentially Abundant Taxa
Some taxa exhibited significant differences (p < 0.05) in abundance between health status cohorts. At the phylum level, two phyla were less abundant in the type 2 diabetes cohort than other cohorts (Supplementary Table S4). In particular, Tenericutes was less abundant in type 2 diabetes participants compared to all other cohorts, and Firmicutes was less abundant in type 2 diabetes participants compared to healthy and prediabetes/healthy participants. Proteobacteria were more abundant in healthy and prediabetes/healthy participants (Supplementary Table S4). No phyla exhibited abundance differences for the remaining cohort comparisons (prediabetes to healthy, prediabetes/healthy to healthy, and prediabetes/healthy to prediabetes). F/B ratios were significantly higher for type 2 diabetes than for any of the three other cohorts (healthy controls: p = 0.01; prediabetes/healthy: p < 0.001; prediabetes: p = 0.01; Supplementary Table S1). There were no significant differences in F/B ratios in any other comparisons (prediabetes to healthy: p = 0.35, prediabetes/healthy to healthy: p = 0.06, and prediabetes/healthy to prediabetes: p = 0.57).
At the genus level, several genera were significantly more abundant for the healthy cohort than for participants in type 2 diabetes (5 genera) and prediabetes (3 genera) cohorts (p < 0.05, Supplementary Table S5). In contrast, Streptococcus and (Bacteroides) genera were significantly less abundant in healthy individuals than in prediabetes/healthy and prediabetes participants, respectively. We also observed that one genus (Lactococcus) was less abundant and two (Selenomonas and Megasphaera) were more abundant in prediabetes/healthy participants compared to prediabetes participants. Prediabetes participants had five genera more abundant than in type 2 diabetes participants, while type 2 diabetes participants had four genera more abundant than prediabetes participants (Supplementary Table S5). There were three genera more abundant in type 2 diabetes as compared to prediabetes/healthy and four genera less abundant between the two cohorts (Supplementary Table S5).
At the OTU level, 55 distinct taxa were found to differ in abundance across cohorts, with 39 of those taxa found to be significantly different in more than one comparison, and 16 taxa unique to a single comparison. Of these 55 OTUs, 60% were identified to species level (n = 15) or strain level (n = 18). The number of differentially abundant OTUs ranged between 10 and 33 for the six pairwise comparisons of treatment cohorts (Figure 3, Supplementary Table S6, and description in Supplementary Materials). When calculating β-diversity using only strains that were differentially abundant across one or more pairwise comparisons, NMDS primarily distinguished healthy from diabetic participants (Supplementary Figure S2b).
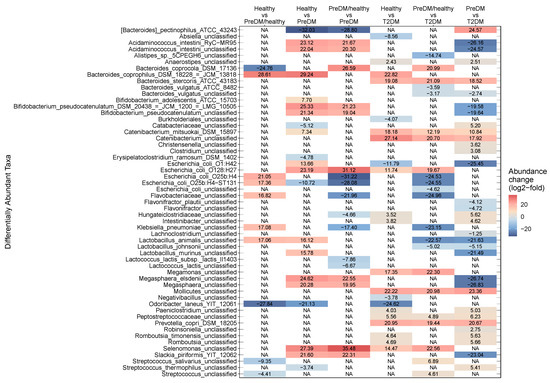
Figure 3.
Observed fold change magnitudes for all 55 differentially abundant taxa present in all comparisons of health status groups. Columns indicate each comparison, and rows show taxonomic assignment for each OTU. Color indicates the log2-fold abundance change in each taxon for each comparison. For positive log2-fold change, the health status category before “vs” in each comparison label is more abundant than the category after the “vs”, and vice versa for negative log2-fold change. For example, when comparing “Healthy vs T2DM,” a positive value indicates a taxon that is more abundant in Healthy, and a negative value indicates a taxon that is more abundant in T2DM. T2DM: Type 2 diabetes; PreDM: prediabetes.
4. Discussion
In the present study, the fecal microbiota of healthy, prediabetes/healthy, prediabetes, and type 2 diabetes individuals were explored using long-read technology to sequence 16S-ITS-23S amplicons, and taxonomic classification was performed using a recently published reference database. The gut microbiota structure of all individuals was found to be mainly composed of Bacteroidetes, Firmicutes, Proteobacteria, and Actinobacteria. To our knowledge, we identified more strain-level microbial differences associated with type 2 diabetes progression than previous studies were able to find due to methodical limitations resulting in a lack of species- and strain-level resolution. As with many previous studies, we noted bacteria that may act as opportunistic pathogens as they are more abundant in the diabetic group than the glucose tolerant groups. Further, our study revealed that some taxa are significantly correlated with healthy individuals and likely infer a protective or probiotic role within the host, which corroborates findings from other studies that show the importance of these microbial markers to health.
4.1. Probiotic Taxa
We identified several taxa that were positively associated with healthy status and that could be candidates for probiotic development. Bifidobacterium pseudocatenulatum (unclassified and strain DSM 20438) was higher in abundance with healthy status and, similarly, B. adolescentis ATCC 15703 shifts to a greater abundance in the healthy group as compared to prediabetes/healthy. There is strong support for Bifidobacterium’s protective role in type 2 diabetes in previous studies. Many papers report a negative association between this genus and type 2 diabetes (see [3]), with only one paper reporting conflicting results [20]. Certain species, such as B. adolescentis, B. bifidum, B. pseudocatenulatum, B. longum, and B. dentium, have been found to have a negative association with type 2 diabetes patients treated with the antihyperglycaemic agent metformin [21]. In animal studies testing for probiotic effects, several species/strains from this genus (B. bifidum, B. longum, B. infantis, B. animalis, B. pseudocatenulatum CECT 7765, and B. breve) showed improvement of glucose tolerance [9,22,23,24,25], with some species (B. adolescentis) being superior in alleviating type 2 diabetes symptoms compared to other species (B. bifidum; [26]). Our results support the notion that Bifidobacterium naturally inhabiting the human gut may play a protective role in type 2 diabetes and indicate that two strains, B. pseudocatenulatum DSM 20438 and B. adolescentis ATCC 15703, have the potential for new probiotic agents.
We also found Prevotella copri DSM 18205 to be more abundant as health increases. However, there have been contradictory results regarding the pathophysiological role of P. copri. This species is correlated with IR in humans and in fat-fed mice [27], and has been found to be elevated in type 2 diabetes [28]. Direct inoculation of rats with P. copri showed an improved glucose homeostasis [29]. This inconsistent pattern may be due to the existence of several P. copri clades or host diet-dependent effects [30]. Given that these microbes were more abundant in glucose tolerant groups, they may be possible candidates for probiotic use in type 2 diabetes prevention and treatment. However, risk, efficacy, and dosage for each strain should be fully explored through science-based clinical studies on targeted populations and pre-market approvals should be established [31].
We found that Romboutsia, a butyrate producing bacteria genus, and R. timonensis also increased in healthy and prediabetes individuals as compared to the type 2 diabetes group. Similarly, other studies found Romboutsia to be inversely associated with IR, type 2 diabetes, and gestational diabetes mellitus [32,33,34]. Additionally, studies have shown that Romboutsia is negatively correlated with fasting glucose, insulin, and high-density lipoprotein cholesterol; and positively correlated with indicators of obesity [35]. Contrary to these findings, one study found a significantly lower relative abundance of R. timonensis in type 2 diabetes participants as compared to healthy controls [36]. Further, Hu et al. (2019) reported that Romboutsia was reduced in the diabetic rats [37]. Therefore, Romboutsia may be a candidate genus for predicting and treating obesity and related metabolic disorders; however, more species and strain-specific knowledge is required to define utilities of these microbes in humans.
4.2. Pathogenic Taxa
In this study, we found several taxa that increased in abundance with progression from prediabetes to type 2 diabetes, indicating a possible pathogenic effect. Odoribacter laneus YIT 12061 was found to be significantly more abundant in prediabetes/healthy, prediabetes, and type 2 diabetes than in healthy controls. This species is a succinate-consuming bacterium and has been associated with a number of diseases, including colitis in a mouse model [38]. Odoribacter has been found to be negatively associated with the expression of intestinal epithelial tight junction proteins, suggesting a link with impairing mucosal barrier function in rats [39]. However, Huber-Ruano et al. (2022) showed that one strain of O. laneus, DSM 22474 A, has beneficial anti-inflammatory and metabolic properties in obese mice [40]. Collectively, our results strongly suggest that the effects of this species may be strain-specific and indicate the need of comprehensive evaluation before pursuing Odoribacter strains for treating type 2 diabetes in humans.
Another genus and species that increased in abundance and could be involved in the development of type 2 diabetes were Flavonifractor plautii and an unclassified Flavonifractor species, which were more abundant in type 2 diabetes participants than prediabetes/healthy participants. This pattern supports previous findings showing that the genus Flavonifractor decreased in prediabetes and increased in type 2 diabetes [41], and was associated with a lower insulin sensitivity and higher prevalence of dysglycemia [42]. F. plautii, specifically, was lower in patients with prediabetes and higher in patients with diabetes [41,43]. One possible mechanism underlying the association of Flavonifractor with type 2 diabetes progression is that this genus may increase oxidative stress and trigger inflammatory cytokines in plasma [44,45]. These cytokines are associated with type 2 diabetes by impairing insulin signal transduction and triggering IR [46]. To support this, possible treatments such as the oral administration of Sanghuangporous vaninii SVE have been shown to improve body weight, glycolipid metabolism, and inflammation-related parameters through modulating gut microbiota including decreasing Flavonifractor [47].
4.3. Species- and Strain-Dependent Associations
We identified stain-dependent type 2 diabetes associations within the Escherichia genus. Escherichia coli O128:H27 were more enriched in healthy and prediabetes/healthy than in prediabetes and type 2 diabetes. In contrast, E. coli O25b:H4 and E. coli O25b:H4-ST131 were more abundant in type 2 diabetes and prediabetes than in prediabetes/healthy; however, both were more abundant in healthy than prediabetes/healthy. Following a similar pattern, E. coli O1:H42 was more abundant in type 2 diabetes than prediabetes and healthy, but also more abundant in healthy as compared to prediabetes. We discovered an unclassified strain of E. coli was more abundant in type 2 diabetes than in prediabetes/healthy. Previous studies have positively associated Escherichia increasing in abundance with progression from normal glucose tolerance to type 2 diabetes [48,49,50] and with effects of metformin therapy [21,51]. Our results support the findings that E. coli may play a role in the pathogenesis of type 2 diabetes as well as metformin mechanisms of action. However, our work illuminates the essence of E. coli strain specificity when considering their functional use as antidiabetic targets for humans.
Within the genus Bacteroides, B. coprophilus DSM 18228 = JCM 13818, and B. stercoris ATCC 43183 were more abundant as health increases; however, B. vulgatus (unclassified strain and strain ATCC 8482) were higher in type 2 diabetes participants. Moreover, (Bacteroides) pectinophilus ATCC 43243 was unique to prediabetes and B. coprocola DSM 17136 to prediabetes/healthy individuals. According to Gurung et al. (2020), many previous studies have found Bacteroides to be either negatively or positively associated with type 2 diabetes [3]. Our study suggests that these inconsistencies could be driven by species and strain-specific effects, which would not have been distinguished in previous studies due to limitations of taxonomic classification below the genus level with traditional partial-16S sequencing. Alternatively, other confounding variables could be involved. For example, the studies that found positive associations with disease noted that participants had taken antidiabetic treatments, such as metformin, which have been shown to result in functional microbial shifts [51,52]. This could account for the apparent inconsistency in this study as well as previous studies. There could be several other confounding variables resulting in these inconsistencies, such as underlying host genetics, diet, physical activity level, medication use, and sequencing techniques. Nonetheless, our study indicates that species specificity of Bacteroides is important when considering the utility of antidiabetic properties and as a beneficial role on glucose metabolism in humans.
We identified three Lactobacillus species that were differentially abundant in our association analysis. Lactobacillus animalis and L. murinus are more abundant in both the two extreme phenotypes, healthy and type 2 diabetes, as compared to the prediabetes/healthy and prediabetes participants, whereas L. johnsonii was associated with type 2 diabetes. Although L. animalis and L. murinus were found to be highly abundant in healthy individuals, they both were also highly abundant in type 2 diabetes, so are not likely candidates for probiotics. The highly diverse Lactobacillus genus, which is frequently detected and reported in type 2 diabetes association studies, has the highest number of taxa with probiotic potential found in the human gut to date [3]. The effects of this genus on type 2 diabetes seem to be species-specific. For example, three species, L. acidophilus [53], L. gasseri [54], L. salivarius [54], were significantly more abundant in type 2 diabetes participants, while L. amylovorus [51] was decreased in type 2 diabetes participants. These previous results, as well as the results of our study suggests that species and strain specificity from this genus impact the functionality of host metabolism. Collectively, these differences in association patterns across species and strains reported herein highlight the significance of using high-resolution taxonomic methods to identify specific species and strain linking the microbiome with type 2 diabetes and other illnesses.
4.4. Microbial Diversity
Our study found mean richness and diversity measures were lower in participants with type 2 diabetes than all other participants, but higher in prediabetes/healthy and prediabetes than in healthy individuals. However, these differences were non-significant across comparisons, which may be due to limited sample size. Although α-diversity has been proposed as a biomarker in general health and disease, there is currently controversy regarding the association between diversity and type 2 diabetes due to inconsistent findings. For example, a systematic review of 13 studies investigating associations of gut microbiota α-diversity and type 2 diabetes found that an equal number of studies identified a negative correlation (n = 2) and a positive correlation (n = 2), and 9 found no correlation [3]. The lack of significant association for α-diversity with type 2 diabetes in our study supports the idea that gut microbiota richness and diversity may not necessarily decline with the progression of illness. However, many of the study participants classified as prediabetes/healthy and prediabetes were taking antihyperglycaemic agents, such as metformin. These agents have been found to increase the α-diversity of gut microbiota, as well as the reduction of HbA1c and fasting blood glucose concentrations [21,55]. These effects could have increased diversity in prediabetes/healthy and prediabetes participants relative to healthy participants in our study. It has also been noted that colonic transit time [56] and stool consistency [57] may interfere with the ability to use α-diversity as a biomarker in disease status.
Beta diversity, a measure of the similarity or dissimilarity of two communities, was found to statistically differ between healthy, prediabetes/healthy, prediabetes, or type 2 diabetes. According to Gurung et al. (2020), of 8 studies reporting β-diversity, 7 did not find any significant association between microbial β-diversity and type 2 diabetes [3]. Taxa may not have clustered distinctly on a community level as shown in the NMDS plot (Figure 2b) due to host participants living in relatively homogeneous environments [58] including similar host subsistence strategies, geographic location, and ethnicity [59].
4.5. F/B Ratios
The progression of type 2 diabetes may, in part, be due to the transformation of dominant bacteria, and, thus, F/B ratios are often measured to investigate associations between the microbiome and type 2 diabetes. The systematic review by Gurung et al. (2020) found that the B/F ratio did not show consistent associations with type 2 diabetes with six publications that found no association, three with a positive association, and four with a negative association [3]. Our findings showed that F/B ratios are highest in the type 2 diabetes group and significantly different than each of the three other categories. F/B ratios were not significantly different between healthy, prediabetes/healthy, and prediabetes cohorts likely due to the effect of antihyperglycaemic drugs as highlighted above.
4.6. Limitations and Future Directions
This study had several potential limitations. First, sample sizes were relatively small. Second, not all OTUs were resolved to the strain level, indicating that additional strain-level associations may exist in our dataset that could not be detected in our analyses. Thus, further development of the reference database could lead to even greater taxonomic resolution in future studies. Third, most prediabetes and type 2 diabetes participants in this study had taken antihyperglycaemic or anti-hypertensive drugs, and/or probiotics/prebiotics, which may have influenced the bacterial communities. Fourth, we could not control for every possible lifestyle factor or duration of disease in the study design, which leaves the possibility of residual confounding factors. Finally, our cross-sectional study cannot draw a direct link between bacterial taxa and particular metabolites involved in type 2 diabetes. Functional studies will be required to determine whether relevant bioactive metabolites were produced by the bacteria that were determined to be differentially abundant between diabetes participants and healthy participants, and if these metabolites were indeed causal for the disease phenotype. Future studies that integrate metagenomic and metabolomic approaches would greatly contribute to treatment discovery. Furthermore, a complete genome sequence and annotation, especially of genes associated with causal metabolites, is necessary for the registration of new probiotic strains.
5. Conclusions
To our knowledge, we identified more strain-level microbial differences associated with type 2 diabetes progression than previous studies were able to find due to methodical limitations resulting in a lack of species- and strain-level resolution. In addition, we demonstrated that sequencing a longer target region than the traditional partial-16S marker can improve taxonomic resolution and identify species-specific and strain-specific effects that could not otherwise have been discovered. The results of this work enhance our understanding of the influence of changes in human fecal microbiota on the pathogenesis of diabetes, and contribute insights into potential new microbiome biomarkers for preventative and/or therapeutic strategies for type 2 diabetes.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/microorganisms11030758/s1, Table S1: Sample metadata; Table S2: Percentage of each phylum per individual; Table S3: Number of known and unclassified taxa; Table S4: Fold change of differentially abundant taxa at phyla level; Table S5: Fold change of differentially abundant taxa at genera level; Table S6: Fold change of all differentially abundant taxa; Table S7: Sample ID and taxonomic identification of each raw read; Figure S1: Rarefaction curves; Figure S2: Microbial beta diversity using discriminatory taxa.
Author Contributions
O.B.B., C.A.V. and R.G. proposed analyzing the shifts of microbiota in prediabetes and type 2 diabetes, and received grant funding. C.A.V., M.A. and R.G. recruited participants. C.A.V. and M.A. collected participant data. S.A.H., C.A.V., D.D.N., K.R.A. and O.B.B. designed the study and planned the analyses. S.A.H. interpreted the results and wrote the manuscript. D.D.N., A.A. and O.B.B. performed sample preparations including DNA libraries. S.A.H. performed the statistical and bioinformatics analyses. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the National Institutes of Health, NIDDK Diabetic Complications Consortium (5U24DK115255-04), and University of Idaho, College of Science Seed Grant Program. Data collection and analyses performed by the University of Idaho IIDS Genomics and Bioinformatics Resources Core were supported in part by NIH COBRE grant (P30GM103324).
Institutional Review Board Statement
The study was conducted in accordance with the Declaration of Helsinki and approved by the Institutional Review Board (or Ethics Committee) of the University of Idaho (protocol code 20-098).
Informed Consent Statement
Informed consent was obtained from all subjects involved in the study.
Data Availability Statement
The data presented in this study are openly available under BioProject accession number PRJNA930056. The SRA accession number for the reads are SAMN32982567 to SAMN32982614.
Acknowledgments
We are grateful to the participants in these cohorts. We thank University of Idaho students Gillian Glivar, Ryan Pike, Morgan Flynn, and J. Bailey for assisting with sample collection; and Rachel Wiedenmann and Katy Lawler for graphics support.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; or in the decision to publish the results.
Abbreviations
| A | alpha |
| β | beta |
| CCS | circular consensus sequencing |
| F/B | Firmicutes/Bacteroidetes |
| HbA1c | hemoglobin A1c |
| IR | insulin resistance |
| ITS | internal transcribed spacer |
| NMDS | non-metric multidimensional scaling |
| OTU | operational taxonomic unit |
References
- Cresci, G.A.; Bawden, E. Gut Microbiome. Nutr. Clin. Pract. 2015, 30, 734–746. [Google Scholar] [CrossRef]
- Larsen, N.; Vogensen, F.K.; van den Berg, F.W.J.; Nielsen, D.S.; Andreasen, A.S.; Pedersen, B.K.; Al-Soud, W.A.; Sørensen, S.J.; Hansen, L.H.; Jakobsen, M. Gut Microbiota in Human Adults with Type 2 Diabetes Differs from Non-Diabetic Adults. PLoS ONE 2010, 5, e9085. [Google Scholar] [CrossRef]
- Gurung, M.; Li, Z.; You, H.; Rodrigues, R.; Jump, D.B.; Morgun, A.; Shulzhenko, N. Role of Gut Microbiota in Type 2 Diabetes Pathophysiology. EBioMedicine 2020, 51, 102590. [Google Scholar] [CrossRef]
- Allin, K.H.; Tremaroli, V.; Caesar, R.; Jensen, B.A.H.; Damgaard, M.T.F.; Bahl, M.I.; Licht, T.R.; Hansen, T.H.; Nielsen, T.; Dantoft, T.M.; et al. Aberrant Intestinal Microbiota in Individuals with Prediabetes. Diabetologia 2018, 61, 810–820. [Google Scholar] [CrossRef]
- Zhang, L.; Chu, J.; Hao, W.; Zhang, J.; Li, H.; Yang, C.; Yang, J.; Chen, X.; Wang, H. Gut Microbiota and Type 2 Diabetes Mellitus: Association, Mechanism, and Translational Applications. Mediators Inflamm. 2021, 2021, 5110276. [Google Scholar] [CrossRef]
- Iatcu, C.O.; Steen, A.; Covasa, M. Gut Microbiota and Complications of Type-2 Diabetes. Nutrients 2021, 14, 166. [Google Scholar] [CrossRef]
- Fijan, S. Microorganisms with Claimed Probiotic Properties: An Overview of Recent Literature. Int. J. Environ. Res. Public. Health 2014, 11, 4745–4767. [Google Scholar] [CrossRef]
- Fei, N.; Zhao, L. An Opportunistic Pathogen Isolated from the Gut of an Obese Human Causes Obesity in Germfree Mice. ISME J. 2013, 7, 880–884. [Google Scholar] [CrossRef]
- Aoki, R.; Kamikado, K.; Suda, W.; Takii, H.; Mikami, Y.; Suganuma, N.; Hattori, M.; Koga, Y. A Proliferative Probiotic Bifidobacterium Strain in the Gut Ameliorates Progression of Metabolic Disorders via Microbiota Modulation and Acetate Elevation. Sci. Rep. 2017, 7, 43522. [Google Scholar] [CrossRef]
- Graf, J.; Ledala, N.; Caimano, M.J.; Jackson, E.; Gratalo, D.; Fasulo, D.; Driscoll, M.D.; Coleman, S.; Matson, A.P. High-Resolution Differentiation of Enteric Bacteria in Premature Infant Fecal Microbiomes Using a Novel RRNA Amplicon. mBio 2021, 12, e03656-20. [Google Scholar] [CrossRef]
- Nguyen, N.-P.; Warnow, T.; Pop, M.; White, B. A Perspective on 16S RRNA Operational Taxonomic Unit Clustering Using Sequence Similarity. Npj Biofilms Microbiomes 2016, 2, 16004. [Google Scholar] [CrossRef]
- Davis, N.M.; Proctor, D.M.; Holmes, S.P.; Relman, D.A.; Callahan, B.J. Simple Statistical Identification and Removal of Contaminant Sequences in Marker-Gene and Metagenomics Data. Microbiome 2018, 6, 226. [Google Scholar] [CrossRef]
- Salter, S.J.; Cox, M.J.; Turek, E.M.; Calus, S.T.; Cookson, W.O.; Moffatt, M.F.; Turner, P.; Parkhill, J.; Loman, N.J.; Walker, A.W. Reagent and Laboratory Contamination Can Critically Impact Sequence-Based Microbiome Analyses. BMC Biol. 2014, 12, 87. [Google Scholar] [CrossRef]
- Karstens, L.; Asquith, M.; Davin, S.; Fair, D.; Gregory, W.T.; Wolfe, A.J.; Braun, J.; McWeeney, S. Controlling for Contaminants in Low-Biomass 16S RRNA Gene Sequencing Experiments. mSystems 2019, 4, e00290-19. [Google Scholar] [CrossRef]
- Oksanen, J.; Simpson, G.; Blanchet, F.G.; Kindt, R.; Legendre, P.; Minchin, P.R.; O’Hara, R.B.; Solymos, P.; Stevens, M.H.H.; Szoecs, E.; et al. Vegan: Community Ecology Package. R Package Version 2022, 2, 321–326. [Google Scholar]
- R Core Team. R: A Language and Environment for Statistical Computing; R Core Team: Vienna, Austria, 2020. [Google Scholar]
- McMurdie, P.J.; Holmes, S. Phyloseq: An R Package for Reproducible Interactive Analysis and Graphics of Microbiome Census Data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated Estimation of Fold Change and Dispersion for RNA-Seq Data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- McMurdie, P.J.; Holmes, S. Waste Not, Want Not: Why Rarefying Microbiome Data Is Inadmissible. PLOS Comput. Biol. 2014, 10, e1003531. [Google Scholar] [CrossRef]
- Sasaki, M.; Ogasawara, N.; Funaki, Y.; Mizuno, M.; Iida, A.; Goto, C.; Koikeda, S.; Kasugai, K.; Joh, T. Transglucosidase Improves the Gut Microbiota Profile of Type 2 Diabetes Mellitus Patients: A Randomized Double-Blind, Placebo-Controlled Study. BMC Gastroenterol. 2013, 13, 81. [Google Scholar] [CrossRef]
- Wu, H.; Esteve, E.; Tremaroli, V.; Khan, M.T.; Caesar, R.; Mannerås-Holm, L.; Ståhlman, M.; Olsson, L.M.; Serino, M.; Planas-Fèlix, M.; et al. Metformin Alters the Gut Microbiome of Individuals with Treatment-Naive Type 2 Diabetes, Contributing to the Therapeutic Effects of the Drug. Nat. Med. 2017, 23, 850–858. [Google Scholar] [CrossRef]
- Kikuchi, K.; Ben Othman, M.; Sakamoto, K. Sterilized Bifidobacteria Suppressed Fat Accumulation and Blood Glucose Level. Biochem. Biophys. Res. Commun. 2018, 501, 1041–1047. [Google Scholar] [CrossRef]
- Le, T.K.C.; Hosaka, T.; Nguyen, T.T.; Kassu, A.; Dang, T.O.; Tran, H.B.; Pham, T.P.; Tran, Q.B.; Le, T.H.H.; Pham, X.D. Bifidobacterium Species Lower Serum Glucose, Increase Expressions of Insulin Signaling Proteins, and Improve Adipokine Profile in Diabetic Mice. Biomed. Res. 2015, 36, 63–70. [Google Scholar] [CrossRef]
- Moya-Pérez, A.; Neef, A.; Sanz, Y. Bifidobacterium Pseudocatenulatum CECT 7765 Reduces Obesity-Associated Inflammation by Restoring the Lymphocyte-Macrophage Balance and Gut Microbiota Structure in High-Fat Diet-Fed Mice. PLoS ONE 2015, 10, e0126976. [Google Scholar] [CrossRef]
- Wang, J.; Tang, H.; Zhang, C.; Zhao, Y.; Derrien, M.; Rocher, E.; van-Hylckama Vlieg, J.E.; Strissel, K.; Zhao, L.; Obin, M.; et al. Modulation of Gut Microbiota during Probiotic-Mediated Attenuation of Metabolic Syndrome in High Fat Diet-Fed Mice. ISME J. 2015, 9, 1–15. [Google Scholar] [CrossRef]
- Qian, X.; Si, Q.; Lin, G.; Zhu, M.; Lu, J.; Zhang, H.; Wang, G.; Chen, W. Bifidobacterium Adolescentis Is Effective in Relieving Type 2 Diabetes and May Be Related to Its Dominant Core Genome and Gut Microbiota Modulation Capacity. Nutrients 2022, 14, 2479. [Google Scholar] [CrossRef]
- Pedersen, C.; Gallagher, E.; Horton, F.; Ellis, R.J.; Ijaz, U.Z.; Wu, H.; Jaiyeola, E.; Diribe, O.; Duparc, T.; Cani, P.D.; et al. Host–Microbiome Interactions in Human Type 2 Diabetes Following Prebiotic Fibre (Galacto-Oligosaccharide) Intake. Br. J. Nutr. 2016, 116, 1869–1877. [Google Scholar] [CrossRef]
- Leite, A.Z.; Rodrigues, N.D.C.; Gonzaga, M.I.; Paiolo, J.C.C.; de Souza, C.A.; Stefanutto, N.A.V.; Omori, W.P.; Pinheiro, D.G.; Brisotti, J.L.; Matheucci Junior, E.; et al. Detection of Increased Plasma Interleukin-6 Levels and Prevalence of Prevotella Copri and Bacteroides Vulgatus in the Feces of Type 2 Diabetes Patients. Front. Immunol. 2017, 8, 1107. [Google Scholar] [CrossRef]
- Péan, N.; Le Lay, A.; Brial, F.; Wasserscheid, J.; Rouch, C.; Vincent, M.; Myridakis, A.; Hedjazi, L.; Dumas, M.-E.; Grundberg, E.; et al. Dominant Gut Prevotella Copri in Gastrectomised Non-Obese Diabetic Goto–Kakizaki Rats Improves Glucose Homeostasis through Enhanced FXR Signalling. Diabetologia 2020, 63, 1223–1235. [Google Scholar] [CrossRef]
- Tett, A.; Huang, K.D.; Asnicar, F.; Fehlner-Peach, H.; Pasolli, E.; Karcher, N.; Armanini, F.; Manghi, P.; Bonham, K.; Zolfo, M.; et al. The Prevotella Copri Complex Comprises Four Distinct Clades Underrepresented in Westernized Populations. Cell Host Microbe 2019, 26, 666–679.e7. [Google Scholar] [CrossRef]
- Grover, S.; Rashmi, H.M.; Srivastava, A.K.; Batish, V.K. Probiotics for Human Health –New Innovations and Emerging Trends. Gut Pathog. 2012, 4, 15. [Google Scholar] [CrossRef]
- Chen, Z.; Radjabzadeh, D.; Chen, L.; Kurilshikov, A.; Kavousi, M.; Ahmadizar, F.; Ikram, M.A.; Uitterlinden, A.G.; Zhernakova, A.; Fu, J.; et al. Association of Insulin Resistance and Type 2 Diabetes With Gut Microbial Diversity: A Microbiome-Wide Analysis From Population Studies. JAMA Netw. Open 2021, 4, e2118811. [Google Scholar] [CrossRef]
- Deng, J.; Zhong, J.; Long, J.; Zou, X.; Wang, D.; Song, Y.; Zhou, K.; Liang, Y.; Huang, R.; Wei, X.; et al. Hypoglycemic Effects and Mechanism of Different Molecular Weights of Konjac Glucomannans in Type 2 Diabetic Rats. Int. J. Biol. Macromol. 2020, 165, 2231–2243. [Google Scholar] [CrossRef]
- Han, L.; Li, T.; Du, M.; Chang, R.; Zhan, B.; Mao, X. Beneficial Effects of Potentilla Discolor Bunge Water Extract on Inflammatory Cytokines Release and Gut Microbiota in High-Fat Diet and Streptozotocin-Induced Type 2 Diabetic Mice. Nutrients 2019, 11, 670. [Google Scholar] [CrossRef]
- Zeng, B.; Lai, Z.; Sun, L.; Zhang, Z.; Yang, J.; Li, Z.; Lin, J.; Zhang, Z. Structural and Functional Profiles of the Gut Microbial Community in Polycystic Ovary Syndrome with Insulin Resistance (IR-PCOS): A Pilot Study. Res. Microbiol. 2019, 170, 43–52. [Google Scholar] [CrossRef]
- Oshim, I.; Agbakoba, N.; Oguejiofor, O.; Anukam, K. Gut Microbiota Compositions and Modulation of Bacterial Metabolic Functional Genes in Type -2 Diabetes Mellitus Individuals at Nnewi, Anambra State, Nigeria. J. Med. Lab. Sci. 2020, 30, 136–150. [Google Scholar] [CrossRef]
- Hu, R.; Zeng, F.; Wu, L.; Wan, X.; Chen, Y.; Zhang, J.; Liu, B. Fermented Carrot Juice Attenuates Type 2 Diabetes by Mediating Gut Microbiota in Rats. Food Funct. 2019, 10, 2935–2946. [Google Scholar] [CrossRef]
- Yoshimura, T.; McLean, M.H.; Dzutsev, A.K.; Yao, X.; Chen, K.; Huang, J.; Gong, W.; Zhou, J.; Xiang, Y.; Badger, J.H.; et al. The Antimicrobial Peptide CRAMP Is Essential for Colon Homeostasis by Maintaining Microbiota Balance. J. Immunol. 2018, 200, 2174–2185. [Google Scholar] [CrossRef]
- Geng, S.; Yang, L.; Cheng, F.; Zhang, Z.; Li, J.; Liu, W.; Li, Y.; Chen, Y.; Bao, Y.; Chen, L.; et al. Gut Microbiota Are Associated With Psychological Stress-Induced Defections in Intestinal and Blood–Brain Barriers. Front. Microbiol. 2020, 10, 3067. [Google Scholar] [CrossRef]
- Huber-Ruano, I.; Calvo, E.; Mayneris-Perxachs, J.; Rodríguez-Peña, M.-M.; Ceperuelo-Mallafré, V.; Cedó, L.; Núñez-Roa, C.; Miro-Blanch, J.; Arnoriaga-Rodríguez, M.; Balvay, A.; et al. Orally Administered Odoribacter Laneus Improves Glucose Control and Inflammatory Profile in Obese Mice by Depleting Circulating Succinate. Microbiome 2022, 10, 135. [Google Scholar] [CrossRef]
- Wu, H.; Tremaroli, V.; Schmidt, C.; Lundqvist, A.; Olsson, L.M.; Krämer, M.; Gummesson, A.; Perkins, R.; Bergström, G.; Bäckhed, F. The Gut Microbiota in Prediabetes and Diabetes: A Population-Based Cross-Sectional Study. Cell Metab. 2020, 32, 379–390.e3. [Google Scholar] [CrossRef]
- Cui, J.; Ramesh, G.; Wu, M.; Jensen, E.T.; Crago, O.; Bertoni, A.G.; Gao, C.; Hoffman, K.L.; Sheridan, P.A.; Wong, K.E.; et al. Butyrate-Producing Bacteria and Insulin Homeostasis: The Microbiome and Insulin Longitudinal Evaluation Study (MILES). Diabetes 2022, 71, 2438–2446. [Google Scholar] [CrossRef] [PubMed]
- Gacesa, R.; Kurilshikov, A.; Vich Vila, A.; Sinha, T.; Klaassen, M.A.Y.; Bolte, L.A.; Andreu-Sánchez, S.; Chen, L.; Collij, V.; Hu, S.; et al. Environmental Factors Shaping the Gut Microbiome in a Dutch Population. Nature 2022, 604, 732–739. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Cheng, H.; Liu, Y.; Xue, M.; Liang, H. Red Yeast Rice Ameliorates High-Fat Diet-Induced Atherosclerosis in Apoe−/− Mice in Association with Improved Inflammation and Altered Gut Microbiota Composition. Food Funct. 2019, 10, 3880–3889. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Mao, J.; Zhou, L.; Xiong, X.; Deng, Y. The Imbalance of Gut Microbiota and Its Correlation with Plasma Inflammatory Cytokines in Pemphigus Vulgaris Patients. Scand. J. Immunol. 2019, 90, e12799. [Google Scholar] [CrossRef]
- Phosat, C.; Panprathip, P.; Chumpathat, N.; Prangthip, P.; Chantratita, N.; Soonthornworasiri, N.; Puduang, S.; Kwanbunjan, K. Elevated C-Reactive Protein, Interleukin 6, Tumor Necrosis Factor Alpha and Glycemic Load Associated with Type 2 Diabetes Mellitus in Rural Thais: A Cross-Sectional Study. BMC Endocr. Disord. 2017, 17, 44. [Google Scholar] [CrossRef]
- Huang, Z.-R.; Zhao, L.-Y.; Zhu, F.-R.; Liu, Y.; Xiao, J.-Y.; Chen, Z.-C.; Lv, X.-C.; Huang, Y.; Liu, B. Anti-Diabetic Effects of Ethanol Extract from Sanghuangporous Vaninii in High-Fat/Sucrose Diet and Streptozotocin-Induced Diabetic Mice by Modulating Gut Microbiota. Foods 2022, 11, 974. [Google Scholar] [CrossRef] [PubMed]
- Diener, C.; Reyes-Escogido, M.D.L.; Jimenez-Ceja, L.M.; Matus, M.; Gomez-Navarro, C.M.; Chu, N.D.; Zhong, V.; Tejero, M.E.; Alm, E.; Resendis-Antonio, O.; et al. Progressive Shifts in the Gut Microbiome Reflect Prediabetes and Diabetes Development in a Treatment-Naive Mexican Cohort. Front. Endocrinol. 2021, 11, 602326. [Google Scholar] [CrossRef]
- Ghaemi, F.; Fateh, A.; Sepahy, A.A.; Zangeneh, M.; Ghanei, M.; Siadat, S.D. Intestinal Microbiota Composition in Iranian Diabetic, Pre-Diabetic and Healthy Individuals. J. Diabetes Metab. Disord. 2020, 19, 1199–1203. [Google Scholar] [CrossRef]
- Zhong, H.; Ren, H.; Lu, Y.; Fang, C.; Hou, G.; Yang, Z.; Chen, B.; Yang, F.; Zhao, Y.; Shi, Z.; et al. Distinct Gut Metagenomics and Metaproteomics Signatures in Prediabetics and Treatment-Naïve Type 2 Diabetics. EBioMedicine 2019, 47, 373–383. [Google Scholar] [CrossRef]
- Forslund, K.; Hildebrand, F.; Nielsen, T.; Falony, G.; Le Chatelier, E.; Sunagawa, S.; Prifti, E.; Vieira-Silva, S.; Gudmundsdottir, V.; Krogh Pedersen, H.; et al. Disentangling Type 2 Diabetes and Metformin Treatment Signatures in the Human Gut Microbiota. Nature 2015, 528, 262–266. [Google Scholar] [CrossRef]
- Cao, T.T.B.; Wu, K.-C.; Hsu, J.-L.; Chang, C.-S.; Chou, C.; Lin, C.-Y.; Liao, Y.-M.; Lin, P.-C.; Yang, L.-Y.; Lin, H.-W. Effects of Non-Insulin Anti-Hyperglycemic Agents on Gut Microbiota: A Systematic Review on Human and Animal Studies. Front. Endocrinol. 2020, 11, 573891. [Google Scholar] [CrossRef] [PubMed]
- Graessler, J.; Qin, Y.; Zhong, H.; Zhang, J.; Licinio, J.; Wong, M.-L.; Xu, A.; Chavakis, T.; Bornstein, A.B.; Ehrhart-Bornstein, M.; et al. Metagenomic Sequencing of the Human Gut Microbiome before and after Bariatric Surgery in Obese Patients with Type 2 Diabetes: Correlation with Inflammatory and Metabolic Parameters. Pharm. J. 2013, 13, 514–522. [Google Scholar] [CrossRef] [PubMed]
- Karlsson, F.H.; Tremaroli, V.; Nookaew, I.; Bergström, G.; Behre, C.J.; Fagerberg, B.; Nielsen, J.; Bäckhed, F. Gut Metagenome in European Women with Normal, Impaired and Diabetic Glucose Control. Nature 2013, 498, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Almugadam, B.S.; Liu, Y.; Chen, S.; Wang, C.; Shao, C.; Ren, B.; Tang, L. Alterations of Gut Microbiota in Type 2 Diabetes Individuals and the Confounding Effect of Antidiabetic Agents. J. Diabetes Res. 2020, 2020, 7253978. [Google Scholar] [CrossRef]
- Roager, H.M.; Hansen, L.B.S.; Bahl, M.I.; Frandsen, H.L.; Carvalho, V.; Gøbel, R.J.; Dalgaard, M.D.; Plichta, D.R.; Sparholt, M.H.; Vestergaard, H.; et al. Colonic Transit Time Is Related to Bacterial Metabolism and Mucosal Turnover in the Gut. Nat. Microbiol. 2016, 1, 1–9. [Google Scholar] [CrossRef]
- Falony, G.; Joossens, M.; Vieira-Silva, S.; Wang, J.; Darzi, Y.; Faust, K.; Kurilshikov, A.; Bonder, M.J.; Valles-Colomer, M.; Vandeputte, D.; et al. Population-Level Analysis of Gut Microbiome Variation. Science 2016, 352, 560–564. [Google Scholar] [CrossRef]
- Rothschild, D.; Weissbrod, O.; Barkan, E.; Kurilshikov, A.; Korem, T.; Zeevi, D.; Costea, P.I.; Godneva, A.; Kalka, I.N.; Bar, N.; et al. Environment Dominates over Host Genetics in Shaping Human Gut Microbiota. Nature 2018, 555, 210–215. [Google Scholar] [CrossRef]
- Gupta, V.K.; Paul, S.; Dutta, C. Geography, Ethnicity or Subsistence-Specific Variations in Human Microbiome Composition and Diversity. Front. Microbiol. 2017, 8, 1162. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).