
Comparative Analysis of the Symbiotic Microbiota in the Chinese Mitten Crab (Eriocheir sinensis): Microbial Structure, Co-Occurrence Patterns, and Predictive Functions

 and
and {kind=link}
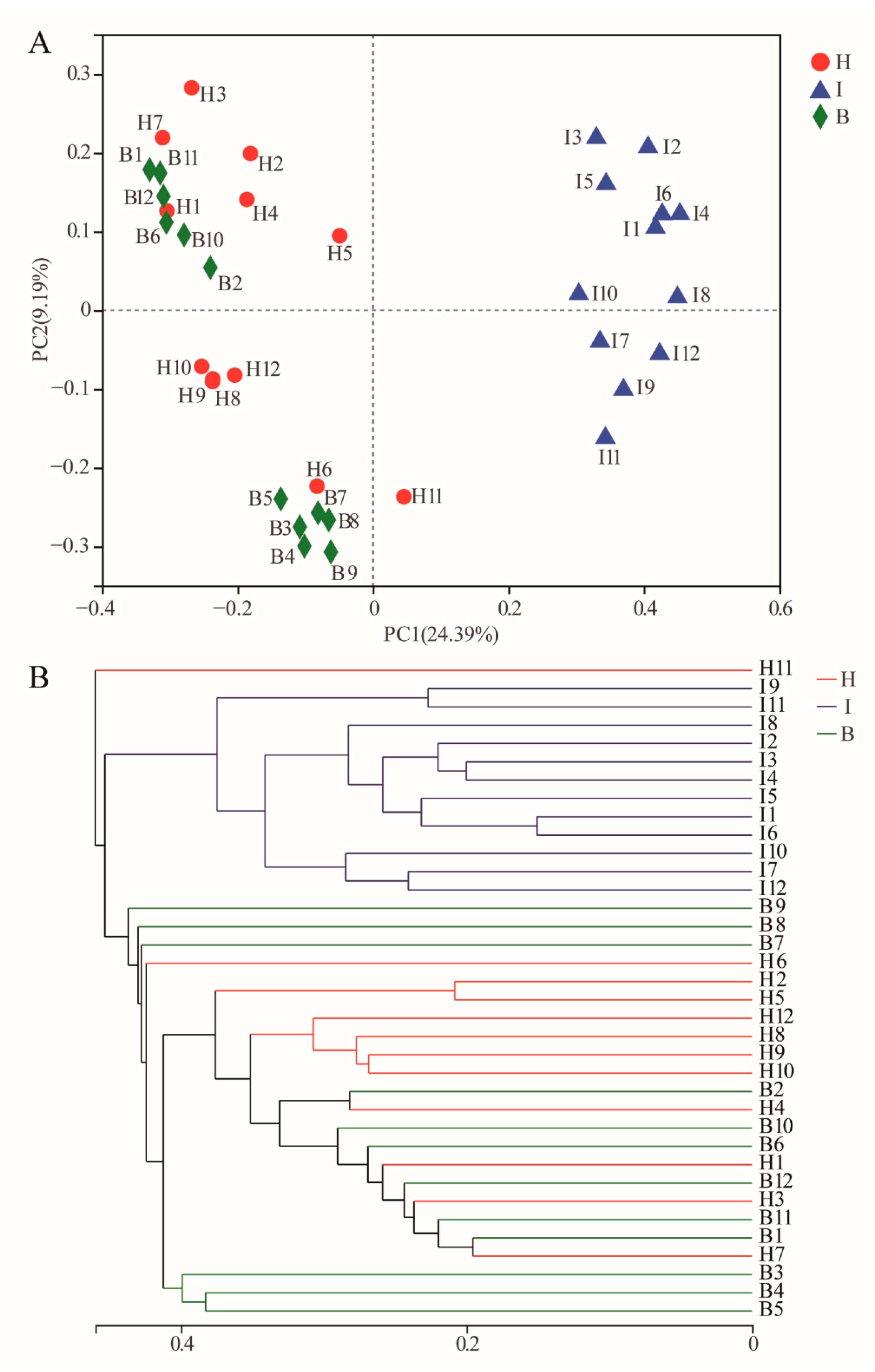{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection and Preparation
2.2. DNA Extraction
2.3. 16S rRNA Gene High-Throughput Sequencing
2.4. High-Throughput Sequencing Data Analysis Processing
2.5. Bioinformatics and Statistical Analysis
3. Results
3.1. Diversity and Structure of Bacterial Communities in Different Sites
3.2. Taxonomic Composition of the Bacterial Communities in Different Sites
3.3. Differences of the Bacterial Communities at Different Sites
3.4. Analysis of Bacterial Co-Occurrence Networks at Different Sites
3.5. Functional Prediction of the Bacterial Communities
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- McFall-Ngai, M.; Hadfield, M.G.; Bosch, T.C.G.; Carey, H.V.; Domazet-Loso, T.; Douglas, A.E.; Dubilier, N.; Eberl, G.; Fukami, T.; Gilbert, S.F.; et al. Animals in a bacterial world, a new imperative for the life sciences. Proc. Natl. Acad. Sci. USA 2013, 110, 3229–3236. [Google Scholar] [CrossRef] [PubMed]
- Bikel, S.; Valdez-Lara, A.; Cornejo-Granados, F.; Rico, K.; Canizales-Quinteros, S.; Soberon, X.; Del Pozo-Yauner, L.; Ochoa-Leyva, A. Combining metagenomics, metatranscriptomics and viromics to explore novel microbial interactions: Towards a systems-level understanding of human microbiome. Comput. Struct. Biotechnol. J. 2015, 13, 390–401. [Google Scholar] [CrossRef] [PubMed]
- Brestoff, J.R.; Artis, D. Commensal bacteria at the interface of host metabolism and the immune system. Nat. Immunol. 2013, 14, 676–684. [Google Scholar] [CrossRef] [PubMed]
- Rungrassamee, W.; Klanchui, A.; Maibunkaew, S.; Chaiyapechara, S.; Jiravanichpaisal, P.; Karoonuthaisiri, N. Characterization of Intestinal Bacteria in Wild and Domesticated Adult Black Tiger Shrimp (Penaeus monodon). PLoS ONE 2014, 9, e91853. [Google Scholar] [CrossRef]
- Verner-Jeffreys, D.W.; Shields, R.J.; Bricknell, I.R.; Birkbeck, T.H. Changes in the gut-associated microflora during the development of Atlantic halibut (Hippoglossus hippoglossus L.) larvae in three British hatcheries. Aquaculture 2003, 219, 21–42. [Google Scholar] [CrossRef]
- Gallo, R.L.; Nakatsuji, T. Microbial Symbiosis with the Innate Immune Defense System of the Skin. J. Investig. Dermatol. 2011, 131, 1974–1980. [Google Scholar] [CrossRef]
- Ceccaldi, H.J. Anatomy and physiology of digestive tract of crustaceans decapods reared in aquaculture. IFREMER Actes De Colloq. 1990, 26, 243–259. [Google Scholar]
- Leano, E.M.; Lavilla-Pitogo, C.R.; Paner, M.G. Bacterial flora in the hepatopancreas of pond-reared Penaeus monodon juveniles with luminous vibriosis. Aquaculture 1998, 164, 367–374. [Google Scholar] [CrossRef]
- Jiang, H.; Li, F.; Zhang, J.; Zhang, J.; Huang, B.; Yu, Y.; Xiang, J. Comparison of Protein Expression Profiles of the Hepatopancreas in Fenneropenaeus chinensis Challenged with Heat-inactivated Vibrio anguillarum and White Spot Syndrome Virus. Mar. Biotechnol. 2014, 16, 111–123. [Google Scholar] [CrossRef]
- Potgieter, M.; Bester, J.; Kell, D.B.; Pretorius, E. The dormant blood microbiome in chronic, inflammatory diseases. Fems Microbiol. Rev. 2015, 39, 567–591. [Google Scholar] [CrossRef]
- Wang, X.-W.; Wang, J.-X. Crustacean hemolymph microbiota: Endemic, tightly controlled, and utilization expectable. Mol. Immunol. 2015, 68, 404–411. [Google Scholar] [CrossRef] [PubMed]
- Dittel, A.I.; Epifanio, C.E. Invasion biology of the Chinese mitten crab Eriochier sinensis: A brief review. J. Exp. Mar. Biol. Ecol. 2009, 374, 79–92. [Google Scholar] [CrossRef]
- Cornejo-Granados, F.; Lopez-Zavala, A.A.; Gallardo-Becerra, L.; Mendoza-Vargas, A.; Sanchez, F.; Vichido, R.; Brieba, L.G.; Viana, M.T.; Sotelo-Mundo, R.R.; Ochoa-Leyva, A. Microbiome of Pacific Whiteleg shrimp reveals differential bacterial community composition between Wild, Aquacultured and AHPND/EMS outbreak conditions. Sci. Rep. 2017, 7, 11783. [Google Scholar] [CrossRef] [PubMed]
- Jagadeesan, B.; Gerner-Smidt, P.; Allard, M.W.; Leuillet, S.; Winkler, A.; Xiao, Y.; Chaffron, S.; Van der Vossen, J.; Tang, S.; Katase, M.; et al. The use of next generation sequencing for improving food safety: Translation into practice. Food Microbiol. 2019, 79, 96–115. [Google Scholar] [CrossRef]
- Stackebrandt, E.; Goebel, B.M. A Place for DNA-DNA Reassociation and 16S Ribosomal-RNA Sequence-Analysis in the Present Species Definition in Bacteriology. Int. J. Syst. Bacteriol. 1994, 44, 846–849. [Google Scholar] [CrossRef]
- Edgar, R.C. UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 2013, 10, 996–998. [Google Scholar] [CrossRef]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef] [PubMed]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef]
- Chen, Y.; Wang, Z.P.; Liu, L.L.; Zhao, H.B.; Wu, P. Stress-responses of microbial population and activity in activated sludge under long-term ciprofloxacin exposure. J. Environ. Manag. 2021, 281, 111896. [Google Scholar] [CrossRef]
- Hammer, O.; Harper, D.A.T.; Ryan, P.D. PAST: Paleontological statistics software package for education and data analysis. Palaeontol. Electron. 2001, 4, 9. [Google Scholar]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate—A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B-Stat. Methodol. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Newman, M.E.J. Modularity and community structure in networks. Proc. Natl. Acad. Sci. USA 2006, 103, 8577–8582. [Google Scholar] [CrossRef]
- Clements, K.D.; Angert, E.R.; Montgomery, W.L.; Choat, J.H. Intestinal microbiota in fishes: What’s known and what’s not. Mol. Ecol. 2014, 23, 1891–1898. [Google Scholar] [CrossRef]
- David, L.A.; Maurice, C.F.; Carmody, R.N.; Gootenberg, D.B.; Button, J.E.; Wolfe, B.E.; Ling, A.V.; Devlin, A.S.; Varma, Y.; Fischbach, M.A.; et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature 2014, 505, 559–563. [Google Scholar] [CrossRef]
- Suzuki, A.; Segawa, T.; Sawa, S.; Nishitani, C.; Ueda, K.; Itou, T.; Asahina, K.; Suzuki, M. Comparison of the gut microbiota of captive common bottlenose dolphins Tursiops truncatus in three aquaria. J. Appl. Microbiol. 2019, 126, 31–39. [Google Scholar] [CrossRef]
- Xiong, J.; Dai, W.; Qiu, Q.; Zhu, J.; Yang, W.; Li, C. Response of host-bacterial colonization in shrimp to developmental stage, environment and disease. Mol. Ecol. 2018, 27, 3686–3699. [Google Scholar] [CrossRef]
- Fu, C.; Cui, Z.; Shi, X.; Liu, J.; Jiang, Y.; Zhang, R. Effects of dietary glyceryl monolaurate supplementation on growth performance, non-specific immunity, antioxidant status and intestinal microflora of Chinese mitten crabs. Fish Shellfish Immunol. 2022, 125, 65–73. [Google Scholar] [CrossRef]
- Zhang, R.; Shi, X.; Liu, J.; Jiang, Y.; Wu, Y.; Xu, Y.; Yang, C. The effects of bamboo leaf flavonoids on growth performance, immunity, antioxidant status, and intestinal microflora of Chinese mitten crabs (Eriocheir sinensis). Anim. Feed Sci. Technol. 2022, 288, 115297. [Google Scholar] [CrossRef]
- Zhang, B.-Y.; Yao, Q.; Zhang, D.-M.; Wang, N.; Liu, H.-J.; Wan, J.-W.; Chen, Y.-K.; Wang, Q.-J.; Guo, Z.-X. Comparative study on growth, digestive function and intestinal microbial composition of female Chinese mitten crab Eriocheir sinensis selected at different growth stages in rice-crab culture systems. Aquaculture 2022, 554, 738120. [Google Scholar] [CrossRef]
- Shen, Z.; Kumar, D.; Liu, X.; Yan, B.; Fang, P.; Gu, Y.; Li, M.; Xie, M.; Yuan, R.; Feng, Y.; et al. Metatranscriptomic Analysis Reveals an Imbalance of Hepatopancreatic Flora of Chinese Mitten Crab Eriocheir sinensis with Hepatopancreatic Necrosis Disease. Biol. Basel 2021, 10, 462. [Google Scholar] [CrossRef]
- Freese, H.M.; Schink, B. Composition and Stability of the Microbial Community inside the Digestive Tract of the Aquatic Crustacean Daphnia magna. Microb. Ecol. 2011, 62, 882–894. [Google Scholar] [CrossRef]
- Ke, L.I.; Tianling, Z.; Yun, T.; Jianjun, Y. Bacterial community structure in intestine of the white shrimp, Litopenaeus vannamei. Acta Microbiol. Sin. 2007, 47, 649–653. [Google Scholar]
- Sizemore, R.K.; Tubiash, H.S.; Lovelace, T.E.; Colwell, R.R. Bacterial-Flora of Hemolymph of Blue-Crab, Callinectes-Sapidus—Numerical Taxonomy. Appl. Microbiol. 1975, 29, 393–399. [Google Scholar] [CrossRef]
- Wu, Z.; Zhang, Q.; Lin, Y.; Hao, J.; Wang, S.; Zhang, J.; Li, A. Taxonomic and Functional Characteristics of the Gill and Gastrointestinal Microbiota and Its Correlation with Intestinal Metabolites in NEW GIFT Strain of Farmed Adult Nile Tilapia (Oreochromis niloticus). Microorganisms 2021, 9, 617. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, M.; Zheng, H.; Ye, H.; Zhang, X.; Li, S. Source of hemolymph microbiota and their roles in the immune system of mud crab. Dev. Comp. Immunol. 2020, 102, 103470. [Google Scholar] [CrossRef]
- Smriga, S.; Sandin, S.A.; Azam, F. Abundance, diversity, and activity of microbial assemblages associated with coral reef fish guts and feces. Fems Microbiol. Ecol. 2010, 73, 31–42. [Google Scholar] [CrossRef]
- Gomez, D.; Sunyer, J.O.; Salinas, I. The mucosal immune system of fish: The evolution of tolerating commensals while fighting pathogens. Fish Shellfish Immunol. 2013, 35, 1729–1739. [Google Scholar] [CrossRef]
- Karlsson, F.H.; Ussery, D.W.; Nielsen, J.; Nookaew, I. A Closer Look at Bacteroides: Phylogenetic Relationship and Genomic Implications of a Life in the Human Gut. Microb. Ecol. 2011, 61, 473–485. [Google Scholar] [CrossRef]
- Ventura, M.; Canchaya, C.; Tauch, A.; Chandra, G.; Fitzgerald, G.F.; Chater, K.F.; van Sinderen, D. Genomics of Actinobacteria: Tracing the evolutionary history of an ancient phylura. Microbiol. Mol. Biol. Rev. 2007, 71, 495–548. [Google Scholar] [CrossRef]
- Wang, C.H.; Zhou, Y.F.; Lv, D.W.; Ge, Y.; Li, H.; You, Y. Change in the intestinal bacterial community structure associated with environmental microorganisms during the growth of Eriocheir sinensis. Microbiologyopen 2019, 8, e00727. [Google Scholar] [CrossRef]
- Sun, Y.; Chen, Z.; Yang, J.; Mutanda, I.; Li, S.; Zhang, Q.; Zhang, Y.; Zhang, Y.; Wang, Y. Pathway-specific enzymes from bamboo and crop leaves biosynthesize anti-nociceptive C-glycosylated flavones. Commun. Biol. 2020, 3, 110. [Google Scholar] [CrossRef]
- Kostanjsek, R.; Strus, J.; Avgustin, G. “Candidatus Bacilloplasma,” a novel lineage of Mollicutes associated with the hindgut wall of the terrestrial isopod Porcellio scaber (Crustacea: Isopoda). Appl. Environ. Microbiol. 2007, 73, 5566–5573. [Google Scholar] [CrossRef]
- Dong, J.; Li, X.D.; Zhang, R.Y.; Zhao, Y.Y.; Wu, G.F.; Liu, J.L.; Zhu, X.C.; Li, L. Comparative analysis of the intestinal bacterial community and expression of gut immunity genes in the Chinese Mitten Crab (Eriocheir sinensis). Amb Express 2018, 8, 25–36. [Google Scholar] [CrossRef]
- Fraune, S.; Zimmer, M. Host-specificity of environmentally transmitted Mycoplasma-like isopod symbionts. Environ. Microbiol. 2008, 10, 2497–2504. [Google Scholar] [CrossRef]
- Prayitno, S.B.; Sarwan; Sarjito. The Diversity of Gut Bacteria Associated with Milkfish (Chanos chanos Forsksal) from Northern Coast of Central Java, Indonesia. In Proceedings of the International Conference on Tropical and Coastal Region Eco-Development(ICTCRED), Semarang, Indonesia, 11–13 August 2014; pp. 375–384. [Google Scholar]
- Agler, M.T.; Ruhe, J.; Kroll, S.; Morhenn, C.; Kim, S.-T.; Weigel, D.; Kemen, E.M. Microbial Hub Taxa Link Host and Abiotic Factors to Plant Microbiome Variation. PLoS Biol. 2016, 14, e1002352. [Google Scholar] [CrossRef]
- Berry, D.; Widder, S. Deciphering microbial interactions and detecting keystone species with co-occurrence networks. Front. Microbiol. 2014, 5, 219. [Google Scholar] [CrossRef]
- Fuhrman, J.A. Microbial community structure and its functional implications. Nature 2009, 459, 193–199. [Google Scholar] [CrossRef]
- Sun, W.W.; Zhang, X.X.; Wan, W.S.; Wang, S.Q.; Wen, X.B.; Zheng, H.P.; Zhang, Y.L.; Li, S.K. Tumor necrosis factor receptor-associated factor 6 (TRAF6) participates in anti-lipopolysaccharide factors (ALFs) gene expression in mud crab. Dev. Comp. Immunol. 2017, 67, 361–376. [Google Scholar] [CrossRef]
- Jousset, A.; Schulz, W.; Scheu, S.; Eisenhauer, N. Intraspecific genotypic richness and relatedness predict the invasibility of microbial communities. Isme J. 2011, 5, 1108–1114. [Google Scholar] [CrossRef]
- Rungrassamee, W.; Klanchui, A.; Chaiyapechara, S.; Maibunkaew, S.; Tangphatsornruang, S.; Jiravanichpaisal, P.; Karoonuthaisiri, N. Bacterial Population in Intestines of the Black Tiger Shrimp (Penaeus monodon) under Different Growth Stages. PLoS ONE 2013, 8, e60802. [Google Scholar] [CrossRef]
- Mouchet, M.A.; Bouvier, C.; Bouvier, T.; Troussellier, M.; Escalas, A.; Mouillot, D. Genetic difference but functional similarity among fish gut bacterial communities through molecular and biochemical fingerprints. Fems Microbiol. Ecol. 2012, 79, 568–580. [Google Scholar] [CrossRef] [PubMed]
- LeaMaster, B.R.; Walsh, W.A.; Brock, J.A.; Fujioka, R.S. Cold stress-induced changes in the aerobic heterotrophic gastrointestinal tract bacterial flora of red hybrid tilapia. J. Fish Biol. 1997, 50, 770–780. [Google Scholar] [CrossRef]
- Bakke, I.; Coward, E.; Andersen, T.; Vadstein, O. Selection in the host structures the microbiota associated with developing cod larvae (Gadus morhua). Environ. Microbiol. 2015, 17, 3914–3924. [Google Scholar] [CrossRef] [PubMed]
- Curtis, M.M.; Hu, Z.; Klimko, C.; Narayanan, S.; Deberardinis, R.; Sperandio, V. The Gut Commensal Bacteroides thetaiotaomicron Exacerbates Enteric Infection through Modification of the Metabolic Landscape. Cell Host Microbe 2014, 16, 759–769. [Google Scholar] [CrossRef]
- Freilich, M.A.; Wieters, E.; Broitman, B.R.; Marquet, P.A.; Navarrete, S.A. Species co-occurrence networks: Can they reveal trophic and non-trophic interactions in ecological communities? Ecology 2018, 99, 690–699. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Schlaeppi, K.; van der Heijden, M.G.A. Keystone taxa as drivers of microbiome structure and functioning. Nat. Rev. Microbiol. 2018, 16, 567–576. [Google Scholar] [CrossRef]
- Tsolekile, N.; Nelana, S.; Oluwafemi, O.S. Porphyrin as Diagnostic and Therapeutic Agent. Molecules 2019, 24, 2669. [Google Scholar] [CrossRef]
- Shi, X.-S.; Meng, L.-H.; Li, X.; Wang, D.-J.; Zhou, X.-W.; Du, F.-Y.; Wang, B.-G.; Li, X.-M. Polyketides and Terpenoids with Potent Antibacterial Activities from the Artemisia argyi-Derived Fungus Trichoderma koningiopsis QA-3. Chem. Biodivers. 2020, 17, e2000566. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, J.; Zhang, Q.; Zhang, T.; Wang, S.; Hao, J.; Wu, Z.; Li, A. Comparative Analysis of the Symbiotic Microbiota in the Chinese Mitten Crab (Eriocheir sinensis): Microbial Structure, Co-Occurrence Patterns, and Predictive Functions. Microorganisms 2023, 11, 544. https://doi.org/10.3390/microorganisms11030544
Yang J, Zhang Q, Zhang T, Wang S, Hao J, Wu Z, Li A. Comparative Analysis of the Symbiotic Microbiota in the Chinese Mitten Crab (Eriocheir sinensis): Microbial Structure, Co-Occurrence Patterns, and Predictive Functions. Microorganisms. 2023; 11(3):544. https://doi.org/10.3390/microorganisms11030544
Chicago/Turabian StyleYang, Jicheng, Qianqian Zhang, Tanglin Zhang, Shuyi Wang, Jingwen Hao, Zhenbing Wu, and Aihua Li. 2023. "Comparative Analysis of the Symbiotic Microbiota in the Chinese Mitten Crab (Eriocheir sinensis): Microbial Structure, Co-Occurrence Patterns, and Predictive Functions" Microorganisms 11, no. 3: 544. https://doi.org/10.3390/microorganisms11030544
APA StyleYang, J., Zhang, Q., Zhang, T., Wang, S., Hao, J., Wu, Z., & Li, A. (2023). Comparative Analysis of the Symbiotic Microbiota in the Chinese Mitten Crab (Eriocheir sinensis): Microbial Structure, Co-Occurrence Patterns, and Predictive Functions. Microorganisms, 11(3), 544. https://doi.org/10.3390/microorganisms11030544