

Comparative Genomic Analysis of Enterococci across Sectors of the One Health Continuum
 , , , ,
, , , , 
Abstract
1. Introduction
2. Methodology
2.1. Enterococcus Recovery from Swine Feces and Whole Genome Sequencing
2.2. Collection of Enterococcus faecium and Enterococcus faecalis Genomes
2.3. Genome Assembly and Data Analysis
3. Results
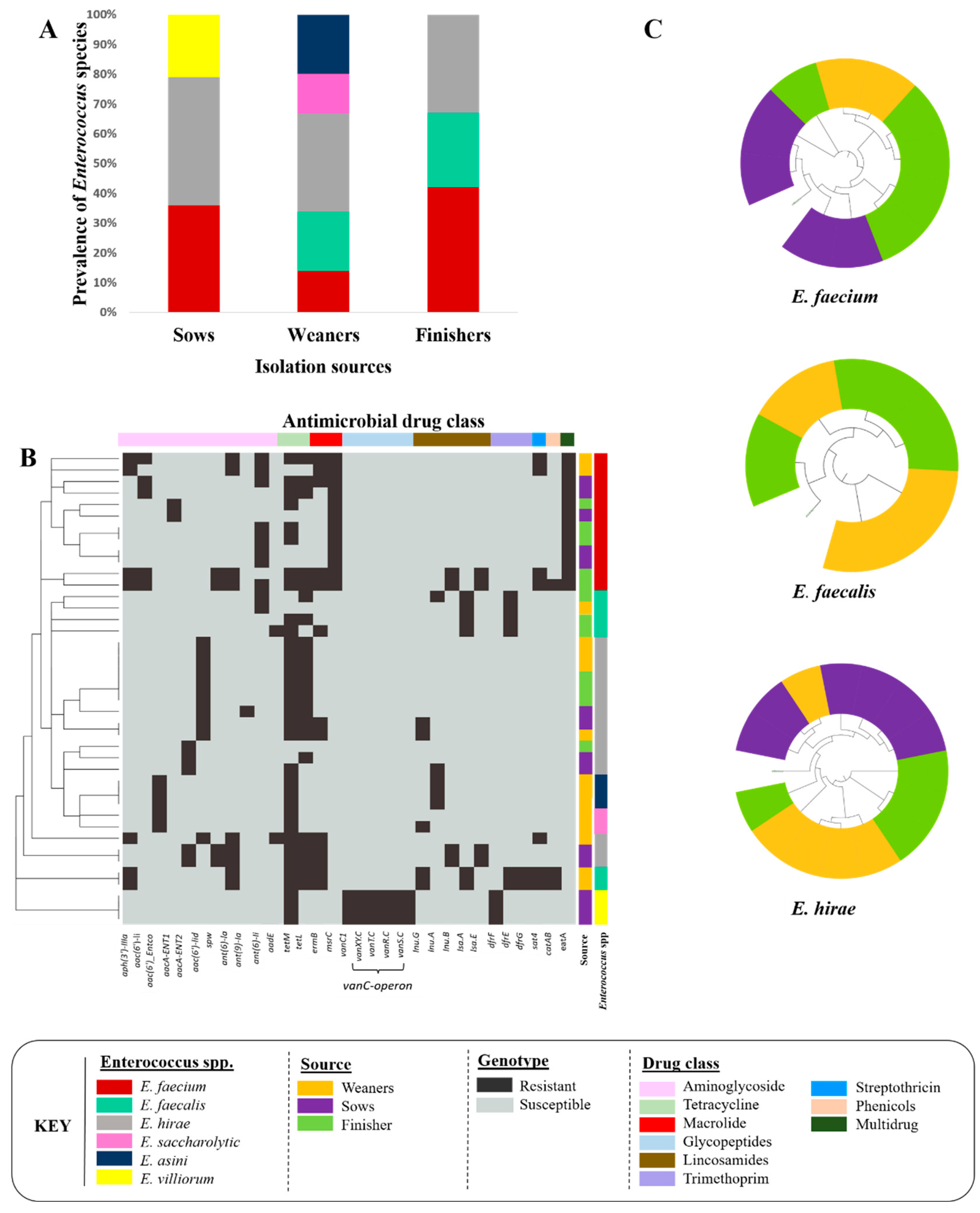
3.1. Enterococci Recovered from Swine Feces
3.1.1. Species Identification
3.1.2. Genome Characterization
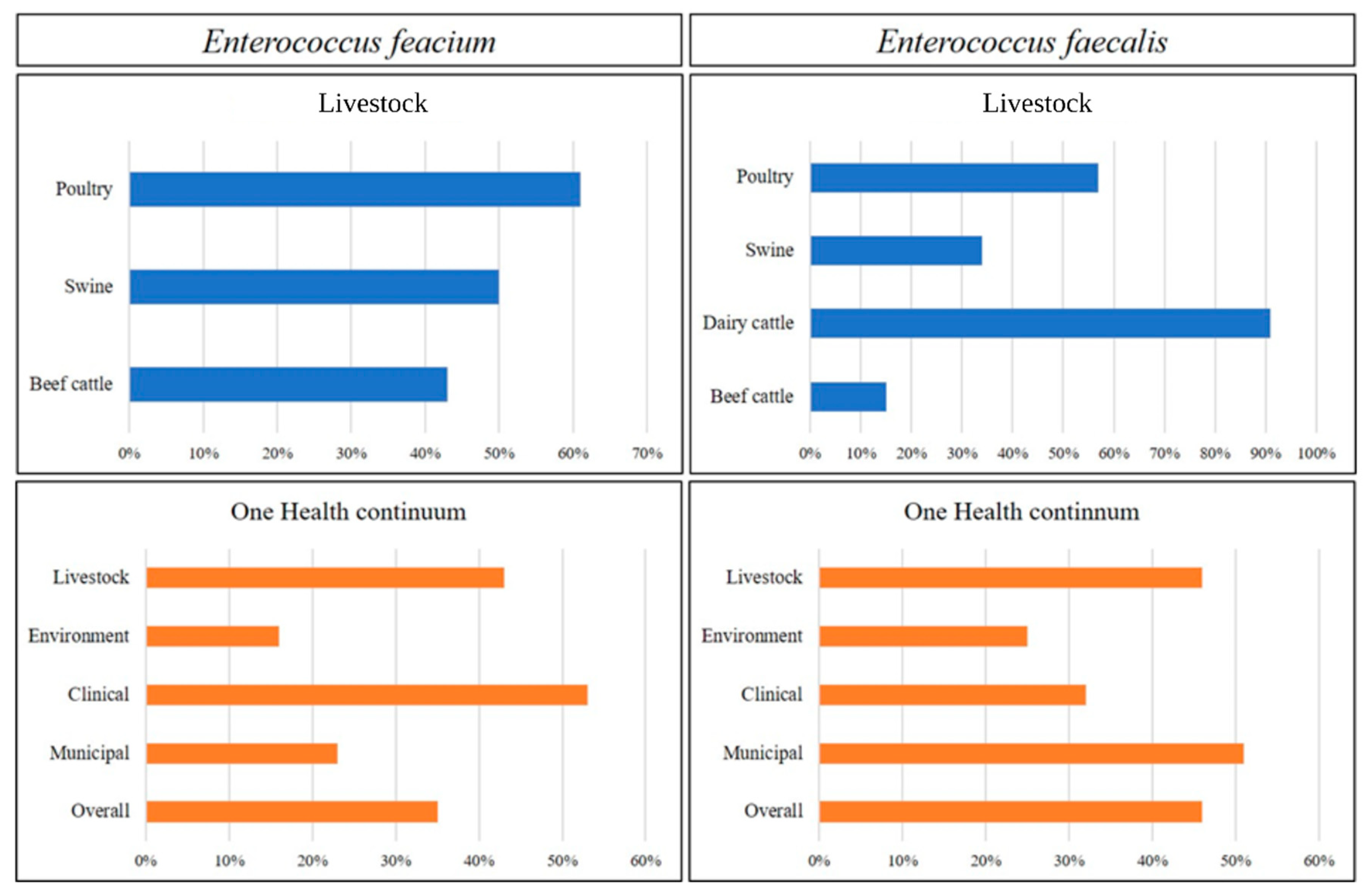
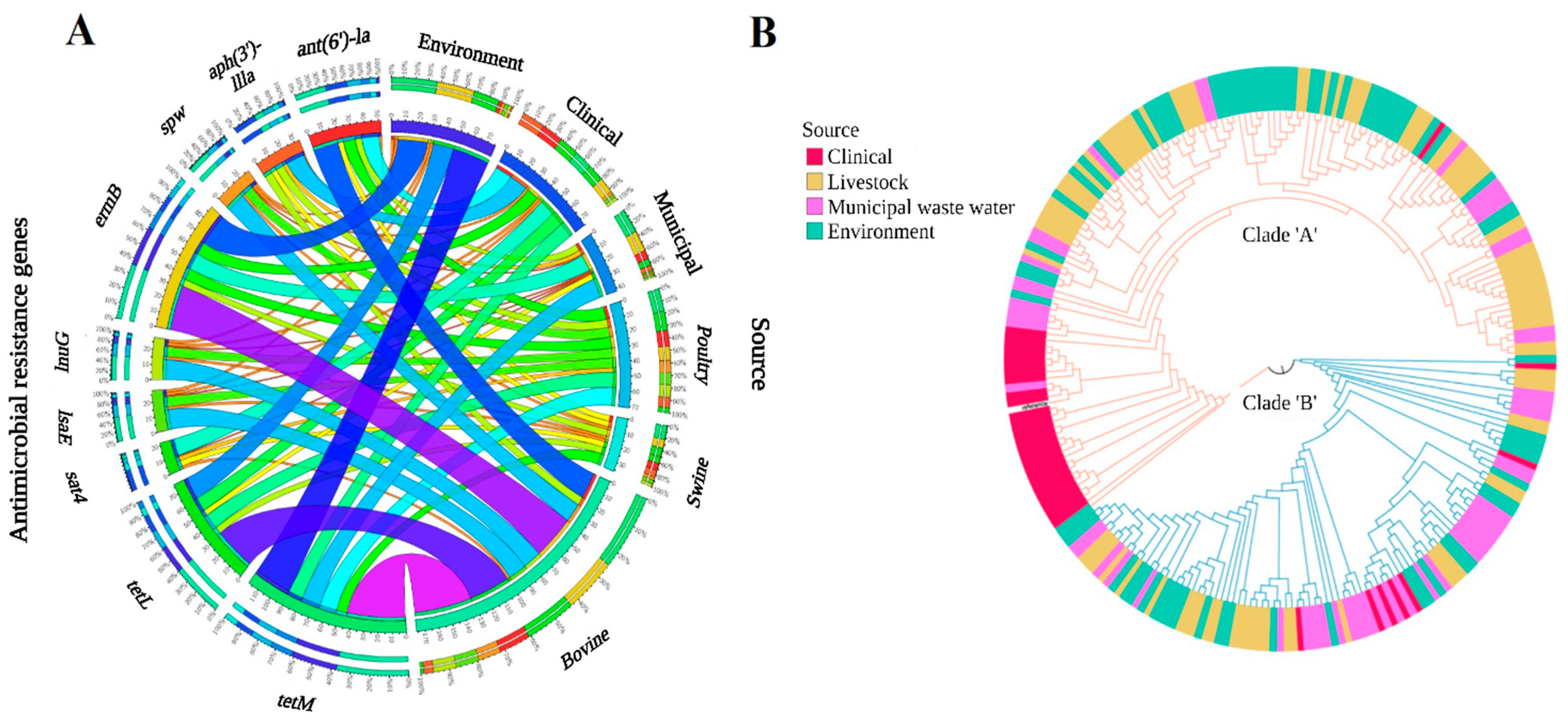
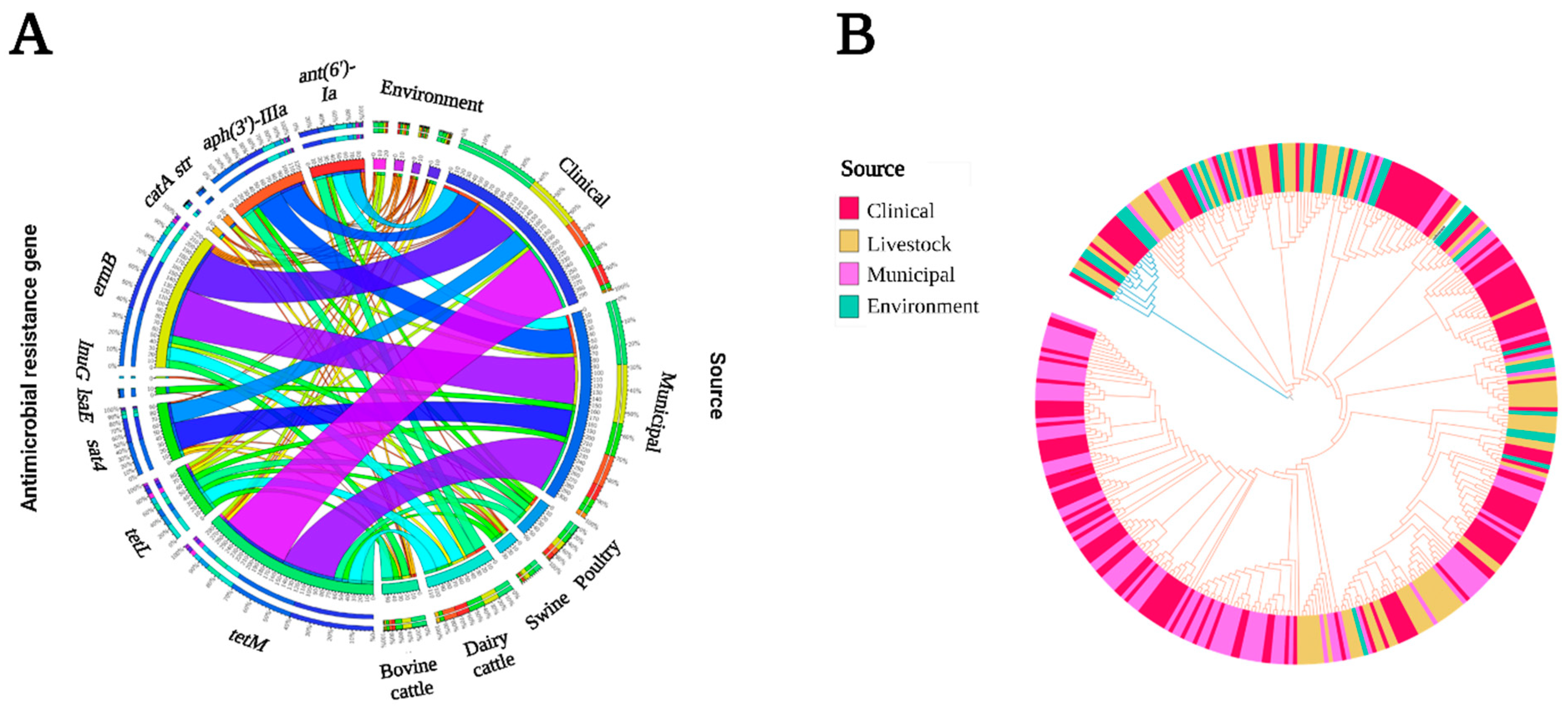
3.2. Comparative Genomic Analysis of E. faecalis and E. faecium across the One Health Continuum
3.2.1. Livestock Production
3.2.2. One Health Continuum
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Baquero, F. Threats of antibiotic resistance: An obliged reappraisal. Int. Microbiol. 2021, 24, 499–506. [Google Scholar] [CrossRef]
- White, A.; Hughes, J.M. Critical Importance of a One Health Approach to Antimicrobial Resistance. EcoHealth 2019, 16, 404–409. [Google Scholar] [CrossRef]
- Lechner, I.C.; Freivogel, K.; Stärk, K.; Visschers, V. Exposure Pathways to Antimicrobial Resistance at the Human-Animal Interface—A Qualitative Comparison of Swiss Expert and Consumer Opinions. Front. Public Health 2020, 8, 345. [Google Scholar] [CrossRef]
- Michael, C.A.; Dominey-Howes, D.; Labbate, M. The antimicrobial resistance crisis: Causes, consequences, and management. Front. Public Health 2014, 2, 145. [Google Scholar] [CrossRef]
- Dunn, S.J.; Connor, C.; McNally, A. The evolution and transmission of multi-drug resistant Escherichia coli and Klebsiella pneumoniae: The complexity of clones and plasmids. Curr. Opin. Microbiol. 2019, 51, 51–56. [Google Scholar] [CrossRef]
- Lebreton, F.; Willems, R.J.L.; Gilmore, M.S. Enterococcus Diversity, Origins in Nature, and Gut Colonization. In En-Terococci: From Commensals to Leading Causes of Drug Resistant Infection; Gilmore, M.S., Clewell, D.B., Ike, Y., Shankar, N., Eds.; Massachusetts Eye and Ear Infirmary: Boston, MA, USA, 2014. [Google Scholar]
- García-Solache, M.; Rice, L.B. The Enterococcus: A Model of Adaptability to Its Environment. Clin. Microbiol. Rev. 2019, 32, e00058-18. [Google Scholar] [CrossRef]
- Poulin-Laprade, D.; Brouard, J.-S.; Gagnon, N.; Turcotte, A.; Langlois, A.; Matte, J.J.; Carrillo, C.D.; Zaheer, R.; McAllister, T.A.; Topp, E.; et al. Resistance Determinants and Their Genetic Context in Enterobacteria from a Longitudinal Study of Pigs Reared under Various Husbandry Conditions. Appl. Environ. Microbiol. 2021, 87, e02612-20. [Google Scholar] [CrossRef]
- Zaheer, R.; Cook, S.R.; Barbieri, R.; Goji, N.; Cameron, A.; Petkau, A.; Polo, R.O.; Tymensen, L.; Stamm, C.; Song, J.; et al. Surveillance of Enterococcus spp. Reveals Distinct Species and Antimicrobial Resistance Diversity across a One-Health Continuum. Sci. Rep. 2020, 10, 3937. [Google Scholar] [CrossRef]
- Rehman, M.A.; Yin, X.; Zaheer, R.; Goji, N.; Amoako, K.K.; McAllister, T.; Pritchard, J.; Topp, E.; Diarra, M.S. Genotypes and Phenotypes of Enterococci Isolated from Broiler Chickens. Front. Sustain. Food Syst. 2018, 2, 83. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. Spades: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. Quast: Quality Assessment Tool for Genome Assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef] [PubMed]
- Seemann, T. Prokka: Rapid Prokaryotic Genome Annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Zheng, D.; Zhou, S.; Chen, L.; Yang, J. VFDB 2022: A general classification scheme for bacterial virulence factors. Nucleic Acids Res. 2021, 50, D912–D917. [Google Scholar] [CrossRef] [PubMed]
- Robertson, J.; Nash, J.H.E. MOB-suite: Software tools for clustering, reconstruction and typing of plasmids from draft assemblies. Microb. Genom. 2018, 4, e000206. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef]
- Hung, W.-W. Using Groel as the Target for Identification of Enterococcus faecium Clades and 7 Clinically Relevant En-terococcus Species. J. Microbiol. Immunol. Infect. 2019, 52, 255–264. [Google Scholar] [CrossRef]
- Jolley, K.A.; Bray, J.E.; Maiden, M.C.J. Open-Access Bacterial Population Genomics: BIGSdb Software, the PubMLST.org Website and Their Applications [Version 1; Peer Review: 2 Approved]. Wellcome Open Res. 2018, 3, 124. [Google Scholar] [CrossRef]
- Jackson, C.R.; Lombard, J.E.; Dargatz, D.A.; Fedorka-Cray, P.J. Prevalence, species distribution and antimicrobial resistance of enterococci isolated from US dairy cattle. Lett. Appl. Microbiol. 2010, 52, 41–48. [Google Scholar] [CrossRef]
- Diarra, M.S.; Rempel, H.; Champagne, J.; Masson, L.; Pritchard, J.; Topp, E. Distribution of Antimicrobial Resistance and Virulence Genes in Enterococcus spp. and Characterization of Isolates from Broiler Chickens. Appl. Environ. Microbiol. 2010, 76, 8033–8043. [Google Scholar] [CrossRef]
- Li, X.; Alvarez, V.; Harper, W.J.; Wang, H.H. Persistent, Toxin-Antitoxin System-Independent, Tetracycline Resistance-Encoding Plasmid from a Dairy Enterococcus faecium Isolate. Appl. Environ. Microbiol. 2011, 77, 7096–7103. [Google Scholar] [PubMed]
- Carson, C.A.; Reid-Smith, R.; Irwin, R.J.; Martin, W.S.; McEwen, S.A. Antimicrobial Use on 24 Beef Farms in Ontario. Can. J. Vet. Res. 2008, 72, 109–118. [Google Scholar] [PubMed]
- CARSS. Canadian Antimicrobial Resistance Surveillance System (CARSS) Report 2022. Available online: https://www.canada.ca/en/public-health/services/publications/drugs-health-products/canadian-antimicrobial-resistance-surveillance-system-report-2022.html#a4.1 (accessed on 25 January 2023).
- Bartoloni, A.; Pallecchi, L.; Rodríguez, H.; Fernandez, C.; Mantella, A.; Bartalesi, F.; Strohmeyer, M.; Kristiansson, C.; Gotuzzo, E.; Paradisi, F.; et al. Antibiotic Resistance in a Very Remote Amazonas Community. Int. J. Antimicrob. Agents 2009, 33, 125–129. [Google Scholar] [PubMed]
- Nejidat, A.; Diaz-Reck, D.; Gelfand, I.; Zaady, E. Persistence and spread of tetracycline resistance genes and microbial community variations in the soil of animal corrals in a semi-arid planted forest. FEMS Microbiol. Ecol. 2021, 97, fiab106. [Google Scholar] [CrossRef]
- Stefańska, I.; Kwiecień, E.; Kizerwetter-Świda, M.; Chrobak-Chmiel, D.; Rzewuska, M. Tetracycline, Macrolide and Lincosamide Resistance in Streptococcus canis Strains from Companion Animals and Its Genetic Determinants. Antibiotics 2022, 11, 1034. [Google Scholar]
- Agga, G.E.; Kasumba, J.; Loughrin, J.H.; Conte, E.D. Anaerobic Digestion of Tetracycline Spiked Livestock Manure and Poultry Litter Increased the Abundances of Antibiotic and Heavy Metal Resistance Genes. Front. Microbiol. 2020, 11, 614424. [Google Scholar] [CrossRef]
- Amachawadi, R.G.; Scott, H.M.; Alvarado, C.A.; Mainini, T.R.; Vinasco, J.; Drouillard, J.S.; Nagaraja, T.G. Occurrence of the Transferable Copper Resistance Gene tcrB among Fecal Enterococci of U.S. Feedlot Cattle Fed Copper-Supplemented Diets. Appl. Environ. Microbiol. 2013, 79, 4369–4375. [Google Scholar]
- Yazdankhah, S.; Rudi, K.; Bernhoft, A. Zinc and copper in animal feed—Development of resistance and co-resistance to antimicrobial agents in bacteria of animal origin. Microb. Ecol. Health Dis. 2014, 25, 25862. [Google Scholar] [CrossRef]
- Hodel-Christian, S.L.; Murray, B.E. Characterization of the gentamicin resistance transposon Tn5281 from Enterococcus faecalis and comparison to staphylococcal transposons Tn4001 and Tn4031. Antimicrob. Agents Chemother. 1991, 35, 1147–1152. [Google Scholar] [CrossRef]
- Mlynarczyk-Bonikowska, B.; Kowalewski, C.; Krolak-Ulinska, A.; Marusza, W. Molecular Mechanisms of Drug Resistance in Staphylococcus aureus. Int. J. Mol. Sci. 2022, 23, 8088. [Google Scholar] [CrossRef]
- Thomas, W.D., Jr.; Archer, G.L. Mobility of Gentamicin Resistance Genes from Staphylococci Isolated in the United States: Identification of Tn4031, a Gentamicin Resistance Transposon from Staphylococcus epidermidis. Antimicrob. Agents Chemother. 1989, 33, 1335–1341. [Google Scholar] [CrossRef] [PubMed]
- Szakacs, T.A.; Kalan, L.; McConnell, M.J.; Eshaghi, A.; Shahinas, D.; McGeer, A.; Wright, G.D.; Low, D.E.; Patel, S.N. Outbreak of Vancomycin-Susceptible Enterococcus faecium Containing the Wild-Type vanA Gene. J. Clin. Microbiol. 2014, 52, 1682–1686. [Google Scholar] [CrossRef] [PubMed]
- Palmer, K.L.; Godfrey, P.; Griggs, A.; Kos, V.N.; Zucker, J.; Desjardins, C.; Cerqueira, G.; Gevers, D.; Walker, S.; Wortman, J.; et al. Comparative Genomics of Enterococci: Variation in Enterococcus faecalis, Clade Structure in E. faecium, and Defining Characteristics of E. gallinarum and E. casseliflavus. mBio 2012, 3, e00318-11. [Google Scholar] [CrossRef] [PubMed]
- Sanderson, H.; Gray, K.L.; Manuele, A.; Maguire, F.; Khan, A.; Liu, C.; Rudrappa, C.N.; Nash, J.H.E.; Robertson, J.; Bessonov, K.; et al. Exploring the Mobilome and Resistome of Enterococcus faecium in a One Health Context across Two Continents. bioRxiv, 2022; preprint. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sources of Genome | Number of Genome | Antimicrobial Usage | Location (Year of Sample Collection) | Reference | ||
|---|---|---|---|---|---|---|
| E. faecium (n = 246) | E. faecalis (n = 376) | |||||
| Municipal waste water (MW) | 56 | 110 | - | Alberta (March 2014–April 2016) | [9] | |
| Clinical isolates (CL) | 36 | 149 | - | |||
| Livestock (LS) | Bovine cattle | 57 | 33 | Conventional (tetracycline, macrolides), natural (antibiotic-free) | ||
| Dairy cattle | - | 22 | NA | Ontario (2004) | This study | |
| Swine | - | 06 | NA | |||
| 12 | 06 | Conventional (penicillin), antibiotic-free (organic, certified-humane, AGRO-COM) | Quebec (2017–2018) | |||
| Poultry | 23 | 09 | Bambermycin, bacitracin, salinomycin, and β-lactams | British Colombia (2005–2008) | [10] | |
| - | 05 | NA | Ontario (2004) | This study | ||
| Environment (EV) | Natural water sources | 46 | 19 | - | Alberta (March 2014–April 2016) | [9] |
| River water | 16 | 07 | - | Ontario (2004) | This study | |
| Domestic animals | - | 03 | NA | |||
| Wild animals | - | 07 | - | |||
| Enterococcal Species | *& Antimicrobial Resistance Genes Profile (Number of Genomes) | Plasmids (Accession Number) (Total) | Antimicrobial Resistance Genes Found on Plasmid | Virulence Genes |
|---|---|---|---|---|
| E. faecalis | aph(3′)-IIIa, ant(6)-la, tetL, tetM, ermB, lnu(G), dfrG, sat4, catA8 (n = 2) | pBEE99 (NC_013533) (n = 2) | All ARGs |
|
| tetL, tetM (n = 1) | pSWS47 (NC_022618.1) (n = 1) | All ARGs | ||
| aadE, tetM, ermB (n = 1) | None | None | ||
| tetL, lnu(A) (n = 1) | None | None | ||
| E. faecium | aph(3′)-IIIa, spw, ant(6)-Ia, tetL, tetM, ermB, lnu(B), lsa(E), sat4, catA8 (n = 1) | pM7M2 (NC_016009) (n = 4) | tetL, tetM |
|
| aph(3′)-IIIa, ant(6)-Ia, tetL, tetM, ermB, sat4 (n = 1) | ||||
| tetL, tetM, ermB (n = 1) | ||||
| tetL, tetM (n = 1) | ||||
| aph(3′)-IIIa, spw, ant(6)-Ia, tetL, tetM, ermB, lnu(B), lsa(E), sat4 (n = 1) | pLAG (KY264168.1) (n = 1) | ant(6)-Ia, tetM, tetL, lnu(B), lsa(E) | ||
| aph(3′)-IIIa, ant(6)-Ia, ermB, sat4 (n = 1) | None | None | ||
| tetM (n = 3) | None | None | ||
| E. hirae | aph(3′)-IIIa, ant(6)-Ia, aadE, tetL, tetM, ermB, sat4 (n = 1) | p3 (CP006623) (n = 1) | aph(3′)-IIIa, ant(6)-Ia, ermB, sat4 |
|
| pBC16 (U32369) (n = 1) | tetM | |||
| spw, ant(6)-Ia, tetL, tetM, ermB, lnuB, lsaE (n = 2) | pEf37BA (MG957432) (n = 2) | All ARGs | ||
| tetL, tetM, ermB, lnuG (n = 2) | pDO1 (CP003584) (n = 2) | tetL, tetM, ermB | ||
| ant(9)-Ia, tetL, tetM (n = 1) | pM7M2 (NC_016009) (n = 1) | tetL, tetM | ||
| tetL, tetM (n = 7) | pM7M2 (NC_016009) (n = 3) | tetL, tetM | ||
| pCTN1046 (CP007650) (n = 1) | tetM | |||
| pBC16 (U32369) (n = 1) | tetL | |||
| tetM, lnuA (n = 1) | (CP029969) (n = 1) | lnu(A) | ||
| E. asini | tetM, lnuG (n = 1) | None | None |
|
| tetM (n = 1) | None | None | ||
| E. villorum | tetM, lsaA (n = 3) | None | None | None |
| E. saccharolyticus | tetM (n = 3) | None | None |
|
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zaidi, S.-e.-Z.; Zaheer, R.; Poulin-Laprade, D.; Scott, A.; Rehman, M.A.; Diarra, M.; Topp, E.; Domselaar, G.V.; Zovoilis, A.; McAllister, T.A. Comparative Genomic Analysis of Enterococci across Sectors of the One Health Continuum. Microorganisms 2023, 11, 727. https://doi.org/10.3390/microorganisms11030727
Zaidi S-e-Z, Zaheer R, Poulin-Laprade D, Scott A, Rehman MA, Diarra M, Topp E, Domselaar GV, Zovoilis A, McAllister TA. Comparative Genomic Analysis of Enterococci across Sectors of the One Health Continuum. Microorganisms. 2023; 11(3):727. https://doi.org/10.3390/microorganisms11030727
Chicago/Turabian StyleZaidi, Sani-e-Zehra, Rahat Zaheer, Dominic Poulin-Laprade, Andrew Scott, Muhammad Attiq Rehman, Moussa Diarra, Edward Topp, Gary Van Domselaar, Athanasios Zovoilis, and Tim A. McAllister. 2023. "Comparative Genomic Analysis of Enterococci across Sectors of the One Health Continuum" Microorganisms 11, no. 3: 727. https://doi.org/10.3390/microorganisms11030727
APA StyleZaidi, S.-e.-Z., Zaheer, R., Poulin-Laprade, D., Scott, A., Rehman, M. A., Diarra, M., Topp, E., Domselaar, G. V., Zovoilis, A., & McAllister, T. A. (2023). Comparative Genomic Analysis of Enterococci across Sectors of the One Health Continuum. Microorganisms, 11(3), 727. https://doi.org/10.3390/microorganisms11030727