
Association Studies on Gut and Lung Microbiomes in Patients with Lung Adenocarcinoma

 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Cohort and Sample Collection
2.2. 16S rRNA Gene Amplicon Sequencing
2.3. Shotgun Metagenome Sequencing
2.4. Phylogenetic Analysis between Gut and Lung Microbiomes
2.5. Statistical Analysis
3. Results
3.1. Characteristics of Study Participants
3.2. The Gut Microbiota of Patients with LADC Exhibited Reduced α-Diversity and Altered Bacterial Composition
3.3. Bacterial Species Found in Both Gut and Lung Microbiota of Patients with LADC
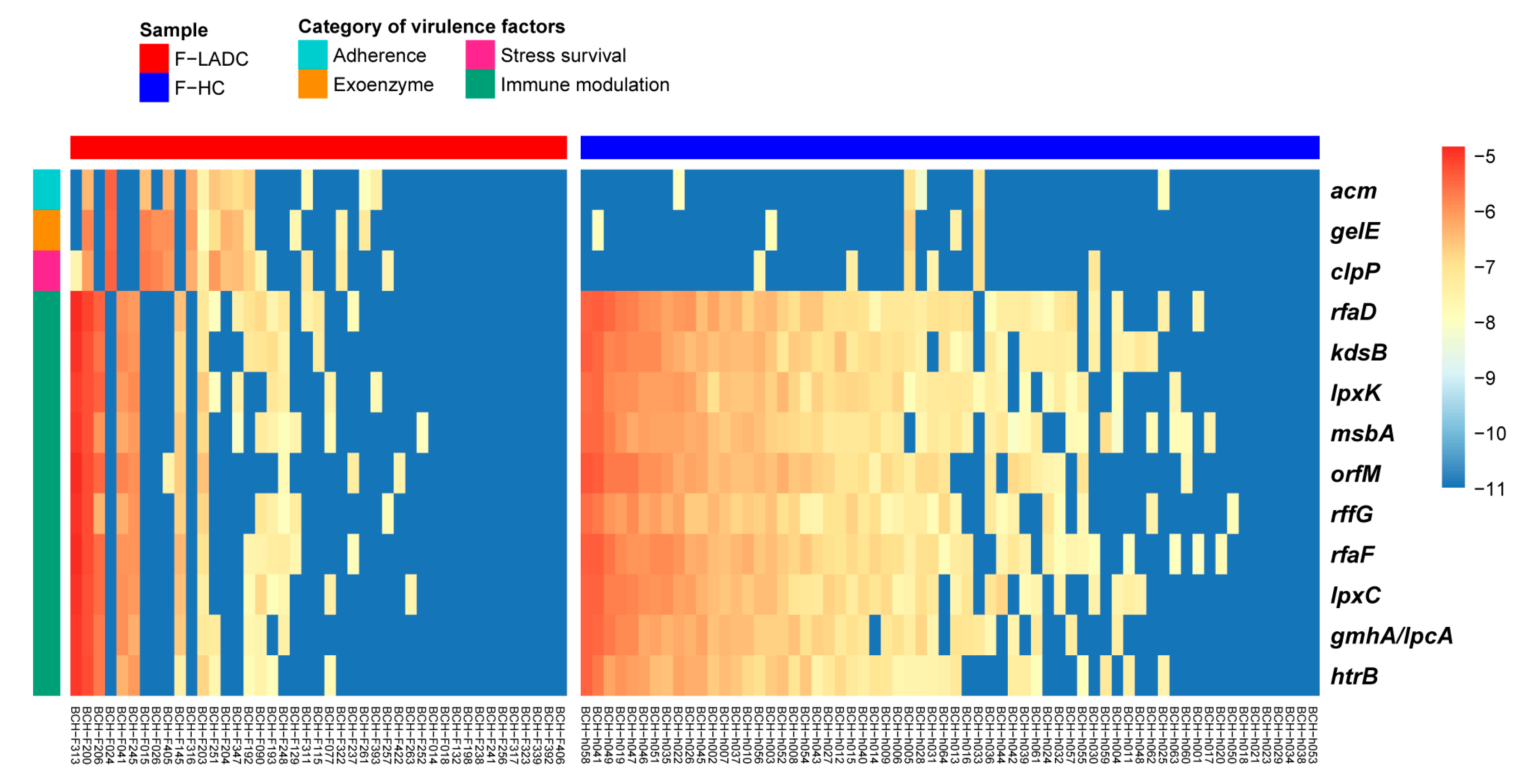
3.4. Comparative Analysis of Metabolic Pathways of Gut Microbiota in Patients with LADC
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Fang, Z.; Xue, Y.; Zhang, J.; Zhu, J.; Gao, R.; Yao, S.; Ye, Y.; Wang, S.; Lin, C.; et al. Specific gut microbiome signature predicts the early-stage lung cancer. Gut Microbes 2020, 11, 1030–1042. [Google Scholar] [CrossRef] [PubMed]
- Zhao, F.; An, R.; Wang, L.; Shan, J.; Wang, X. Specific Gut Microbiome and Serum Metabolome Changes in Lung Cancer Patients. Front. Cell. Infect. Microbiol. 2021, 11, 725284. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Sung, J.Y.; Yong, D.; Chun, J.; Kim, S.Y.; Song, J.H.; Chung, K.S.; Kim, E.Y.; Jung, J.Y.; Kang, Y.A.; et al. Characterization of microbiome in bronchoalveolar lavage fluid of patients with lung cancer comparing with benign mass like lesions. Lung Cancer 2016, 102, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Kim, O.H.; Choi, B.Y.; Kim, D.K.; Kim, N.H.; Rho, J.K.; Sul, W.J.; Lee, S.W. The microbiome of lung cancer tissue and its association with pathological and clinical parameters. Am. J. Cancer Res. 2022, 12, 2350–2362. [Google Scholar]
- Jin, C.; Lagoudas, G.K.; Zhao, C.; Bullman, S.; Bhutkar, A.; Hu, B.; Ameh, S.; Sandel, D.; Liang, X.S.; Mazzilli, S.; et al. Commensal Microbiota Promote Lung Cancer Development via gammadelta T Cells. Cell 2019, 176, 998–1013.e1016. [Google Scholar] [CrossRef]
- Bingula, R.; Filaire, M.; Radosevic-Robin, N.; Bey, M.; Berthon, J.Y.; Bernalier-Donadille, A.; Vasson, M.P.; Filaire, E. Desired Turbulence? Gut-Lung Axis, Immunity, and Lung Cancer. J. Oncol. 2017, 2017, 5035371. [Google Scholar] [CrossRef]
- Wypych, T.P.; Wickramasinghe, L.C.; Marsland, B.J. The influence of the microbiome on respiratory health. Nat. Immunol. 2019, 20, 1279–1290. [Google Scholar] [CrossRef]
- Lu, H.; Gao, N.L.; Tong, F.; Wang, J.; Li, H.; Zhang, R.; Ma, H.; Yang, N.; Zhang, Y.; Wang, Y.; et al. Alterations of the Human Lung and Gut Microbiomes in Non-Small Cell Lung Carcinomas and Distant Metastasis. Microbiol. Spectr. 2021, 9, e0080221. [Google Scholar] [CrossRef]
- Durack, J.; Huang, Y.J.; Nariya, S.; Christian, L.S.; Ansel, K.M.; Beigelman, A.; Castro, M.; Dyer, A.M.; Israel, E.; Kraft, M.; et al. Bacterial biogeography of adult airways in atopic asthma. Microbiome 2018, 6, 104. [Google Scholar] [CrossRef]
- Schneeberger, P.H.H.; Prescod, J.; Levy, L.; Hwang, D.; Martinu, T.; Coburn, B. Microbiota analysis optimization for human bronchoalveolar lavage fluid. Microbiome 2019, 7, 141. [Google Scholar] [CrossRef] [PubMed]
- Sulaiman, I.; Wu, B.G.; Li, Y.; Scott, A.S.; Malecha, P.; Scaglione, B.; Wang, J.; Basavaraj, A.; Chung, S.; Bantis, K.; et al. Evaluation of the airway microbiome in nontuberculous mycobacteria disease. Eur. Respir. J. 2018, 52, 1800810. [Google Scholar] [CrossRef] [PubMed]
- Brierley, J.; O’Sullivan, B.; Asamura, H.; Byrd, D.; Huang, S.H.; Lee, A.; Piñeros, M.; Mason, M.; Moraes, F.Y.; Rösler, W.; et al. Global Consultation on Cancer Staging: Promoting consistent understanding and use. Nat. Rev. Clin. Oncol. 2019, 16, 763–771. [Google Scholar] [CrossRef] [PubMed]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [PubMed]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef]
- Bokulich, N.A.; Kaehler, B.D.; Rideout, J.R.; Dillon, M.; Bolyen, E.; Knight, R.; Huttley, G.A.; Gregory Caporaso, J. Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin. Microbiome 2018, 6, 90. [Google Scholar] [CrossRef]
- Yilmaz, P.; Parfrey, L.W.; Yarza, P.; Gerken, J.; Pruesse, E.; Quast, C.; Schweer, T.; Peplies, J.; Ludwig, W.; Glöckner, F.O. The SILVA and “All-species Living Tree Project (LTP)” taxonomic frameworks. Nucleic Acids Res. 2014, 42, D643–D648. [Google Scholar] [CrossRef]
- Li, D.; Liu, C.M.; Luo, R.; Sadakane, K.; Lam, T.W. MEGAHIT: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef]
- Hyatt, D.; Chen, G.L.; Locascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 2010, 11, 119. [Google Scholar] [CrossRef]
- Beghini, F.; McIver, L.J.; Blanco-Míguez, A.; Dubois, L.; Asnicar, F.; Maharjan, S.; Mailyan, A.; Manghi, P.; Scholz, M.; Thomas, A.M.; et al. Integrating taxonomic, functional, and strain-level profiling of diverse microbial communities with bioBakery 3. Elife 2021, 10, e65088. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Parks, D.H.; Tyson, G.W.; Hugenholtz, P.; Beiko, R.G. STAMP: Statistical analysis of taxonomic and functional profiles. Bioinformatics 2014, 30, 3123–3124. [Google Scholar] [CrossRef]
- Liu, B.; Zheng, D.; Zhou, S.; Chen, L.; Yang, J. VFDB 2022: A general classification scheme for bacterial virulence factors. Nucleic Acids Res. 2022, 50, D912–D917. [Google Scholar] [CrossRef]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2—Approximately maximum-likelihood trees for large alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef]
- Dixon, P. VEGAN, a package of R functions for community ecology. J. Veg. Sci. 2003, 14, 927–930. [Google Scholar] [CrossRef]
- Petersen, C.; Round, J.L. Defining dysbiosis and its influence on host immunity and disease. Cell. Microbiol. 2014, 16, 1024–1033. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Wang, S.; Shen, J.; Hu, Q.; Zhang, Y.; Ma, D.; Chai, K. Analysis of Gut Microbiota Composition in Lung Adenocarcinoma Patients with TCM Qi-Yin Deficiency. Am. J. Chin. Med. 2021, 49, 1667–1682. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, H.; Cheng, L.; Wang, Y.; Zhang, Y.K.; Zhao, M.F.; Liang, G.D.; Zhang, M.C.; Li, Y.G.; Zhao, J.B.; Gao, Y.N.; et al. Dysbiosis of the Gut Microbiome in Lung Cancer. Front. Cell. Infect. Microbiol. 2019, 9, 112. [Google Scholar] [CrossRef]
- Zhang, W.Q.; Zhao, S.K.; Luo, J.W.; Dong, X.P.; Hao, Y.T.; Li, H.; Shan, L.; Zhou, Y.; Shi, H.B.; Zhang, Z.Y.; et al. Alterations of fecal bacterial communities in patients with lung cancer. Am. J. Transl. Res. 2018, 10, 3171–3185. [Google Scholar] [PubMed]
- Lim, M.Y.; Hong, S.; Hwang, K.H.; Lim, E.J.; Han, J.Y.; Nam, Y.D. Diagnostic and prognostic potential of the oral and gut microbiome for lung adenocarcinoma. Clin. Transl. Med. 2021, 11, e508. [Google Scholar] [CrossRef]
- Willems, R.J.; Top, J.; van Santen, M.; Robinson, D.A.; Coque, T.M.; Baquero, F.; Grundmann, H.; Bonten, M.J. Global spread of vancomycin-resistant Enterococcus faecium from distinct nosocomial genetic complex. Emerg. Infect. Dis. 2005, 11, 821–828. [Google Scholar] [CrossRef]
- Yu, T.; Li, L.; Zhao, Q.; Wang, P.; Zuo, X. Complete genome sequence of bile-isolated Enterococcus avium strain 352. Gut Pathog. 2019, 11, 16. [Google Scholar] [CrossRef]
- Chia, J.H.; Wu, T.S.; Wu, T.L.; Chen, C.L.; Chuang, C.H.; Su, L.H.; Chang, H.J.; Lu, C.C.; Kuo, A.J.; Lai, H.C.; et al. Clostridium innocuum is a vancomycin-resistant pathogen that may cause antibiotic-associated diarrhoea. Clin. Microbiol. Infect. 2018, 24, 1195–1199. [Google Scholar] [CrossRef] [PubMed]
- Gardiner, B.J.; Tai, A.Y.; Kotsanas, D.; Francis, M.J.; Roberts, S.A.; Ballard, S.A.; Junckerstorff, R.K.; Korman, T.M. Clinical and microbiological characteristics of Eggerthella lenta bacteremia. J. Clin. Microbiol. 2015, 53, 626–635. [Google Scholar] [CrossRef] [PubMed]
- Morrison, D.J.; Preston, T. Formation of short chain fatty acids by the gut microbiota and their impact on human metabolism. Gut Microbes 2016, 7, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Nogal, A.; Valdes, A.M.; Menni, C. The role of short-chain fatty acids in the interplay between gut microbiota and diet in cardio-metabolic health. Gut Microbes 2021, 13, 1897212. [Google Scholar] [CrossRef] [PubMed]
- Ban, H.; Shigemitsu, K.; Yamatsuji, T.; Haisa, M.; Nakajo, T.; Takaoka, M.; Nobuhisa, T.; Gunduz, M.; Tanaka, N.; Naomoto, Y. Arginine and Leucine regulate p70 S6 kinase and 4E-BP1 in intestinal epithelial cells. Int. J. Mol. Med. 2004, 13, 537–543. [Google Scholar] [CrossRef] [PubMed]
- Trefely, S.; Lovell, C.D.; Snyder, N.W.; Wellen, K.E. Compartmentalised acyl-CoA metabolism and roles in chromatin regulation. Mol. Metab. 2020, 38, 100941. [Google Scholar] [CrossRef]
- Ilter, D.; Gomes, A.P. Age-induced metabolic reprogramming underlies cancer progression. Mol. Cell. Oncol. 2021, 8, 1876506. [Google Scholar] [CrossRef]
- Gomes, A.P.; Ilter, D.; Low, V.; Drapela, S.; Schild, T.; Mullarky, E.; Han, J.; Elia, I.; Broekaert, D.; Rosenzweig, A.; et al. Altered propionate metabolism contributes to tumour progression and aggressiveness. Nat. Metab. 2022, 4, 435–443. [Google Scholar] [CrossRef]
- Svensson, R.U.; Parker, S.J.; Eichner, L.J.; Kolar, M.J.; Wallace, M.; Brun, S.N.; Lombardo, P.S.; Van Nostrand, J.L.; Hutchins, A.; Vera, L.; et al. Inhibition of acetyl-CoA carboxylase suppresses fatty acid synthesis and tumor growth of non-small-cell lung cancer in preclinical models. Nat. Med. 2016, 22, 1108–1119. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Hosomi, K.; Kawashima, H.; Chen, Y.A.; Mohsen, A.; Ohno, H.; Konishi, K.; Tanisawa, K.; Kifushi, M.; Kogawa, M.; et al. Dietary Vitamin B1 Intake Influences Gut Microbial Community and the Consequent Production of Short-Chain Fatty Acids. Nutrients 2022, 14, 2078. [Google Scholar] [CrossRef] [PubMed]
- Peterson, C.T.; Rodionov, D.A.; Osterman, A.L.; Peterson, S.N. B Vitamins and Their Role in Immune Regulation and Cancer. Nutrients 2020, 12, 3380. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Xiong, Z.; Sun, L.; Yang, J.; Jin, Q. VFDB 2012 update: Toward the genetic diversity and molecular evolution of bacterial virulence factors. Nucleic Acids Res. 2012, 40, D641–D645. [Google Scholar] [CrossRef]
- Maldonado, R.F.; Sá-Correia, I.; Valvano, M.A. Lipopolysaccharide modification in Gram-negative bacteria during chronic infection. FEMS Microbiol. Rev. 2016, 40, 480–493. [Google Scholar] [CrossRef]
- Park, J.H.; Jeong, S.Y.; Choi, A.J.; Kim, S.J. Lipopolysaccharide directly stimulates Th17 differentiation in vitro modulating phosphorylation of RelB and NF-κB1. Immunol. Lett. 2015, 165, 10–19. [Google Scholar] [CrossRef]
- Yang, Y.; Du, L.; Shi, D.; Kong, C.; Liu, J.; Liu, G.; Li, X.; Ma, Y. Dysbiosis of human gut microbiome in young-onset colorectal cancer. Nat. Commun. 2021, 12, 6757. [Google Scholar] [CrossRef]
- Ogita, T.; Yamamoto, Y.; Mikami, A.; Shigemori, S.; Sato, T.; Shimosato, T. Oral Administration of Flavonifractor plautii Strongly Suppresses Th2 Immune Responses in Mice. Front. Immunol. 2020, 11, 379. [Google Scholar] [CrossRef]
- Alexander, M.; Ang, Q.Y.; Nayak, R.R.; Bustion, A.E.; Sandy, M.; Zhang, B.; Upadhyay, V.; Pollard, K.S.; Lynch, S.V.; Turnbaugh, P.J. Human gut bacterial metabolism drives Th17 activation and colitis. Cell Host Microbe 2022, 30, 17–30. [Google Scholar] [CrossRef]
- Mitchell, J.L.; Hill, S.L. Immune response to Haemophilus parainfluenzae in patients with chronic obstructive lung disease. Clin. Diagn. Lab. Immunol. 2000, 7, 25–30. [Google Scholar] [CrossRef]
- Ananieva, E.A.; Powell, J.D.; Hutson, S.M. Leucine Metabolism in T Cell Activation: mTOR Signaling and Beyond. Adv. Nutr. 2016, 7, 798S–805S. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristic | Total | LADC | Healthy Controls | p Value * |
|---|---|---|---|---|
| (n = 107) | (n = 43) | (n = 64) | ||
| Sample type | ||||
| Feces | 107 | 43 | 64 | |
| BALF | 42 | 42 | NA | |
| Sex | 0.5538 | |||
| Female | 50 (46.7) | 22 (51.2) | 28 (43.8) | |
| Male | 57 (53.3) | 21 (48.8) | 36 (56.2) | |
| Age (years) | <0.001 | |||
| Mean (±SD) | 50 (±13) | 57 (±11) | 46 (±12) | |
| Smoking status | 0.8345 | |||
| Current or recent smoker | 34 (31.8) | 13 (30.2) | 21 (32.8) | |
| Never smoked | 73 (68.2) | 30 (69.8) | 43 (67.2) | |
| Disease stage | ||||
| I | 20 (46.5) | 20 (46.5) | NA | |
| II | 3 (7.0) | 3 (7.0) | NA | |
| III | 10 (23.3) | 10 (23.3) | NA | |
| IV | 10 (23.3) | 10 (23.3) | NA | |
| Distant metastasis | ||||
| Absence | 33 (76.7) | 33 (76.7) | NA | |
| Presence | 10 (23.3) | 10 (23.3) | NA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guo, Y.; Yuan, W.; Lyu, N.; Pan, Y.; Cao, X.; Wang, Y.; Han, Y.; Zhu, B. Association Studies on Gut and Lung Microbiomes in Patients with Lung Adenocarcinoma. Microorganisms 2023, 11, 546. https://doi.org/10.3390/microorganisms11030546
Guo Y, Yuan W, Lyu N, Pan Y, Cao X, Wang Y, Han Y, Zhu B. Association Studies on Gut and Lung Microbiomes in Patients with Lung Adenocarcinoma. Microorganisms. 2023; 11(3):546. https://doi.org/10.3390/microorganisms11030546
Chicago/Turabian StyleGuo, Yaqiong, Wenjie Yuan, Na Lyu, Yuanlong Pan, Xiaoqing Cao, Yuxuan Wang, Yi Han, and Baoli Zhu. 2023. "Association Studies on Gut and Lung Microbiomes in Patients with Lung Adenocarcinoma" Microorganisms 11, no. 3: 546. https://doi.org/10.3390/microorganisms11030546
APA StyleGuo, Y., Yuan, W., Lyu, N., Pan, Y., Cao, X., Wang, Y., Han, Y., & Zhu, B. (2023). Association Studies on Gut and Lung Microbiomes in Patients with Lung Adenocarcinoma. Microorganisms, 11(3), 546. https://doi.org/10.3390/microorganisms11030546