
Rumen and Fecal Microbiota Characteristics of Qinchuan Cattle with Divergent Residual Feed Intake

 ,
, 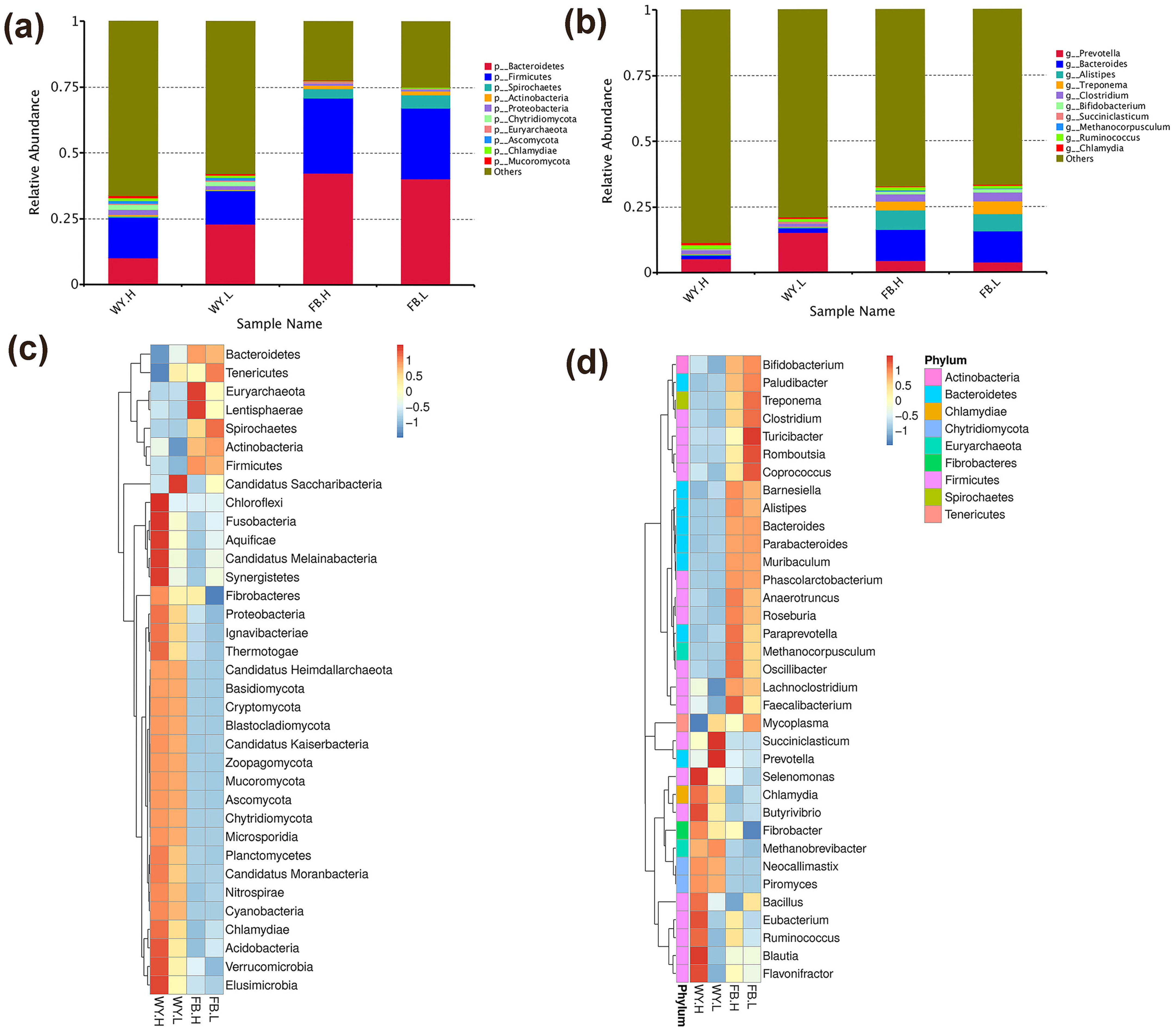{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Collection of Test Animals and Samples
2.2. Sequencing Library Construction and Metagenome Sequencing
2.3. Metagenome Assembling, Gene Prediction and Abundance Analysis
2.4. Classification Annotation and Function Annotation
2.5. Analysis of Intestinal Differential mRNA and Microbial Interactions
3. Results
3.1. Experimental Animal Selection and Sequencing Quality Control
3.2. The Composition of the Microbiota
3.3. Function Analysis
3.4. RFI-Related Host-Gene Expression and Rumen Microbial Association Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Abecia, L.; Martinez-Fernandez, G. Analysis of the Rumen Microbiome and Metabolome to Study the Effect of an Antimethanogenic Treatment Applied in Early Life of Kid Goats. Front. Microbiol. 2018, 9, 02227. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Shi, H. Effect of Limit-Fed Diets with Different Forage to Concentrate Rations on Fecal Bacterial and Archaeal Community Composition in Holstein Heifers. Front. Microbiol. 2018, 9, 00976. [Google Scholar] [CrossRef] [PubMed]
- Nkrumah, J.D.; Okine, E.K. Relationships of feedlot feed efficiency, performance, and feeding behavior with metabolic rate, methane production, and energy partitioning in beef cattle. J. Anim. Sci. 2006, 84, 145–153. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Ye, H. Transcriptome analysis of mRNA and miRNA in skeletal muscle indicates an important network for differential Residual Feed Intake in pigs. Sci. Rep. 2015, 5, 11953. [Google Scholar]
- Ben Shabat, S.K.; Sasson, G. Specific microbiome-dependent mechanisms underlie the energy harvest efficiency of ruminants. ISME J. 2016, 10, 2958–2972. [Google Scholar] [CrossRef]
- Ramayo-Caldas, Y.; Ballester, M. Integrative approach using liver and duodenum RNA-Seq data identifies candidate genes and pathways associated with feed efficiency in pigs. Sci. Rep. 2018, 8, 558. [Google Scholar] [CrossRef]
- McGovern, E.; McGee, M. Investigation into the effect of divergent feed efficiency phenotype on the bovine rumen microbiota across diet and breed. Sci. Rep. 2020, 10, 15317. [Google Scholar] [CrossRef] [PubMed]
- Menezes, A.; Valadares, F.S. 148 Residual feed intake and body weight gain relate to feeding behavior in Nellore bulls: Implications for performance and maintenance requirements. J. Anim. Sci. 2019, 97 (Supplement_3), 149–150. [Google Scholar] [CrossRef]
- Sanna, S.; Kurilshikov, A. Challenges and future directions for studying effects of host genetics on the gut microbiome. Nat. Genet. 2022, 54, 100–106. [Google Scholar] [CrossRef]
- Yang, H.; Wu, J. ABO genotype alters the gut microbiota by regulating GalNAc levels in pigs. Nature 2022, 606, 358–367. [Google Scholar] [CrossRef]
- Li, F.; Li, C. Host genetics influence the rumen microbiota and heritable rumen microbial features associate with feed efficiency in cattle. Microbiome 2019, 7, 92. [Google Scholar] [CrossRef] [PubMed]
- Paz, H.A.; Hales, K.E. Rumen bacterial community structure impacts feed efficiency in beef cattle. J. Anim. Sci. 2018, 96, 1045–1058. [Google Scholar] [CrossRef] [PubMed]
- Bergamaschi, M.; Tiezzi, F. Gut microbiome composition differences among breeds impact feed efficiency in swine. Microbiome 2020, 8, 110. [Google Scholar] [CrossRef]
- Aliakbari, A.; Zemb, O. Microbiability and microbiome-wide association analyses of feed efficiency and performance traits in pigs. Genet. Sel. Evol. 2022, 54, 29. [Google Scholar] [CrossRef]
- Wang, Y.; Zhou, P. Effect of host genetics and gut microbiome on fat deposition traits in pigs. Front. Microbiol. 2022, 13, 925200. [Google Scholar] [CrossRef] [PubMed]
- Zhao, G.; Xiang, Y. Exploring the Possible Link between the Gut Microbiome and Fat Deposition in Pigs. Oxidative Med. Cell. Longev. 2022, 2022, 1098892. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Han, L. Characterization and Duodenal Transcriptome Analysis of Chinese Beef Cattle with Divergent Feed Efficiency Using RNA-Seq. Front. Genet. 2021, 12, 741878. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Karlsson, F.H.; Tremaroli, V. Gut metagenome in European women with normal, impaired and diabetic glucose control. Nature 2013, 498, 99–103. [Google Scholar] [CrossRef]
- Li, J.; Jia, H. An integrated catalog of reference genes in the human gut microbiome. Nat. Biotechnol. 2014, 32, 834–841. [Google Scholar] [CrossRef]
- Jia, B.; Raphenya, A.R. CARD 2017: Expansion and model-centric curation of the comprehensive antibiotic resistance database. Nucleic Acids Res. 2017, 45, D566–D573. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Sato, Y. KEGG mapping tools for uncovering hidden features in biological data. Protein Sci. 2022, 31, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Drula, E.; Garron, M.L. The carbohydrate-active enzyme database: Functions and literature. Nucleic Acids Res. 2022, 50, D571–D577. [Google Scholar] [CrossRef] [PubMed]
- Rothrock, M.J.; Min, B.R. Antibiotic resistance, antimicrobial residues, and bacterial community diversity in pasture-raised poultry, swine, and beef cattle manures. J. Anim. Sci. 2021, 99, skab144. [Google Scholar] [CrossRef]
- De, R.; Mukhopadhyay, A.K. Metagenomic analysis of gut microbiome and resistome of diarrheal fecal samples from Kolkata, India, reveals the core and variable microbiota including signatures of microbial dark matter. Gut Pathog. 2020, 12, 32. [Google Scholar] [CrossRef]
- Lynch, J.B.; Hsiao, E.Y. Microbiomes as sources of emergent host phenotypes. Science 2019, 365, 1405–1409. [Google Scholar] [CrossRef]
- Brussow, H. The relationship between the host genome, microbiome, and host phenotype. Environ. Microbiol. 2020, 22, 1170–1173. [Google Scholar] [CrossRef]
- Yen, S.; Johnson, J.S. Metagenomics: A path to understanding the gut microbiome. Mamm. Genome 2021, 32, 282–296. [Google Scholar] [CrossRef]
- Bubier, J.A.; Chesler, E.J. Host genetic control of gut microbiome composition. Mamm. Genome 2021, 32, 263–281. [Google Scholar] [CrossRef]
- Kim, M.; Kim, J. Investigation of bacterial diversity in the feces of cattle fed different diets. J. Anim. Sci. 2014, 92, 683–694. [Google Scholar] [CrossRef] [PubMed]
- Delgado, B.; Bach, A. Whole rumen metagenome sequencing allows classifying and predicting feed efficiency and intake levels in cattle. Sci. Rep. 2019, 9, 11. [Google Scholar] [CrossRef] [PubMed]
- Noel, S.J.; Olijhoek, D.W. Rumen and Fecal Microbial Community Structure of Holstein and Jersey Dairy Cows as Affected by Breed, Diet, and Residual Feed Intake. Animals 2019, 9, 498. [Google Scholar] [CrossRef] [PubMed]
- Clemmons, B.A.; Martino, C. Rumen Bacteria and Serum Metabolites Predictive of Feed Efficiency Phenotypes in Beef Cattle. Sci. Rep. 2019, 9, 19265. [Google Scholar] [CrossRef]
- McGovern, E.; Kenny, D.A. 16S rRNA Sequencing Reveals Relationship Between Potent Cellulolytic Genera and Feed Efficiency in the Rumen of Bulls. Front. Microbiol. 2018, 9, 1842. [Google Scholar] [CrossRef]
- Ellison, M.J.; Conant, G.C. Diet and feed efficiency status affect rumen microbial profiles of sheep. Small Rumin. Res. 2017, 156, 12–19. [Google Scholar] [CrossRef]
- Shinoda, C.; Yasuda, J. Genetic relationships of feed efficiency and growth traits with carcass traits in Japanese Shorthorn cattle. Anim. Sci. J. 2022, 93, e13691. [Google Scholar] [CrossRef]
- Myer, P.R.; Smith, T.P. Rumen microbiome from steers differing in feed efficiency. PLoS ONE 2015, 10, e0129174. [Google Scholar] [CrossRef]
- McCormack, U.M.; Curiao, T. Exploring a Possible Link between the Intestinal Microbiota and Feed Efficiency in Pigs. Appl. Environ. Microbiol. 2017, 83, 00380. [Google Scholar] [CrossRef]
- Lv, H.; Huang, Y. Microbial Composition in the Duodenum and Ileum of Yellow Broilers with High and Low Feed Efficiency. Front. Microbiol. 2021, 12, 689653. [Google Scholar] [CrossRef]
- McKee, L.S.; La Rosa, S.L. Polysaccharide degradation by the Bacteroidetes: Mechanisms and nomenclature. Environ. Microbiol. Rep. 2021, 13, 559–581. [Google Scholar] [CrossRef] [PubMed]
- Gardiner, G.E.; Metzler-Zebeli, B.U. Impact of Intestinal Microbiota on Growth and Feed Efficiency in Pigs: A Review. Microorganisms 2020, 8, 1886. [Google Scholar] [CrossRef]
- Johnson, D.R.; Lee, T.K. The functional and taxonomic richness of wastewater treatment plant microbial communities are associated with each other and with ambient nitrogen and carbon availability. Environ. Microbiol. 2015, 17, 4851–4860. [Google Scholar] [CrossRef] [PubMed]
- Castro-Ramos, J.J.; Solis-Oba, A. Effect of the initial pH on the anaerobic digestion process of dairy cattle manure. AMB Express 2022, 12, 162. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhang, G. Effects of High Forage/Concentrate Diet on Volatile Fatty Acid Production and the Microorganisms Involved in VFA Production in Cow Rumen. Animals 2020, 10, 223. [Google Scholar] [CrossRef]
- Fu, X.; Liu, Z. Nondigestible carbohydrates, butyrate, and butyrate-producing bacteria. Crit. Rev. Food Sci. Nutr. 2019, 59, S130–S152. [Google Scholar] [CrossRef]
- Chung, Y.W.; Gwak, H.J. Functional dynamics of bacterial species in the mouse gut microbiome revealed by metagenomic and metatranscriptomic analyses. PLoS ONE 2020, 15, e0227886. [Google Scholar] [CrossRef]
- Ran, T.; Jiao, P. Fecal bacterial community of finishing beef steers fed ruminally protected and non-protected active dried yeast. J. Anim. Sci. 2020, 98, skaa058. [Google Scholar] [CrossRef]
- Amat, S.; Lantz, H. Prevotella in Pigs: The Positive and Negative Associations with Production and Health. Microorganisms 2020, 8, 1584. [Google Scholar] [CrossRef]
- Wirth, R.; Kadar, G. The Planktonic Core Microbiome and Core Functions in the Cattle Rumen by Next Generation Sequencing. Front. Microbiol. 2018, 9, 2285. [Google Scholar] [CrossRef]
- Holman, D.B.; Gzyl, K.E. A meta-analysis of the bovine gastrointestinal tract microbiota. FEMS Microbiol. Ecol. 2019, 95, fiz072. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Sun, H. Metagenomics reveals differences in microbial composition and metabolic functions in the rumen of dairy cows with different residual feed intake. Anim. Microbiome 2022, 4, 19. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Hitch, T. Comparative metagenomic and metatranscriptomic analyses reveal the breed effect on the rumen microbiome and its associations with feed efficiency in beef cattle. Microbiome 2019, 7, 6. [Google Scholar] [CrossRef] [PubMed]
- de Freitas, A.S.; de David, D.B. Microbial patterns in rumen are associated with gain of weight in beef cattle. Antonie Van Leeuwenhoek Int. J. Gen. Mol. Microbiol. 2020, 113, 1299–1312. [Google Scholar] [CrossRef] [PubMed]
- Lamendella, R.; Domingo, J.W. Comparative fecal metagenomics unveils unique functional capacity of the swine gut. BMC Microbiol. 2011, 11, 103. [Google Scholar] [CrossRef]
- Xue, M.; Sun, H. Assessment of Rumen Microbiota from a Large Dairy Cattle Cohort Reveals the Pan and Core Bacteriomes Contributing to Varied Phenotypes. Appl. Environ. Microbiol. 2018, 84, 30054362. [Google Scholar] [CrossRef]
- Skarlupka, J.H.; Kamenetsky, M.E. The ruminal bacterial community in lactating dairy cows has limited variation on a day-to-day basis. J. Anim. Sci. Biotechnol. 2019, 10, 66. [Google Scholar] [CrossRef]
- Walker, N.D.; McEwan, N.R. Cloning and functional expression of dipeptidyl peptidase IV from the ruminal bacterium Prevotella albensis M384(T). Microbiology-SGM 2003, 149, 2227–2234. [Google Scholar] [CrossRef]
- Precup, G.; Vodnar, D.C. Gut Prevotella as a possible biomarker of diet and its eubiotic versus dysbiotic roles: A comprehensive literature review. Br. J. Nutr. 2019, 122, 131–140. [Google Scholar] [CrossRef]
- Amaral, W.Z.; Lubach, G.R. Social Influences on Prevotella and the Gut Microbiome of Young Monkeys. Psychosom. Med. 2017, 79, 888–897. [Google Scholar] [CrossRef]
- Wang, J.; Lang, T. Core Gut Bacteria Analysis of Healthy Mice. Front. Microbiol. 2019, 10, 887. [Google Scholar] [CrossRef] [PubMed]
- Lv, X.; Chai, J. The Signature Microbiota Drive Rumen Function Shifts in Goat Kids Introduced to Solid Diet Regimes. Microorganisms 2019, 7, 516. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Ni, J. Effect of green tea and mulberry leaf powders on the gut microbiota of chicken. BMC Vet. Res. 2019, 15, 77. [Google Scholar] [CrossRef]
- Wang, B.; Luo, Y. Impact of feeding regimens on the composition of gut microbiota and metabolite profiles of plasma and feces from Mongolian sheep. J. Microbiol. 2020, 58, 472–482. [Google Scholar] [CrossRef] [PubMed]
- Quan, J.; Wu, Z. Metagenomic Characterization of Intestinal Regions in Pigs with Contrasting Feed Efficiency. Front. Microbiol. 2020, 11, 32. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Huang, X. Uncovering the composition of microbial community structure and metagenomics among three gut locations in pigs with distinct fatness. Sci. Rep. 2016, 6, 27427. [Google Scholar] [CrossRef]
- Xue, M.Y.; Xie, Y.Y. Integrated meta-omics reveals new ruminal microbial features associated with feed efficiency in dairy cattle. Microbiome 2022, 10, 32. [Google Scholar] [CrossRef]
- Stewart, R.D.; Auffret, M.D. Assembly of 913 microbial genomes from metagenomic sequencing of the cow rumen. Nat. Commun. 2018, 9, 870. [Google Scholar] [CrossRef]
- Guan, L.L.; Nkrumah, J.D. Linkage of microbial ecology to phenotype: Correlation of rumen microbial ecology to cattle’s feed efficiency. FEMS Microbiol. Lett. 2008, 288, 85–91. [Google Scholar] [CrossRef]
- Hernandez-Sanabria, E.; Guan, L.L. Correlation of particular bacterial PCR-denaturing gradient gel electrophoresis patterns with bovine ruminal fermentation parameters and feed efficiency traits. Appl. Environ. Microbiol. 2010, 76, 6338–6350. [Google Scholar] [CrossRef]
- Carberry, C.A.; Kenny, D.A. Effect of phenotypic residual feed intake and dietary forage content on the rumen microbial community of beef cattle. Appl. Environ. Microbiol. 2012, 78, 4949–4958. [Google Scholar] [CrossRef] [PubMed]
- Auffret, M.D.; Stewart, R.D. Identification of Microbial Genetic Capacities and Potential Mechanisms Within the Rumen Microbiome Explaining Differences in Beef Cattle Feed Efficiency. Front. Microbiol. 2020, 11, 1229. [Google Scholar] [CrossRef] [PubMed]
- Daghio, M.; Ciucci, F. Correlation of Breed, Growth Performance, and Rumen Microbiota in Two Rustic Cattle Breeds Reared Under Different Conditions. Front. Microbiol. 2021, 12, 652031. [Google Scholar] [CrossRef]
- de Wit, N.J.; IJssennagger, N. Oit1/Fam3D, a gut-secreted protein displaying nutritional status-dependent regulation. J. Nutr. Biochem. 2012, 23, 1425–1433. [Google Scholar] [CrossRef] [PubMed]
- Sommer, F.; Nookaew, I. Site-specific programming of the host epithelial transcriptome by the gut microbiota. Genome Biol. 2015, 16, 62. [Google Scholar] [CrossRef]
- Shah, T.M.; Patel, J.G. Host transcriptome and microbiome interaction modulates physiology of full-sibs broilers with divergent feed conversion ratio. NPJ Biofilms Microbiomes 2019, 5, 24. [Google Scholar] [CrossRef]
- Hou, K.; Wu, Z.X. Microbiota in health and diseases. Signal Transduct. Target. Ther. 2022, 7, 135. [Google Scholar] [CrossRef]
- Zhang, S.; Tang, Z. Dietary Beta-Hydroxy-Beta-Methyl Butyrate Supplementation Inhibits Hepatic Fat Deposition via Regulating Gut Microbiota in Broiler Chickens. Microorganisms 2022, 10, 169. [Google Scholar] [CrossRef]
- Fu, Z.; Yang, H. Ileal Microbiota Alters the Immunity Statues to Affect Body Weight in Muscovy Ducks. Front. Immunol. 2022, 13, 844102. [Google Scholar] [CrossRef]
- Su, Y.; Ge, Y. The digestive and reproductive tract microbiotas and their association with body weight in laying hens. Poult. Sci. 2021, 100, 101422. [Google Scholar] [CrossRef]
- Vetrani, C.; Di Nisio, A. From Gut Microbiota through Low-Grade Inflammation to Obesity: Key Players and Potential Targets. Nutrients 2022, 14, 2103. [Google Scholar] [CrossRef] [PubMed]
- Wessels, A.G. Influence of the Gut Microbiome on Feed Intake of Farm Animals. Microorganisms 2022, 10, 1305. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, X.; Ma, Y.; Yang, C.; Zhao, Z.; Ding, Y.; Zhang, Y.; Wang, P.; Zhao, L.; Li, C.; Su, Z.; et al. Rumen and Fecal Microbiota Characteristics of Qinchuan Cattle with Divergent Residual Feed Intake. Microorganisms 2023, 11, 358. https://doi.org/10.3390/microorganisms11020358
Zhou X, Ma Y, Yang C, Zhao Z, Ding Y, Zhang Y, Wang P, Zhao L, Li C, Su Z, et al. Rumen and Fecal Microbiota Characteristics of Qinchuan Cattle with Divergent Residual Feed Intake. Microorganisms. 2023; 11(2):358. https://doi.org/10.3390/microorganisms11020358
Chicago/Turabian StyleZhou, Xiaonan, Ying Ma, Chaoyun Yang, Zhiyan Zhao, Yanling Ding, Yanfeng Zhang, Pengfei Wang, Lei Zhao, Chenglong Li, Zonghua Su, and et al. 2023. "Rumen and Fecal Microbiota Characteristics of Qinchuan Cattle with Divergent Residual Feed Intake" Microorganisms 11, no. 2: 358. https://doi.org/10.3390/microorganisms11020358
APA StyleZhou, X., Ma, Y., Yang, C., Zhao, Z., Ding, Y., Zhang, Y., Wang, P., Zhao, L., Li, C., Su, Z., Wang, X., Ming, W., Zeng, L., & Kang, X. (2023). Rumen and Fecal Microbiota Characteristics of Qinchuan Cattle with Divergent Residual Feed Intake. Microorganisms, 11(2), 358. https://doi.org/10.3390/microorganisms11020358