
Two-Dose Ceftiofur Treatment Increases Cephamycinase Gene Quantities and Fecal Microbiome Diversity in Dairy Cows Diagnosed with Metritis
 , ,
, ,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethical Statement
2.2. Study Design
2.3. Animal Sample Collection and Sample Processing
2.4. Quantitative Real-Time PCR
2.4.1. Total Community DNA Extraction
2.4.2. AMR Genes and Standard Curve Generation
2.4.3. qPCR Reaction Setup and Gene Copy Number Analysis
2.4.4. Statistical Analysis
2.5. 16s rRNA Amplicon Gene Sequencing and Bioinformatic Analysis
2.5.1. Total Community DNA Extraction and Library Preparation
2.5.2. Bioinformatic Analyses
3. Results
3.1. Quantification of blaCMY-2, blaCTX-M, mphA and qnrB19
3.1.1. Descriptive Statistics
3.1.2. Detection of blaCMY-2, blaCTX-M, mphA and qnrB19 Genes
3.2. Bacterial 16S Metagenomics
3.2.1. Descriptive Statistics
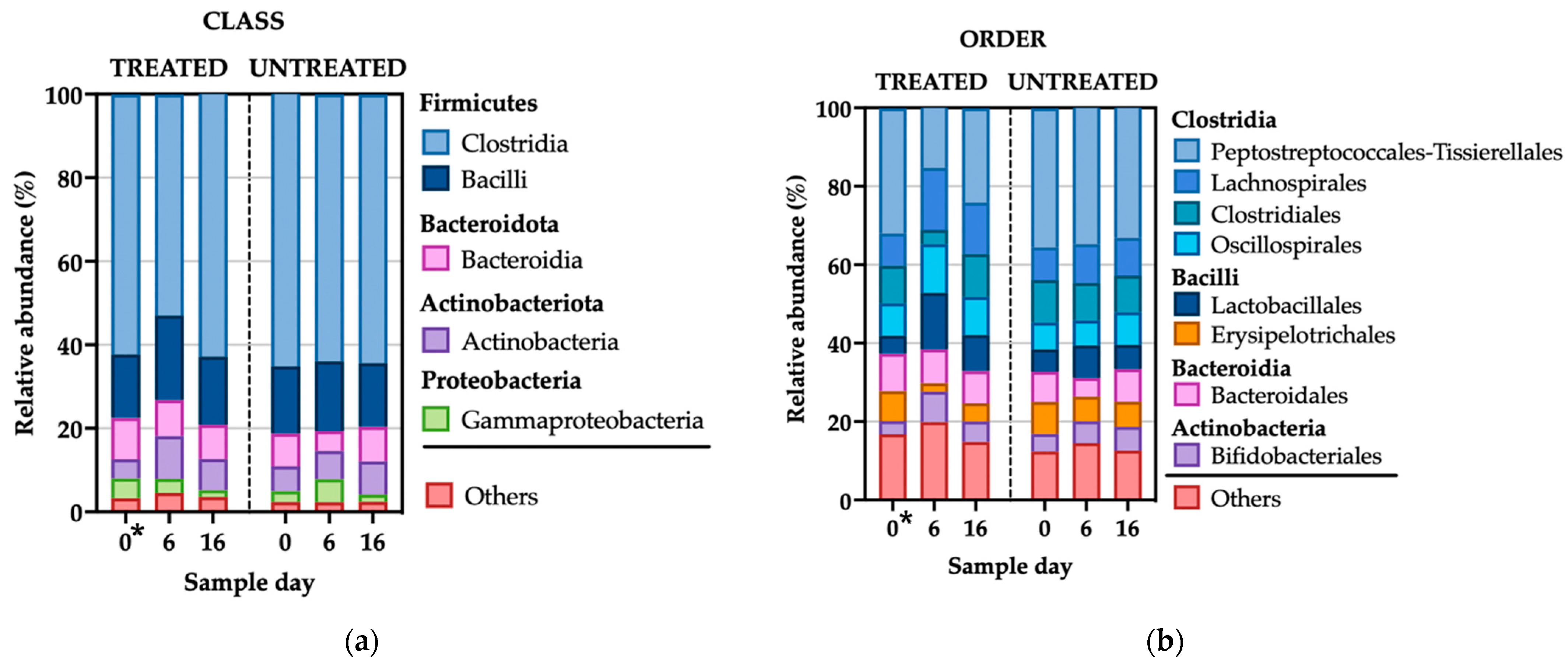
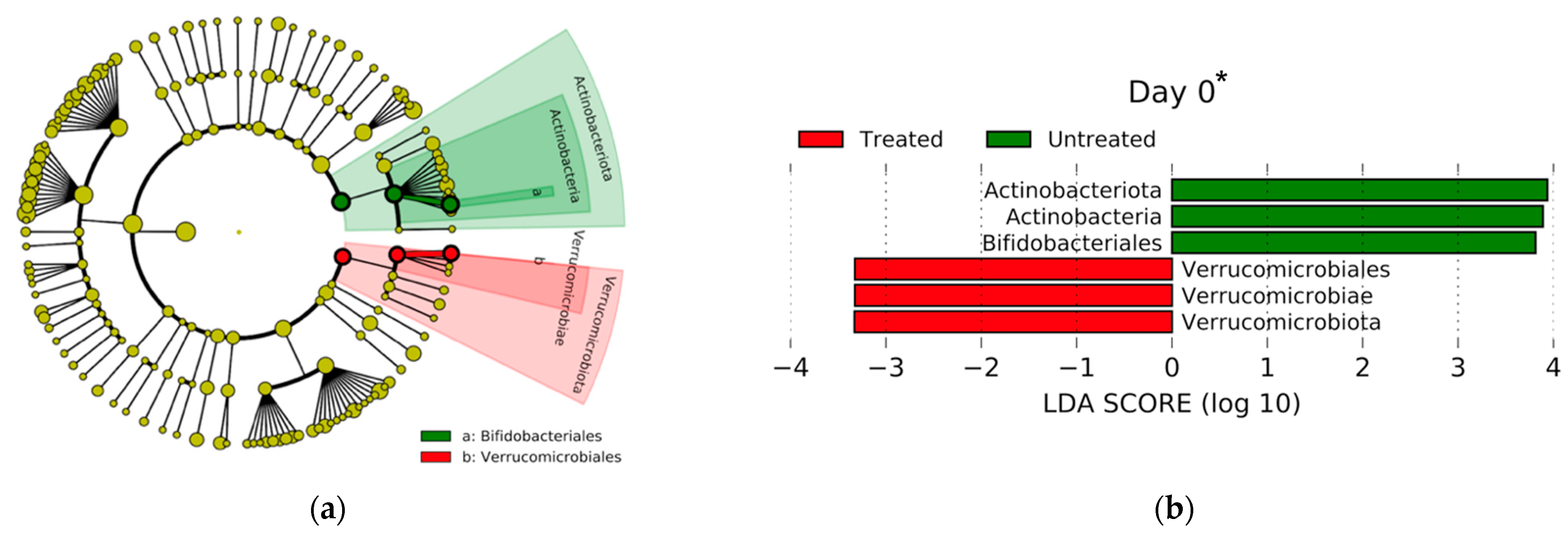
3.2.2. Microbial Compositional Analyses
3.2.3. Alpha Diversity
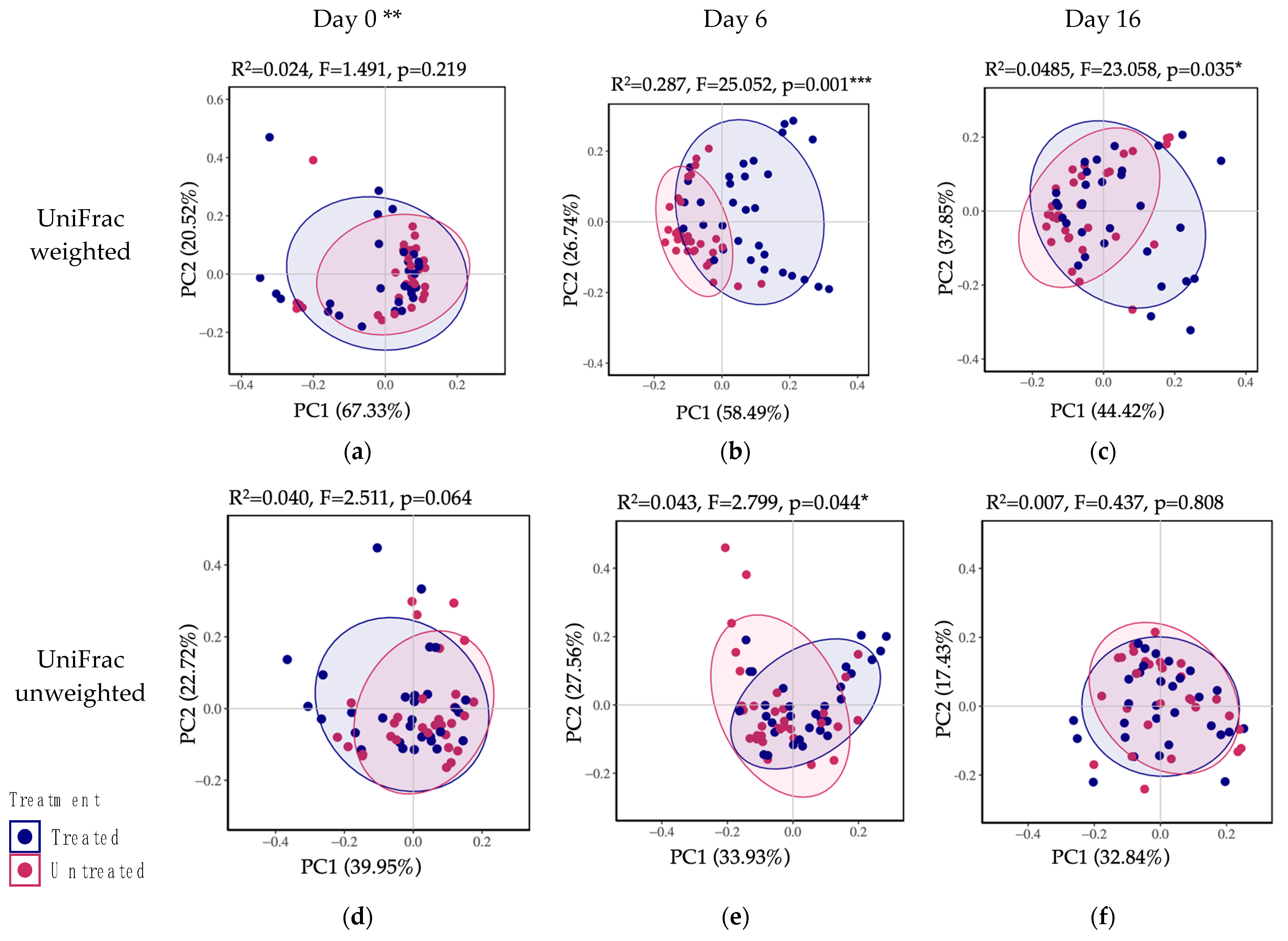
3.2.4. Beta Diversity (Community Comparison)
4. Discussion
4.1. Two-Dose CCFA Favors the Selection of the blaCMY-2 Gene
4.2. The Feces from Treated Animals Presented a Significantly Higher Microbial Diversity
4.3. Limitations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Fleming, A. Classics in infectious diseases: On the antibacterial action of cultures of a penicillium, with special reference to their use in the isolation of B. influenzae by Alexander Fleming. Br. J. Exp. Pathol. 1929, 10, 226–236, reprinted in Rev. Infect. Dis. 1980, 2, 129–139. [Google Scholar]
- Prestinaci, F.; Pezzotti, P.; Pantosti, A. Antimicrobial resistance: A global multifaceted phenomenon. Pathog. Glob. Health 2015, 109, 309–318. [Google Scholar] [CrossRef] [PubMed]
- CDC. Antibiotic Resistance Threats in the United States; U.S. Department of Health and Human Services: Atlanta, GA, USA, 2019.
- O’Neil, J. Tackling Drug-Resistant Infections Globally: Final Report and Recommendations; Government of the United Kingdom: London, UK, 2016; Volume 1, p. 84.
- Castanheira, M.; Simner, P.J.; Bradford, P.A. Extended-spectrum beta-lactamases: An update on their characteristics, epidemiology and detection. JAC Antimicrob. Resist. 2021, 3, dlab092. [Google Scholar] [CrossRef]
- Davies, J.; Davies, D. Origins and evolution of antibiotic resistance. Microbiol. Mol. Biol. Rev. 2010, 74, 417–433. [Google Scholar] [CrossRef] [PubMed]
- Antimicrobial Resistance, C. Global burden of bacterial antimicrobial resistance in 2019: A systematic analysis. Lancet 2022, 399, 629–655. [Google Scholar] [CrossRef]
- Basavaraju, M.; Gunashree, B.S. Escherichia coli: An Overview of Main Characteristics; IntechOpen: London, UK, 2022. [Google Scholar] [CrossRef]
- Poirel, L.; Madec, J.Y.; Lupo, A.; Schink, A.K.; Kieffer, N.; Nordmann, P.; Schwarz, S. Antimicrobial Resistance in Escherichia coli. Microbiol. Spectr. 2018, 6, 4. [Google Scholar] [CrossRef]
- Braz, V.S.; Melchior, K.; Moreira, C.G. Escherichia coli as a Multifaceted Pathogenic and Versatile Bacterium. Front. Cell Infect. Microbiol. 2020, 10, 548492. [Google Scholar] [CrossRef]
- Huddleston, J.R. Horizontal gene transfer in the human gastrointestinal tract: Potential spread of antibiotic resistance genes. Infect. Drug Resist. 2014, 7, 167–176. [Google Scholar] [CrossRef]
- Rozwandowicz, M.; Brouwer, M.S.M.; Fischer, J.; Wagenaar, J.A.; Gonzalez-Zorn, B.; Guerra, B.; Mevius, D.J.; Hordijk, J. Plasmids carrying antimicrobial resistance genes in Enterobacteriaceae. J. Antimicrob. Chemother. 2018, 73, 1121–1137. [Google Scholar] [CrossRef]
- Jacoby, G.A. AmpC beta-lactamases. Clin. Microbiol. Rev. 2009, 22, 161–182. [Google Scholar] [CrossRef]
- Xiang, Y.; Wu, F.; Chai, Y.; Xu, X.; Yang, L.; Tian, S.; Zhang, H.; Li, Y.; Yang, C.; Liu, H.; et al. A new plasmid carrying mphA causes prevalence of azithromycin resistance in enterotoxigenic Escherichia coli serogroup O6. BMC Microbiol. 2020, 20, 247. [Google Scholar] [CrossRef]
- Stanton, I.C.; Murray, A.K.; Zhang, L.; Snape, J.; Gaze, W.H. Evolution of antibiotic resistance at low antibiotic concentrations including selection below the minimal selective concentration. Commun. Biol. 2020, 3, 467. [Google Scholar] [CrossRef] [PubMed]
- Phuc Nguyen, M.C.; Woerther, P.L.; Bouvet, M.; Andremont, A.; Leclercq, R.; Canu, A. Escherichia coli as reservoir for macrolide resistance genes. Emerg. Infect. Dis. 2009, 15, 1648–1650. [Google Scholar] [CrossRef]
- Worthington, R.J.; Melander, C. Overcoming resistance to beta-lactam antibiotics. J. Org. Chem. 2013, 78, 4207–4213. [Google Scholar] [CrossRef]
- Bush, K.; Bradford, P.A. beta-Lactams and beta-Lactamase Inhibitors: An Overview. Cold Spring Harb. Perspect. Med. 2016, 6, a025247. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, J.; Guarner, F.; Bustos Fernandez, L.; Maruy, A.; Sdepanian, V.L.; Cohen, H. Antibiotics as Major Disruptors of Gut Microbiota. Front. Cell Infect. Microbiol. 2020, 10, 572912. [Google Scholar] [CrossRef]
- Merenda, V.R.; Lezier, D.; Odetti, A.; Figueiredo, C.C.; Risco, C.A.; Bisinotto, R.S.; Chebel, R.C. Effects of metritis treatment strategies on health, behavior, reproductive, and productive responses of Holstein cows. J. Dairy. Sci. 2021, 104, 2056–2073. [Google Scholar] [CrossRef] [PubMed]
- Perez-Baez, J.; Silva, T.V.; Risco, C.A.; Chebel, R.C.; Cunha, F.; De Vries, A.; Santos, J.E.P.; Lima, F.S.; Pinedo, P.; Schuenemann, G.M.; et al. The economic cost of metritis in dairy herds. J. Dairy. Sci. 2021, 104, 3158–3168. [Google Scholar] [CrossRef] [PubMed]
- Taylor, E.A.; Jordan, E.R.; Garcia, J.A.; Hagevoort, G.R.; Norman, K.N.; Lawhon, S.D.; Pineiro, J.M.; Scott, H.M. Effects of two-dose ceftiofur treatment for metritis on the temporal dynamics of antimicrobial resistance among fecal Escherichia coli in Holstein-Friesian dairy cows. PLoS ONE 2019, 14, e0220068. [Google Scholar] [CrossRef]
- Samtiya, M.; Matthews, K.R.; Dhewa, T.; Puniya, A.K. Antimicrobial Resistance in the Food Chain: Trends, Mechanisms, Pathways, and Possible Regulation Strategies. Foods 2022, 11, 2966. [Google Scholar] [CrossRef]
- Draxxin Cattle Prescribing Information. 2019. Available online: https://www.zoetisus.com/content/pages/products/cattle/Draxxin-resources/Assets/Draxxin-Cattle-Prescribing-Information.pdf (accessed on 1 September 2023).
- Enroflox 100 Package Insert. 2021. Available online: https://www.norbrook.com/media/2873/enroflox-100-package-insert-6405101670i07.pdf (accessed on 1 September 2023).
- Taylor, E.A.; Ossa-Trujillo, C.; Vinasco, J.; Jordan, E.R.; Garcia Buitrago, J.A.; Hagevoort, R.; Norman, K.N.; Lawhon, S.D.; Pineiro, J.M.; Levent, G.; et al. Use of critically important antimicrobial classes early in life may adversely impact bacterial resistance profiles during adult years: Potential co-selection for plasmid-borne fluoroquinolone and macrolide resistance via extended-spectrum beta-lactam use in dairy cattle. Lett. Appl. Microbiol. 2021, 72, 220–224. [Google Scholar] [CrossRef]
- WHO. Critically Important Antimicrobials for Human Medicine, 6th ed.; World Health Organization: Geneva, Switzerland, 2019. [Google Scholar]
- Scott, H.M.; Acuff, G.; Bergeron, G.; Bourassa, M.W.; Gill, J.; Graham, D.W.; Kahn, L.H.; Morley, P.S.; Salois, M.J.; Simjee, S.; et al. Critically important antibiotics: Criteria and approaches for measuring and reducing their use in food animal agriculture. Ann. N. Y. Acad. Sci. 2019, 1441, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Code of Federal Regulations. Chapter I. Food and Drug Administration; Subchapter E. Animal drugs, feeds, and related products; Part 530. Extralabel drug use in animals. In Food and Drugs; 21 CFR 530; Code of Federal Regulations: Washington, DC, USA, 2012. [Google Scholar]
- Brankatschk, R.; Bodenhausen, N.; Zeyer, J.; Burgmann, H. Simple absolute quantification method correcting for quantitative PCR efficiency variations for microbial community samples. Appl. Environ. Microbiol. 2012, 78, 4481–4489. [Google Scholar] [CrossRef] [PubMed]
- Bustin, S.A.; Benes, V.; Garson, J.A.; Hellemans, J.; Huggett, J.; Kubista, M.; Mueller, R.; Nolan, T.; Pfaffl, M.W.; Shipley, G.L.; et al. The MIQE guidelines: Minimum information for publication of quantitative real-time PCR experiments. Clin. Chem. 2009, 55, 611–622. [Google Scholar] [CrossRef] [PubMed]
- Vikram, A.; Rovira, P.; Agga, G.E.; Arthur, T.M.; Bosilevac, J.M.; Wheeler, T.L.; Morley, P.S.; Belk, K.E.; Schmidt, J.W. Impact of “Raised without Antibiotics” Beef Cattle Production Practices on Occurrences of Antimicrobial Resistance. Appl. Environ. Microbiol. 2017, 83, e01682-17. [Google Scholar] [CrossRef]
- Marti, E.; Jofre, J.; Balcazar, J.L. Prevalence of antibiotic resistance genes and bacterial community composition in a river influenced by a wastewater treatment plant. PLoS ONE 2013, 8, e78906. [Google Scholar] [CrossRef]
- Carey, A.M.; Capik, S.F.; Giebel, S.; Nickodem, C.; Pineiro, J.M.; Scott, H.M.; Vinasco, J.; Norman, K.N. Prevalence and Profiles of Antibiotic Resistance Genes mph(A) and qnrB in Extended-Spectrum Beta-Lactamase (ESBL)-Producing Escherichia coli Isolated from Dairy Calf Feces. Microorganisms 2022, 10, 411. [Google Scholar] [CrossRef]
- Klindworth, A.; Pruesse, E.; Schweer, T.; Peplies, J.; Quast, C.; Horn, M.; Glockner, F.O. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 2013, 41, e1. [Google Scholar] [CrossRef]
- Illumina. Illumina 16S Metagenomic Sequencing Library Preparation (Illumina Technical Note 15044223). 2013. Available online: http://support.illumina.com/documents/documentation/chemistry_documentation/16s/16s-metagenomic-library-prep-guide-15044223-b.pdf (accessed on 1 September 2023).
- Ewels, P.; Magnusson, M.; Lundin, S.; Kaller, M. MultiQC: Summarize analysis results for multiple tools and samples in a single report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. 2010. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed on 1 September 2023).
- Estaki, M.; Jiang, L.; Bokulich, N.A.; McDonald, D.; Gonzalez, A.; Kosciolek, T.; Martino, C.; Zhu, Q.; Birmingham, A.; Vazquez-Baeza, Y.; et al. QIIME 2 Enables Comprehensive End-to-End Analysis of Diverse Microbiome Data and Comparative Studies with Publicly Available Data. Curr. Protoc. Bioinformatics 2020, 70, e100. [Google Scholar] [CrossRef]
- Bokulich, N.A.; Kaehler, B.D.; Rideout, J.R.; Dillon, M.; Bolyen, E.; Knight, R.; Huttley, G.A.; Gregory Caporaso, J. Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin. Microbiome 2018, 6, 90. [Google Scholar] [CrossRef] [PubMed]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef]
- Kanwar, N.; Scott, H.M.; Norby, B.; Loneragan, G.H.; Vinasco, J.; Cottell, J.L.; Chalmers, G.; Chengappa, M.M.; Bai, J.; Boerlin, P. Impact of treatment strategies on cephalosporin and tetracycline resistance gene quantities in the bovine fecal metagenome. Sci. Rep. 2014, 4, 5100. [Google Scholar] [CrossRef]
- Alali, W.Q.; Scott, H.M.; Norby, B.; Gebreyes, W.; Loneragan, G.H. Quantification of the bla(CMY-2) in feces from beef feedlot cattle administered three different doses of ceftiofur in a longitudinal controlled field trial. Foodborne Pathog. Dis. 2009, 6, 917–924. [Google Scholar] [CrossRef] [PubMed]
- Bennani, H.; Mateus, A.; Mays, N.; Eastmure, E.; Stark, K.D.C.; Hasler, B. Overview of Evidence of Antimicrobial Use and Antimicrobial Resistance in the Food Chain. Antibiotics 2020, 9, 49. [Google Scholar] [CrossRef]
- Castanheira, S.; Lopez-Escarpa, D.; Pucciarelli, M.G.; Cestero, J.J.; Baquero, F.; Garcia-Del Portillo, F. An alternative penicillin-binding protein involved in Salmonella relapses following ceftriaxone therapy. EBioMedicine 2020, 55, 102771. [Google Scholar] [CrossRef] [PubMed]
- Lewis, G.L.; Fenton, R.J.; Moriyama, E.N.; Loy, J.D.; Moxley, R.A. Association of ISVsa3 with Multidrug Resistance in Salmonella enterica Isolates from Cattle (Bos taurus). Microorganisms 2023, 11, 631. [Google Scholar] [CrossRef]
- Dolejska, M.; Villa, L.; Hasman, H.; Hansen, L.; Carattoli, A. Characterization of IncN plasmids carrying bla CTX-M-1 and qnr genes in Escherichia coli and Salmonella from animals, the environment and humans. J. Antimicrob. Chemother. 2013, 68, 333–339. [Google Scholar] [CrossRef] [PubMed]
- Jacoby, G.A.; Walsh, K.E.; Mills, D.M.; Walker, V.J.; Oh, H.; Robicsek, A.; Hooper, D.C. qnrB, another plasmid-mediated gene for quinolone resistance. Antimicrob. Agents Chemother. 2006, 50, 1178–1182. [Google Scholar] [CrossRef]
- Bauernfeind, A.; Grimm, H.; Schweighart, S. A new plasmidic cefotaximase in a clinical isolate of Escherichia coli. Infection 1990, 18, 294–298. [Google Scholar] [CrossRef]
- Wittum, T.E.; Mollenkopf, D.F.; Daniels, J.B.; Parkinson, A.E.; Mathews, J.L.; Fry, P.R.; Abley, M.J.; Gebreyes, W.A. CTX-M-type extended-spectrum beta-lactamases present in Escherichia coli from the feces of cattle in Ohio, United States. Foodborne Pathog. Dis. 2010, 7, 1575–1579. [Google Scholar] [CrossRef]
- Bauernfeind, A.S.; Schweighart, K. Dornbusch, and H. Giamarellou. A transfer-able cephamycinase in Klebsiella pneumoniae. In Program and Abstracts of the 30th Interscience Conference on Antimicrobial Agents and Chemotherapy; American Society for Microbiology: Washington, DC, USA, 1990. [Google Scholar]
- Fey, P.D.; Safranek, T.J.; Rupp, M.E.; Dunne, E.F.; Ribot, E.; Iwen, P.C.; Bradford, P.A.; Angulo, F.J.; Hinrichs, S.H. Ceftriaxone-resistant salmonella infection acquired by a child from cattle. N. Engl. J. Med. 2000, 342, 1242–1249. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Li, X.; Atwill, E.R.; Aly, S.S.; Williams, D.R.; Su, Z. Dynamic changes in fecal bacterial microbiota of dairy cattle across the production line. BMC Microbiol. 2022, 22, 132. [Google Scholar] [CrossRef]
- Shade, A. Diversity is the question, not the answer. ISME J. 2017, 11, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Wagner, R.D.; Johnson, S.J.; Cerniglia, C.E.; Erickson, B.D. Bovine intestinal bacteria inactivate and degrade ceftiofur and ceftriaxone with multiple beta-lactamases. Antimicrob. Agents Chemother. 2011, 55, 4990–4998. [Google Scholar] [CrossRef]
- Poretsky, R.; Rodriguez, R.L.; Luo, C.; Tsementzi, D.; Konstantinidis, K.T. Strengths and limitations of 16S rRNA gene amplicon sequencing in revealing temporal microbial community dynamics. PLoS ONE 2014, 9, e93827. [Google Scholar] [CrossRef]
- Wichmann, F.; Udikovic-Kolic, N.; Andrew, S.; Handelsman, J. Diverse antibiotic resistance genes in dairy cow manure. mBio 2014, 5, e01017. [Google Scholar] [CrossRef]
- Chambers, L.; Yang, Y.; Littier, H.; Ray, P.; Zhang, T.; Pruden, A.; Strickland, M.; Knowlton, K. Metagenomic Analysis of Antibiotic Resistance Genes in Dairy Cow Feces following Therapeutic Administration of Third Generation Cephalosporin. PLoS ONE 2015, 10, e0133764. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target Gene | Primers and Probes Sequences | Amount (μM) | Annealing Tm (°C) | Product Size (bp) | References |
|---|---|---|---|---|---|
| blaCMY-2 | F 5′-TTCTCCGGGACAACTTGACG-3′ R 5′-GCATCTCCCAGCCTAATCCC-3′ | 1 | 60 | 210 | [32] |
| blaCTX-M | F 5′-CTATGGCACCACCAACGATA-3′ R 5′-ACGGCTTTCTGCCTTAGGTT-3′ | 5 | 60 | 103 | [33] |
| mphA | F-5′GCG AAG GTC GAA CCA GAG-3′ R-5′GAG TCT TCG AGC ATG GGA TAG-3′ P-5′56-FAM/CGA TTC TTG/ZEN/AGCATT GCC AGC ACC/3IABkFQ/-3′ | 2.5 | 60 | 119 | This study |
| qnrB19 | F 5′-CAC ATT ATC GCG TGA CCA ATT-3′ R 5′-GAT GCC TGG TAG CTG TCT AAC-3′ | 1 | 58 | 91 | Modified from [34] |
| 16S rRNA | F 5′-CCTACGGGNGGCWGCAG-3′ R 5′-GACTACHVGGGTATCTA ATCC-3′ | 3.5 | 58 | 464 | [35] |
| Standardized Gene Target (Log10) | Descriptive Statistics | Overall Detections * (N) | Treatment | Sample day | Dairy Farm | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Treated | Untreated | 0 | 6 | 16 | 1 | 2 | 3 | |||
| blaCMY−2 | Observations | 348 | 170 | 178 | 103 | 118 | 127 | 102 | 126 | 120 |
| Mean | −5.73 | −5.40 | −6.08 | −6.19 | −5.56 | −5.52 | −6.21 | −5.95 | −5.10 | |
| Median | −5.65 | −5.36 | −6.13 | −6.30 | −5.61 | −5.49 | −6.29 | −6.04 | −5.13 | |
| 95% CI | −5.84/−5.62 | −5.56/−5.25 | −6.22/−5.93 | −6.41/−5.97 | −5.72/−5.41 | −5.7/−5.34 | −6.45/−5.98 | −6.08/−5.81 | −5.24/−4.95 | |
| Standard error | 0.06 | 0.08 | 0.07 | 0.11 | 0.08 | 0.09 | 0.12 | 0.07 | 0.07 | |
| blaCTX−M | Observations | 352 | 179 | 173 | 113 | 116 | 123 | 117 | 124 | 111 |
| Mean | −6.29 | −6.26 | −6.33 | −6.36 | −6.08 | −6.43 | −6.16 | −6.29 | −6.44 | |
| Median | −6.38 | −6.37 | −6.42 | −6.45 | −6.14 | −6.57 | −6.19 | −6.47 | −6.48 | |
| 95% CI | −6.38/−6.2 | −6.39/−6.12 | −6.45/−6.21 | −6.51/−6.2 | −6.25/−5.92 | −6.57/−6.28 | −6.33/−5.98 | −6.42/−6.16 | −6.6/−6.28 | |
| Standard error | 0.05 | 0.07 | 0.06 | 0.08 | 0.08 | 0.07 | 0.09 | 0.07 | 0.08 | |
| mphA | Observations | 346 | 172 | 174 | 114 | 116 | 116 | 124 | 123 | 99 |
| Mean | −5.04 | −5.00 | −5.07 | −5.11 | −4.93 | −5.08 | −5.01 | −4.85 | −5.30 | |
| Median | −5.15 | −5.09 | −5.18 | −5.16 | −5.03 | −5.26 | −5.11 | −4.91 | −5.48 | |
| 95% CI | −6.01/−5.82 | −5.12/−4.88 | −5.2/−4.95 | −5.24/−4.98 | −5.07/−4.8 | −5.26/−4.89 | −5.17/−4.85 | −4.99/−4.71 | −5.44/−5.17 | |
| Standard error | 0.05 | 0.06 | 0.06 | 0.07 | 0.07 | 0.09 | 0.08 | 0.07 | 0.07 | |
| qnrB19 | Observations | 383 | 191 | 192 | 127 | 128 | 128 | 132 | 132 | 119 |
| Mean | −5.91 | −5.86 | −5.97 | −5.90 | −5.92 | −5.92 | −5.61 | −5.69 | −6.50 | |
| Median | −5.96 | −5.98 | −5.95 | −5.95 | −5.92 | −6.14 | −5.79 | −5.76 | −6.59 | |
| 95% CI | −6.01/−5.82 | −5.99/−5.73 | −6.1/−5.84 | −6.05/−5.75 | −6.07/−5.77 | −6.1/−5.74 | −5.78/−5.45 | −5.82/−5.56 | −6.64/−6.35 | |
| Standard error | 0.05 | 0.07 | 0.07 | 0.08 | 0.08 | 0.09 | 0.08 | 0.07 | 0.07 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ossa-Trujillo, C.; Taylor, E.A.; Sarwar, F.; Vinasco, J.; Jordan, E.R.; Buitrago, J.A.G.; Hagevoort, G.R.; Lawhon, S.D.; Piñeiro, J.M.; Galloway-Peña, J.; et al. Two-Dose Ceftiofur Treatment Increases Cephamycinase Gene Quantities and Fecal Microbiome Diversity in Dairy Cows Diagnosed with Metritis. Microorganisms 2023, 11, 2728. https://doi.org/10.3390/microorganisms11112728
Ossa-Trujillo C, Taylor EA, Sarwar F, Vinasco J, Jordan ER, Buitrago JAG, Hagevoort GR, Lawhon SD, Piñeiro JM, Galloway-Peña J, et al. Two-Dose Ceftiofur Treatment Increases Cephamycinase Gene Quantities and Fecal Microbiome Diversity in Dairy Cows Diagnosed with Metritis. Microorganisms. 2023; 11(11):2728. https://doi.org/10.3390/microorganisms11112728
Chicago/Turabian StyleOssa-Trujillo, Claudia, Ethan A. Taylor, Fatima Sarwar, Javier Vinasco, Ellen R. Jordan, Jose A. García Buitrago, G. Robert Hagevoort, Sara D. Lawhon, Juan M. Piñeiro, Jessica Galloway-Peña, and et al. 2023. "Two-Dose Ceftiofur Treatment Increases Cephamycinase Gene Quantities and Fecal Microbiome Diversity in Dairy Cows Diagnosed with Metritis" Microorganisms 11, no. 11: 2728. https://doi.org/10.3390/microorganisms11112728
APA StyleOssa-Trujillo, C., Taylor, E. A., Sarwar, F., Vinasco, J., Jordan, E. R., Buitrago, J. A. G., Hagevoort, G. R., Lawhon, S. D., Piñeiro, J. M., Galloway-Peña, J., Norman, K. N., & Scott, H. M. (2023). Two-Dose Ceftiofur Treatment Increases Cephamycinase Gene Quantities and Fecal Microbiome Diversity in Dairy Cows Diagnosed with Metritis. Microorganisms, 11(11), 2728. https://doi.org/10.3390/microorganisms11112728