
Diaporthe Species on Palms: Molecular Re-Assessment and Species Boundaries Delimitation in the D. arecae Species Complex




Abstract
1. Introduction
2. Material and Methods
2.1. Specimen Collection, Examination, and Single-Spore Isolation
2.2. Morphological Observation and Characterization
2.3. Sequence Alignment and Phylogenetic Analyses
2.4. Phylogenetic Species Recognition
2.5. Phylogenetic Informativeness Analysis
2.6. Coalescent-Based Species Delimitation Analyses
2.7. Pairwise Homoplasy Index Test and Phylogenetic Network Analyses
2.8. Population Genetic Diversity
2.9. Hierarchical Cluster Analysis of Phenotypic Data
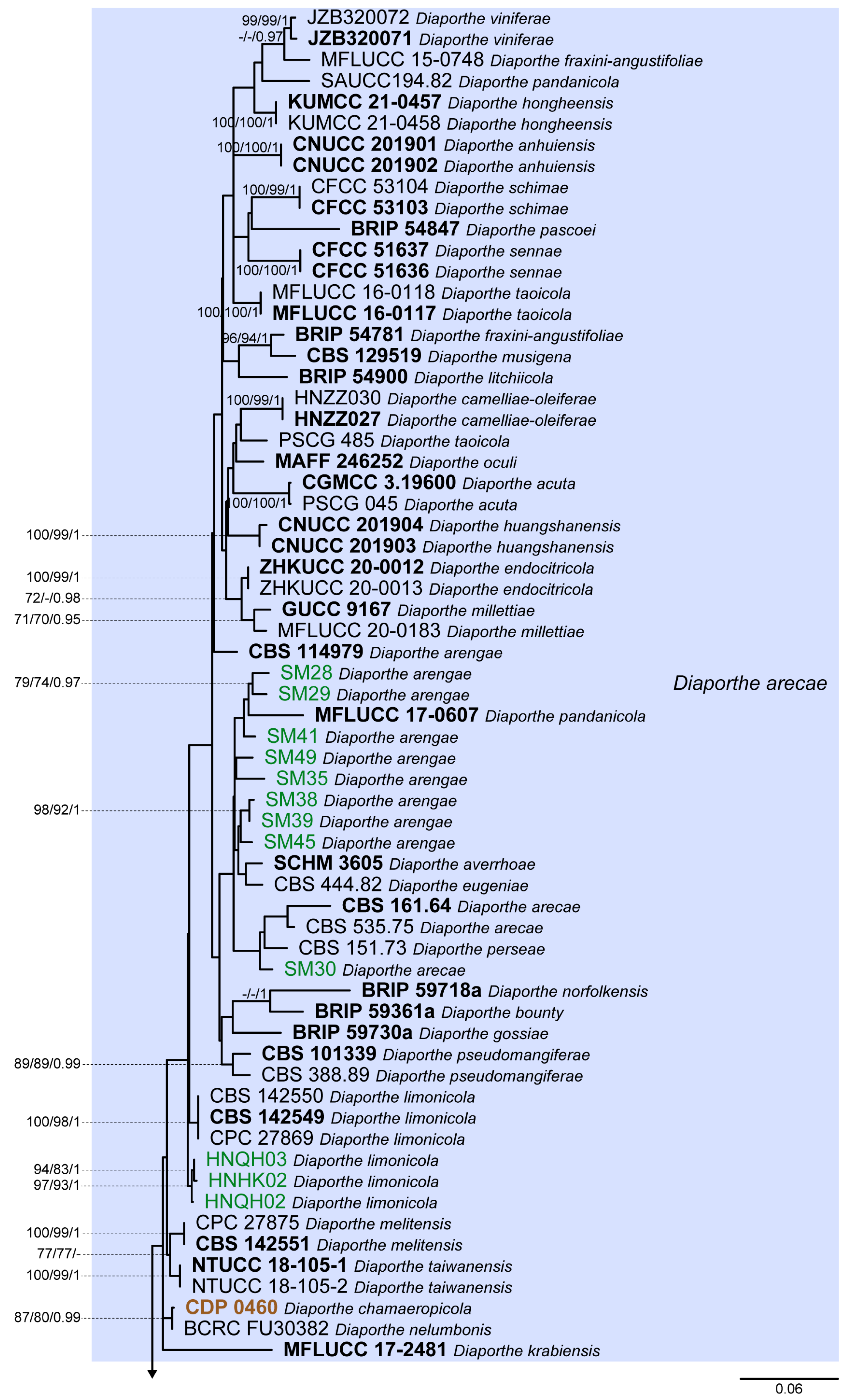
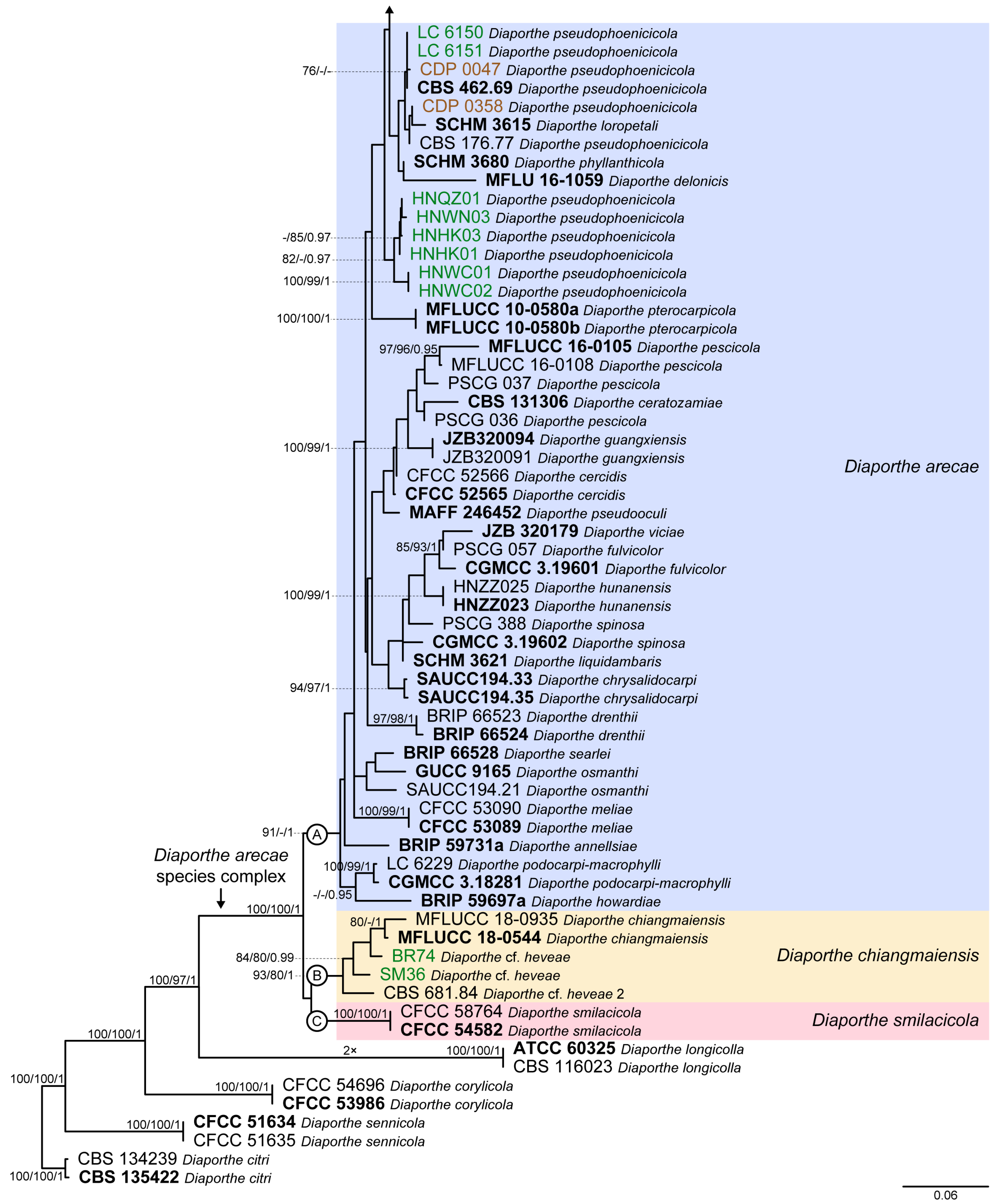
3. Results
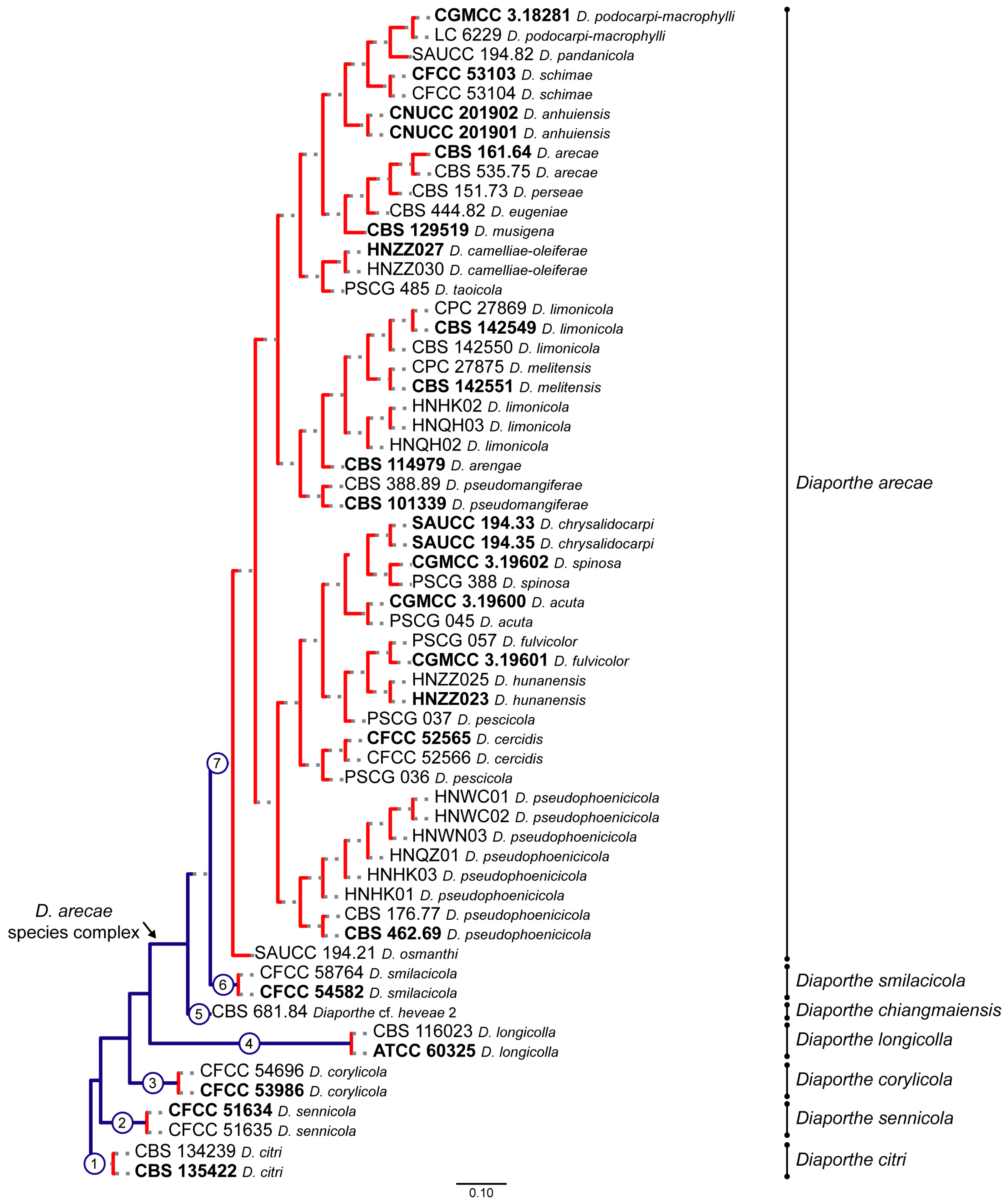
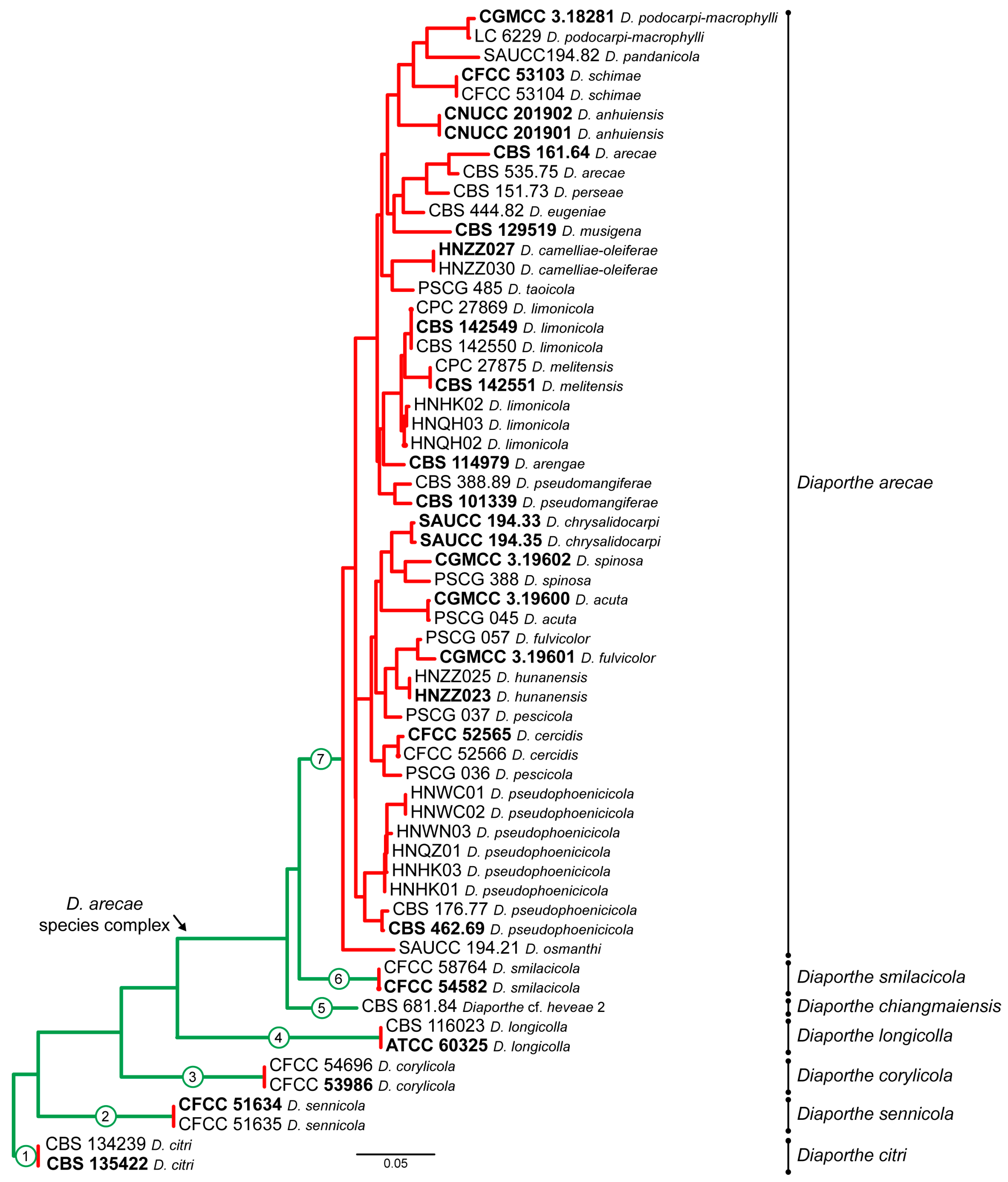
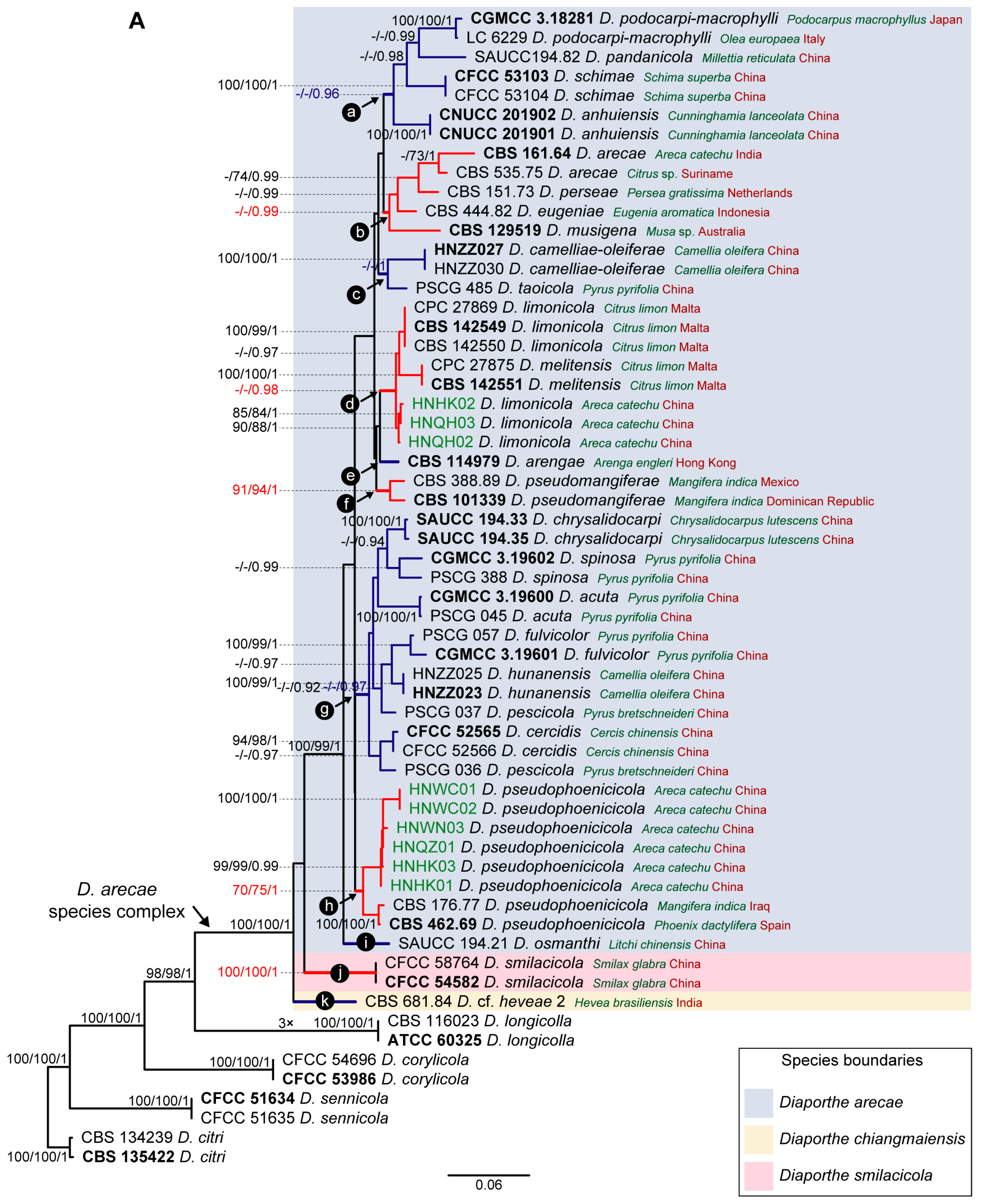
3.1. Preliminary Phylogenetic Analyses
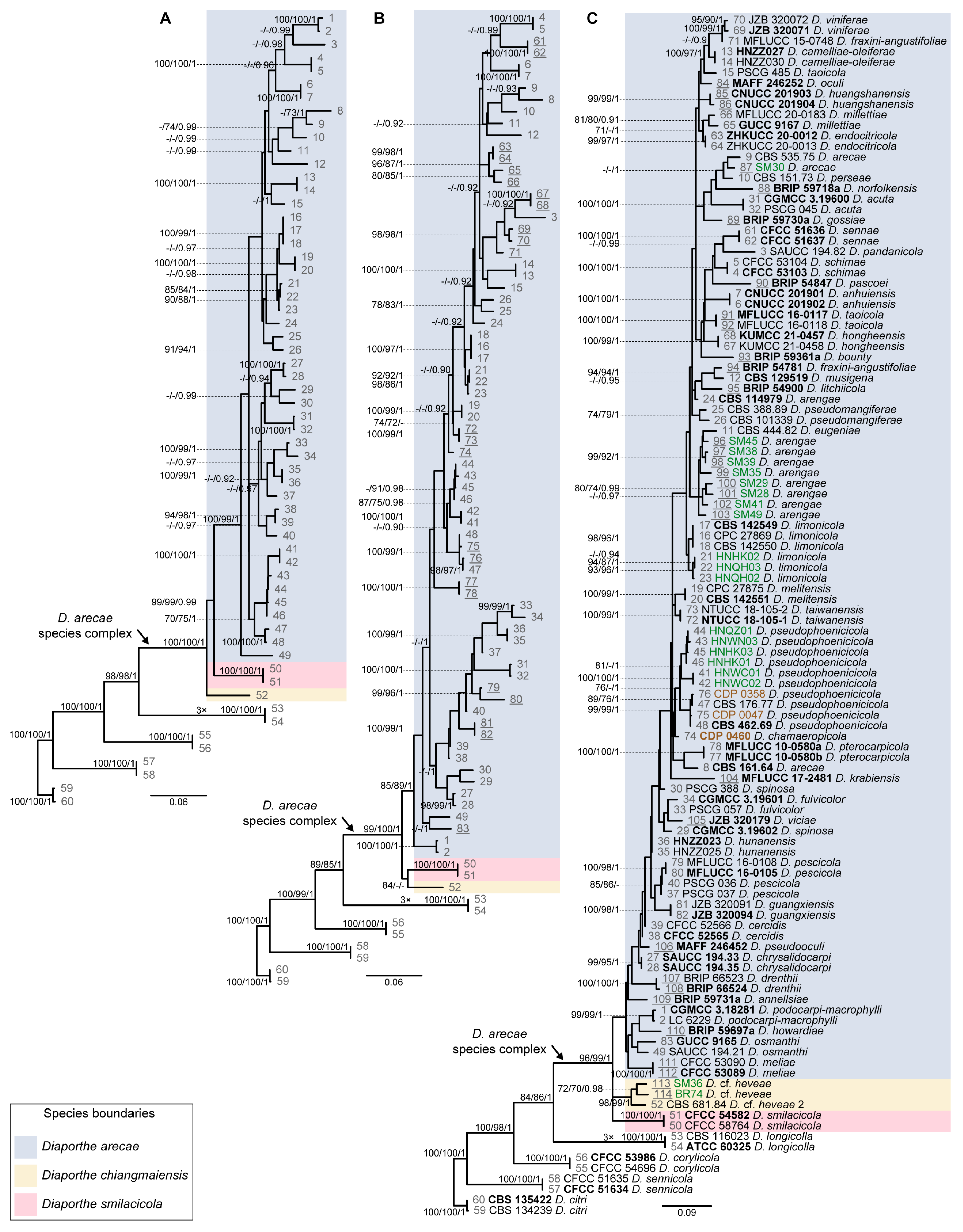
3.2. Species Delimitation Based on the GCPSR Principle
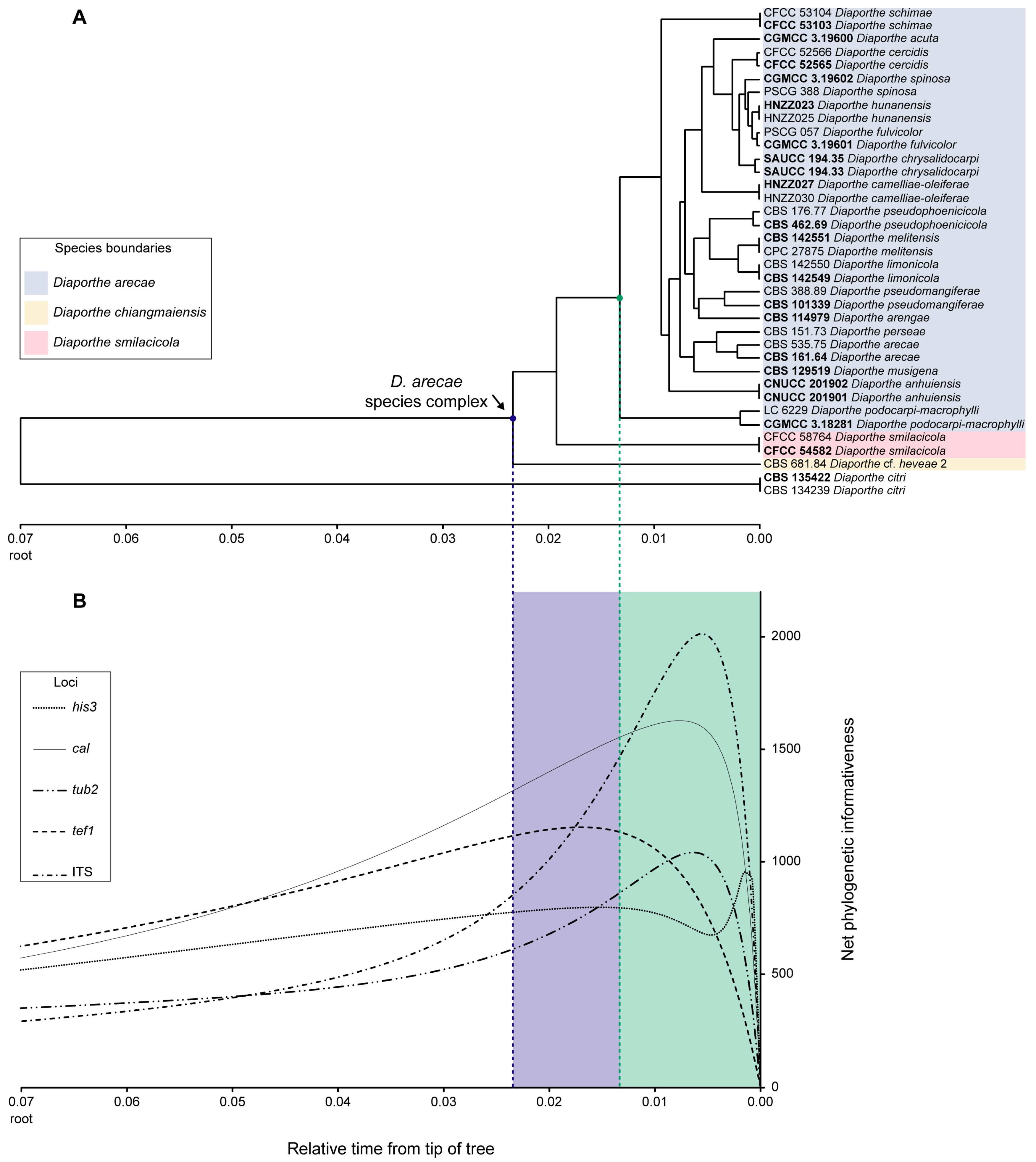
3.3. Phylogenetic Informativeness and Informative Characters of Each Locus
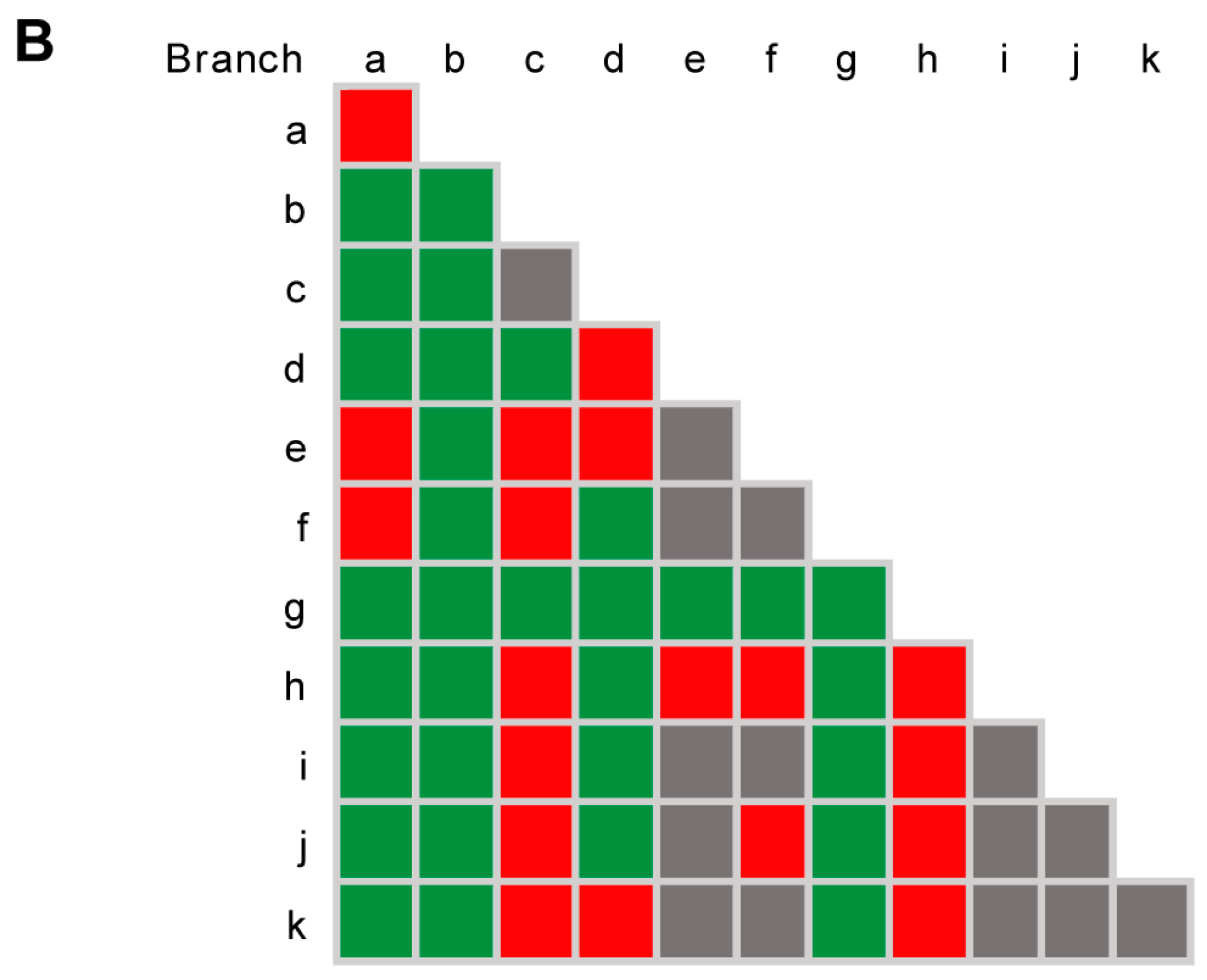
3.4. Species Delimitation Based on Poisson Tree Processes Models
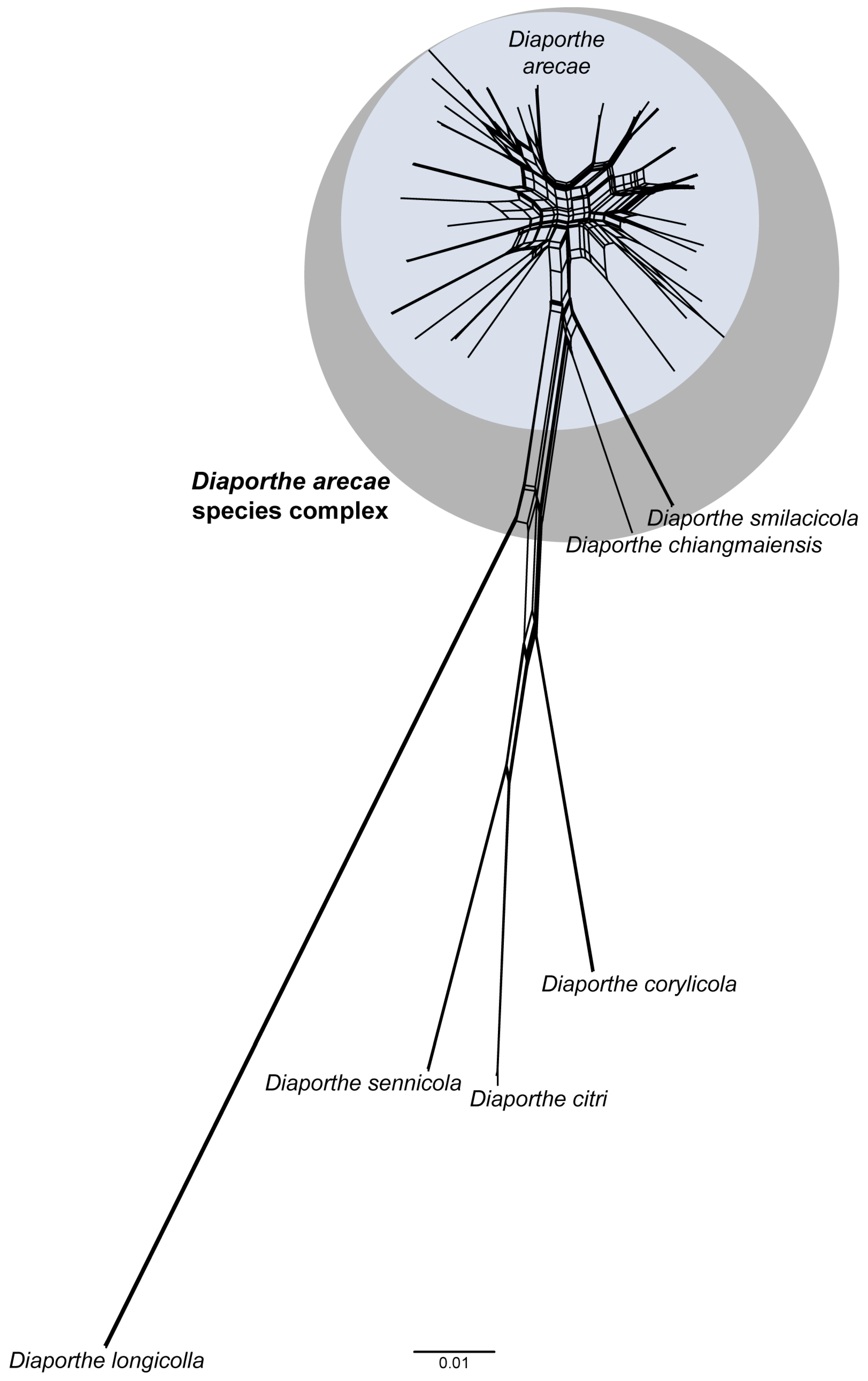
3.5. Pairwise Homoplasy Test and Phylogenetic Network Analyses
3.6. Population Genetic Diversity
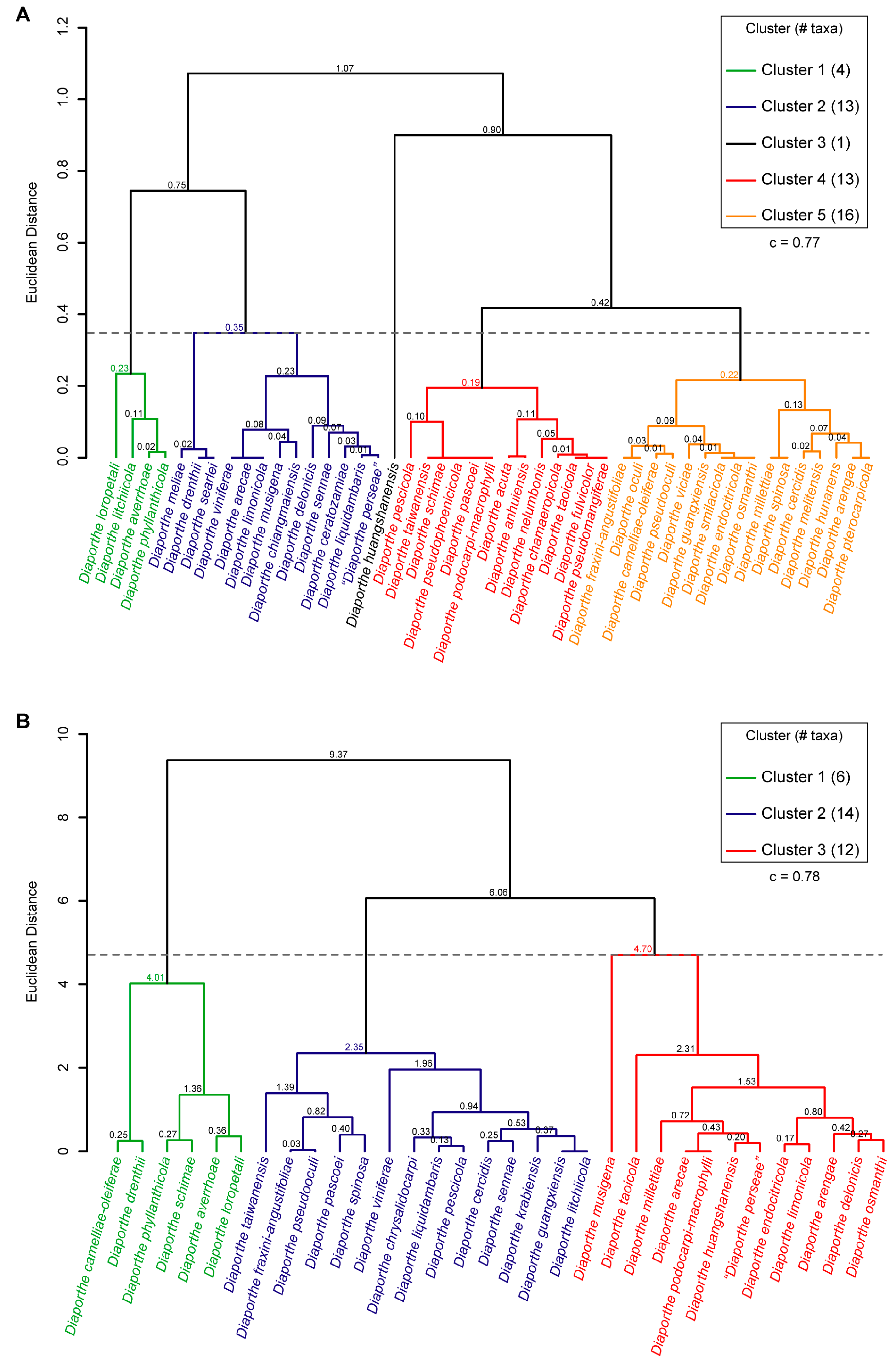
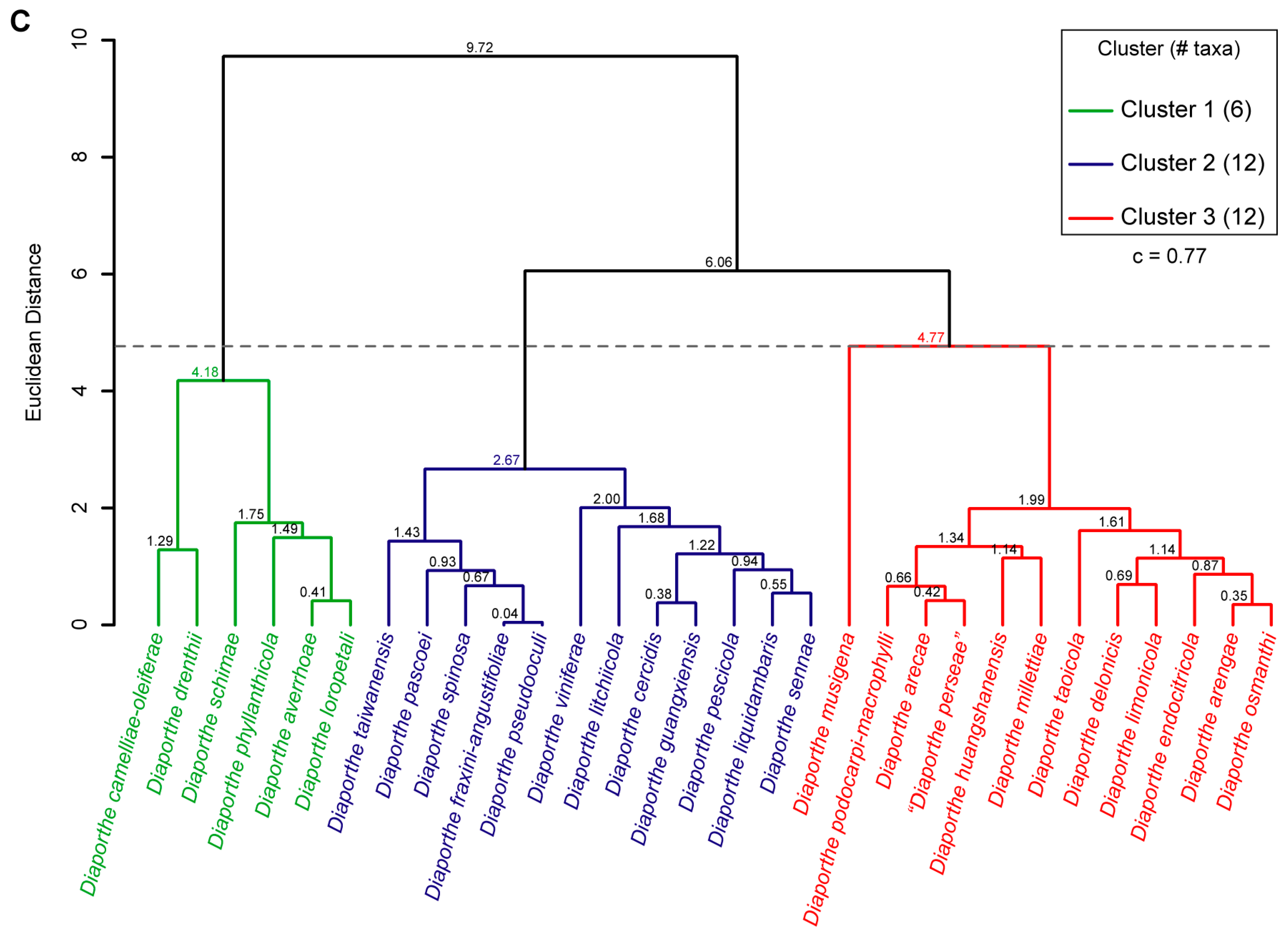
3.7. Hierarchical Cluster Analysis of Phenotypic Data
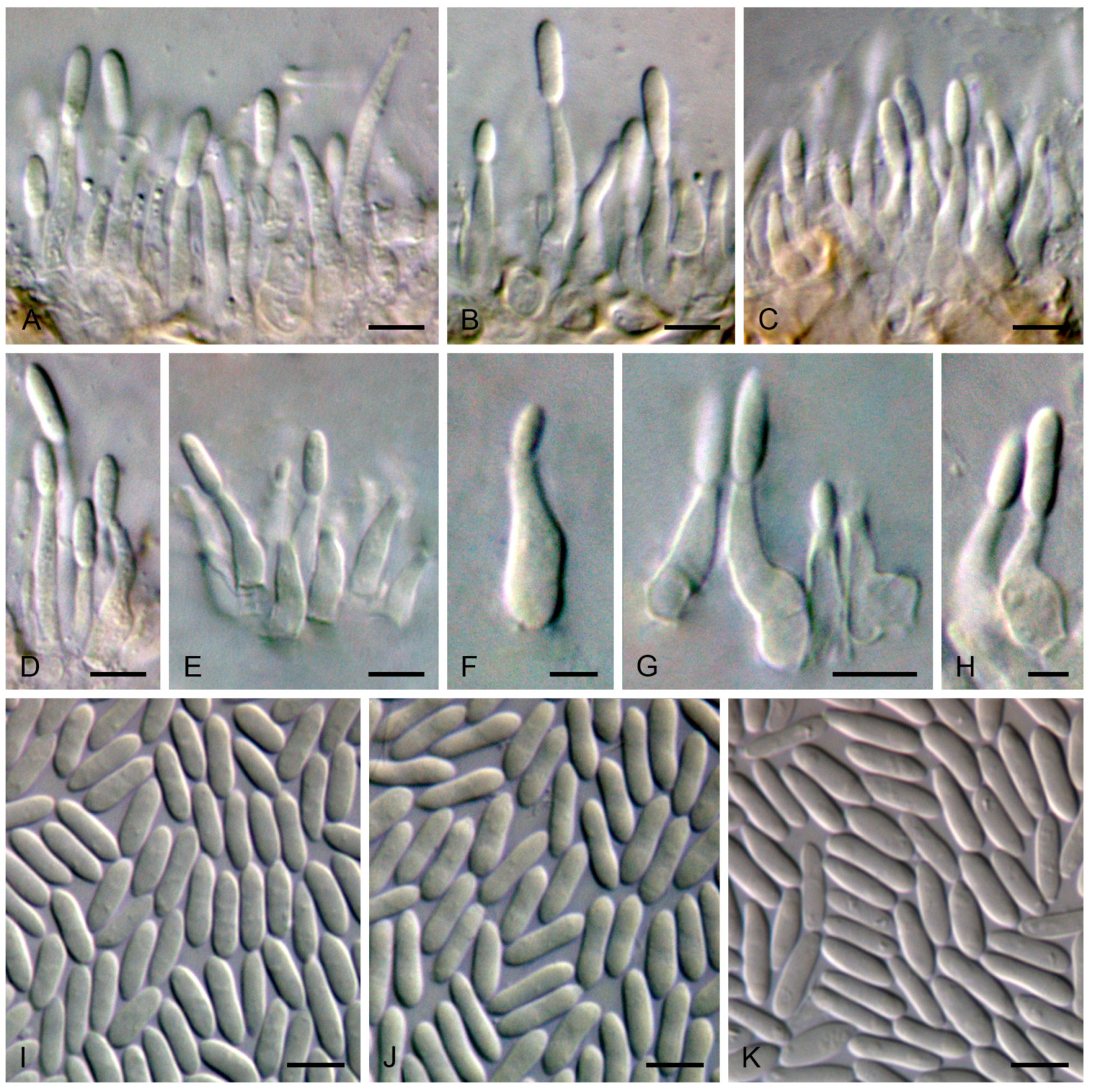
4. Taxonomy
5. Discussion
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Murali, T.S.; Suryanarayanan, T.S.; Geeta, R. Endophytic Phomopsis species: Host range and implications for diversity estimates. Can. J. Microbiol. 2006, 52, 673–680. [Google Scholar] [CrossRef] [PubMed]
- Santos, J.M.; Phillips, A.J.L. Resolving the complex of Diaporthe (Phomopsis) species occurring on Foeniculum vulgare in Portugal. Fungal Divers. 2009, 34, 111–125. [Google Scholar]
- Garcia-Reyne, A.; López-Medrano, F.; Morales, J.M.; Esteban, C.G.; Martín, I.; Eraña, I.; Meiie, Y.; Lalueza, A.; Alastruey-Izquierdo, A.; Rodríguez-Tudela, J.L.; et al. Cutaneous infection by Phomopsis longicolla in a renal transplant recipient from Guinea: First report of human infection by this fungus. Transpl. Infect. Dis. 2011, 13, 204–207. [Google Scholar] [CrossRef] [PubMed]
- Udayanga, D.; Liu, X.; McKenzie, E.H.C.; Chukeatirote, E.; Bahkali, A.H.A.; Hyde, K.D. The genus Phomopsis: Biology, applications, species concepts and names of common phytopathogens. Fungal Divers. 2011, 50, 189–225. [Google Scholar] [CrossRef]
- Hyde, K.D.; Nilsson, R.H.; Alias, S.A.; Ariyawansa, H.A.; Blair, J.E.; Cai, L.; de Cock, A.W.A.M.; Dissanayake, A.J.; Glockling, S.L.; Goonasekara, I.D.; et al. One stop shop: Backbones trees for important phytopathogenic genera: I. Fungal Divers. 2014, 67, 21–125. [Google Scholar] [CrossRef]
- Uecker, F.A. A world list of Phomopsis names with notes on nomenclature, morphology and biology. Mycol. Mem. 1988, 13, 1–231. [Google Scholar]
- Udayanga, D.; Castlebury, L.A.; Rossman, A.Y.; Chukeatirote, E.; Hyde, K.D. Insights into the genus Diaporthe: Phylogenetic species delimitation in the D. eres species complex. Fungal Divers. 2014, 67, 203–229. [Google Scholar] [CrossRef]
- Udayanga, D.; Castlebury, L.A.; Rossman, A.Y.; Hyde, K.D. Species limits in Diaporthe: Molecular re-assessment of D. citri, D. cytosporella, D. foeniculina and D. rudis. Persoonia 2014, 32, 83–101. [Google Scholar] [CrossRef]
- Udayanga, D.; Castlebury, L.A.; Rossman, A.Y.; Chukeatirote, E.; Hyde, K.D. The Diaporthe sojae species complex, phylogenetic re-assessment of pathogens associated with soybean, cucurbits and other field crops. Fungal Biol. 2015, 119, 383–407. [Google Scholar] [CrossRef]
- Guarnaccia, V.; Crous, P.W. Emerging citrus diseases in Europe caused by species of Diaporthe. IMA Fungus 2017, 8, 317–334. [Google Scholar] [CrossRef]
- Guarnaccia, V.; Vitale, A.; Cirvilleri, G.; Aiello, D.; Susca, A.; Epifani, F.; Perrone, G.; Polizzi, G. Characterisation and pathogenicity of fungal species associated with branch cankers and stem-end rot of avocado in Italy. Eur. J. Plant Pathol. 2016, 146, 963–976. [Google Scholar] [CrossRef]
- Guarnaccia, V.; Groenewald, J.Z.; Woodhall, J.; Armengol, J.; Cinelli, T.; Eichmeier, A.; Ezra, D.; Fontaine, F.; Gramaje, D.; Gutierrez-Aguirregabiria, A.; et al. Diaporthe diversity and pathogenicity revealed from a broad survey of grapevine diseases in Europe. Persoonia 2018, 40, 135–153. [Google Scholar] [CrossRef] [PubMed]
- Marin-Felix, Y.; Hernández-Restrepo, M.; Wingfield, M.J.; Akulov, A.; Carnegie, A.J.; Cheewangkoon, R.; Gramaje, D.; Groenewald, J.Z.; Guarnaccia, V.; Halleen, F.; et al. Genera of phytopathogenic fungi: GOPHY 2. Stud. Mycol. 2019, 92, 47–133. [Google Scholar] [CrossRef] [PubMed]
- Mostert, L.; Crous, P.W.; Kang, J.C.; Phillips, A.J.L. Species of Phomopsis and a Libertella sp. occurring on grapevines with specific reference to South Africa: Morphological, cultural, molecular and pathological characterization. Mycologia 2001, 93, 146–167. [Google Scholar] [CrossRef]
- Mostert, L.; Kang, J.C.; Crous, P.W.; Denman, S. Phomopsis saccharata sp. nov., causing a canker and die-back disease of Protea repens in South Africa. Sydowia 2001, 53, 227–235. [Google Scholar]
- van Rensburg, J.C.J.; Lamprecht, S.C.; Groenewald, J.Z.; Castlebury, L.A.; Crous, P.W. Characterization of Phomopsis spp. associated with die-back of rooibos (Aspalathus linearis) in South Africa. Stud. Mycol. 2016, 55, 65–74. [Google Scholar] [CrossRef]
- Diogo, E.L.F.; Santos, J.M.; Phillips, A.J.L. Phylogeny, morphology and pathogenicity of Diaporthe and Phomopsis species on almond in Portugal. Fungal Divers. 2010, 44, 107–115. [Google Scholar] [CrossRef]
- Thompson, S.; Tan, Y.; Young, A.; Neate, S.; Aitken, E.; Shivas, R. Stem cankers on sunflower (Helianthus annuus) in Australia reveal a complex of pathogenic Diaporthe (Phomopsis) species. Persoonia 2011, 27, 80–89. [Google Scholar] [CrossRef]
- Thompson, S.M.; Tan, Y.P.; Shivas, R.G.; Neate, S.M.; Morin, L.; Bissett, A.; Aitken, E.A.B. Green and brown bridges between weeds and crops reveal novel Diaporthe species in Australia. Persoonia 2015, 35, 39–49. [Google Scholar] [CrossRef]
- Díaz, G.A.; Latorre, B.A.; Lolas, M.; Ferrada, E.; Naranjo, P.; Zoffoli, J.P. Identification and characterization of Diaporthe ambigua, D. australafricana, D. novem, and D. rudis causing a postharvest fruit rot in kiwifruit. Plant Dis. 2017, 101, 1402–1410. [Google Scholar] [CrossRef]
- Manawasinghe, I.S.; Dissanayake, A.J.; Li, X.; Liu, M.; Wanasinghe, D.N.; Xu, J.; Zhao, W.; Zhang, W.; Zhou, Y.; Hyde, K.D.; et al. High genetic diversity and species complexity of Diaporthe associated with grapevine dieback in China. Front. Microbiol. 2019, 10, 1936. [Google Scholar] [CrossRef] [PubMed]
- Wingfield, M.J.; de Beer, Z.W.; Slippers, B.; Wingfield, B.D.; Groenewald, J.Z.; Lombard, L.; Crous, P.W. One fungus, one name promotes progressive plant pathology. Mol. Plant Pathol. 2012, 13, 604–613. [Google Scholar] [CrossRef] [PubMed]
- Udayanga, D.; Liu, X.; Crous, P.W.; McKenzie, E.H.C.; Chukeatirote, E.; Hyde, K.D. A multi-locus phylogenetic evaluation of Diaporthe (Phomopsis). Fungal Divers. 2012, 56, 157–171. [Google Scholar] [CrossRef]
- Gomes, R.R.; Glienke, C.; Videira, S.I.R.; Lombard, L.; Groenewald, J.Z.; Crous, P.W. Diaporthe: A genus of endophytic, saprobic and plant pathogenic fungi. Persoonia 2013, 31, 1–41. [Google Scholar] [CrossRef] [PubMed]
- Dissanayake, A.J.; Phillips, A.J.L.; Hyde, K.D.; Yan, J.; Li, X. The current status of species in Diaporthe. Mycosphere 2017, 8, 1106–1156. [Google Scholar] [CrossRef]
- Gao, Y.; Liu, F.; Duan, W.; Crous, P.W.; Cai, L. Diaporthe is paraphyletic. IMA Fungus 2017, 8, 153–187. [Google Scholar] [CrossRef] [PubMed]
- Norphanphoun, C.; Gentekaki, E.; Hongsanan, S.; Jayawardena, R.; Senanayake, I.C.; Manawasinghe, I.S.; Abeywickrama, P.D.; Bhunjun, C.S.; Hyde, K.D. Diaporthe: Formalizing the species-group concept. Mycosphere 2022, 13, 752–819. [Google Scholar] [CrossRef]
- Crous, P.W.; Gams, W.; Stalpers, J.A.; Robert, V.; Stegehuis, G. MycoBank: An online initiative to launch mycology into the 21st century. Stud. Mycol. 2004, 50, 19–22. [Google Scholar]
- Wehmeyer, L.E. The genus Diaporthe Nitschke and its segregates. Univ. Mich. Stud. Sci. Ser. 1993, 9, 1–349. [Google Scholar]
- Santos, J.M.; Correia, V.G.; Phillips, A.J.L. Primers for mating-type diagnosis in Diaporthe and Phomopsis: Their use in teleomorph induction in vitro and biological species definition. Fungal Biol. 2010, 114, 255–270. [Google Scholar] [CrossRef]
- Udayanga, D.; Liu, X.; McKenzie, E.H.C.; Chukeatirote, E.; Hyde, K.D. Multi-locus phylogeny reveals three new species of Diaporthe from Thailand. Cryptogam. Mycol. 2012, 33, 295–309. [Google Scholar] [CrossRef]
- Santos, L.; Alves, A.; Alves, R. Evaluating multi-locus phylogenies for species boundaries determination in the genus Diaporthe. PeerJ 2017, 5, e3120. [Google Scholar] [CrossRef] [PubMed]
- Senanayake, I.C.; Crous, P.W.; Groenewald, J.Z.; Maharachchikumbura, S.S.N.; Jeewon, R.; Phillips, A.J.L.; Bhat, J.D.; Perera, R.H.; Li, Q.R.; Li, W.J.; et al. Families of Diaporthales based on morphological and phylogenetic evidence. Stud. Mycol. 2017, 86, 217–296. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Wang, M.; Damm, U.; Crous, P.W.; Cai, L. Species boundaries in plant pathogenic fungi: A Colletotrichum case study. BMC Plant Biol. 2016, 16, 81. [Google Scholar] [CrossRef]
- Huang, F.; Udayanga, D.; Wang, X.; Hou, X.; Mei, X.; Fu, Y.; Hyde, K.D.; Li, H. Endophytic Diaporthe associated with Citrus: A phylogenetic reassessment with seven new species from China. Fungal Biol. 2015, 119, 331–347. [Google Scholar] [CrossRef]
- Hilário, S.; Gonçalves, M.F.M.; Alves, A. Using genealogical concordance and coalescent-based species delimitation to assess species boundaries in the Diaporthe eres complex. J. Fungi 2021, 25, 507. [Google Scholar] [CrossRef]
- Hilário, S.; Santos, L.; Alves, A. Diaporthe amygdali, a species complex or a complex species? Fungal Biol. 2021, 125, 505–518. [Google Scholar] [CrossRef]
- Srivastava, H.C.; Banu, Z.; Govindarajan, V.S. Fruit rot of areca nut caused by a new fungus. Mycologia 1962, 54, 5–11. [Google Scholar] [CrossRef]
- Tan, Y.P.; Edwards, J.; Grice, K.R.E.; Shivas, R.G. Molecular phylogenetic analysis reveals six new species of Diaporthe from Australia. Fungal Divers. 2013, 61, 251–260. [Google Scholar] [CrossRef]
- Arciuolo, R.; Santos, C.; Soares, C.; Castello, G.; Spigolon, N.; Chiusa, G.; Lima, N.; Battilani, P. Molecular characterization of Diaporthe species associated with hazelnut defects. Front. Plant Sci. 2020, 11, 611655. [Google Scholar] [CrossRef]
- Dissanayake, A.J.; Zhang, W.; Liu, M.; Hyde, K.D.; Zhao, W.S.; Li, X.; Yan, J. Diaporthe species associated with peach tree dieback in Hubei, China. Mycosphere 2017, 8, 533–549. [Google Scholar] [CrossRef]
- Ozawa, K.; Mochizuki, K.; Takagi, D.; Ishida, K.; Sunada, A.; Ohkusu, K.; Kamei, K.; Hashimoto, A.; Tanaka, K. Identification and antifungal sensitivity of two new species of Diaporthe isolated. J. Infect. Chemother. 2019, 25, 96–103. [Google Scholar] [CrossRef]
- Hilário, S.; Santos, L.; Phillips, A.J.L.; Alves, A. Caveats of the internal transcribed spacer region as a barcode to resolve species boundaries in Diaporthe. Fungal Biol. 2022, 126, 54–74. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.W.; Jacobson, D.J.; Kroken, S.; Kasuga, T.; Geiser, D.M.; Hibbett, D.S.; Fisher, M.C. Phylogenetic species recognition and species concepts in fungi. Fungal Genet. Biol. 2000, 31, 21–32. [Google Scholar] [CrossRef] [PubMed]
- Maddison, W.P. Gene trees in species trees. Syst. Biol. 1997, 46, 523–536. [Google Scholar] [CrossRef]
- Kubatko, L.S.; Degnan, J.H. Inconsistency of phylogenetic estimates from concatenated data under coalescence. Syst. Biol. 2007, 56, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Stewart, J.E.; Timmer, L.W.; Lawrence, C.B.; Pryor, B.M.; Peever, T.L. Discord between morphological and phylogenetic species boundaries: Incomplete lineage sorting and recombination results in fuzzy species boundaries in an asexual fungal pathogen. BMC Evol. Biol. 2014, 14, 38. [Google Scholar] [CrossRef]
- Achari, S.R.; Kaur, J.; Dinh, Q.; Mann, R.; Sawbridge, T.; Summerell, B.A.; Edwards, J. Phylogenetic relationship between Australian Fusarium oxysporum isolates and resolving the species complex using the multispecies coalescent model. BMC Genom. 2020, 21, 248. [Google Scholar] [CrossRef]
- Avise, J.C.; Ball, R.M., Jr. Principles of genealogical concordance in species concepts and biological taxonomy. In Oxford Surveys in Evolutionary Biology; Futuyma, D., Antonovics, J., Eds.; Oxford University Press: Oxford, UK, 1990; Volume 7, pp. 45–67. [Google Scholar]
- Carstens, B.C.; Knowles, L.L. Estimating species phylogeny from gene-tree probabilities despite incomplete lineage sorting: An example from Melanoplus grasshoppers. Syst. Biol. 2007, 56, 400–411. [Google Scholar] [CrossRef]
- Degnan, J.H.; Rosenberg, N.A. Gene tree discordance, phylogenetic inference and the multispecies coalescent. Trends Ecol. Evol. 2009, 24, 332–340. [Google Scholar] [CrossRef]
- Kubatko, L.S.; Carstens, B.C.; Knowles, L.L. STEM: Species tree estimation using maximum likelihood for gene trees under coalescence. Bioinformatics 2009, 25, 9–973. [Google Scholar] [CrossRef]
- Eckert, A.J.; Carstens, B.C. Does gene flow destroy phylogenetic signal? The performance of three methods for estimating species phylogenies in the presence of gene flow. Mol. Phylogenet. Evol. 2008, 49, 832–842. [Google Scholar] [CrossRef] [PubMed]
- Rannala, B.; Yang, Z. Species Delimitation. In Phylogenetics in the Genomic Era; Scornavacca, C., Delsuc, F., Galtier, N., Eds.; No Commercial Publisher, 2020; Chapter 5.5; pp. 5.5:1–5.5:18. Available online: https://hal.inria.fr/PGE (accessed on 15 June 2023).
- Chaisiri, C.; Liu, X.; Lin, Y.; Fu, Y.; Zhu, F.; Luo, C. Phylogenetic and haplotype network analyses of Diaporthe eres species in China based on sequences of multiple loci. Biology 2021, 10, 179. [Google Scholar] [CrossRef] [PubMed]
- Rannala, B.; Yang, Z. Bayes estimation of species divergence times and ancestral population sizes using DNA sequences from multiple loci. Genetics 2003, 164, 1645–1656. [Google Scholar] [CrossRef] [PubMed]
- Edwards, S.V. Is a new and general theory of molecular systematics emerging? Evolution 2009, 63, 1–19. [Google Scholar] [CrossRef]
- Liu, L.; Wu, S.; Yu, L. Coalescent methods for estimating species trees from phylogenomic data. J. Syst. Evol. 2015, 53, 380–390. [Google Scholar] [CrossRef]
- Jiao, X.; Flouri, T.; Yang, Z. Multispecies coalescent and its applications to infer species phylogenies and cross-species gene flow. Natl. Sci. Rev. 2021, 8, nwab127. [Google Scholar] [CrossRef]
- Fujita, M.K.; Leaché, A.D.; Burbrink, F.T.; McGuire, J.A.; Moritz, C. Coalescent-based species delimitation in an integrative taxonomy. Trends Ecol. Evol. 2012, 27, 480–488. [Google Scholar] [CrossRef]
- Mirarab, S.; Nakhleh, L.; Warnow, T. Multispecies coalescent: Theory and applications in phylogenetics. Annu. Rev. Ecol. Evol. Syst. 2021, 52, 247–268. [Google Scholar] [CrossRef]
- Bustamante, D.E.; Oliva, M.; Leiva, S.; Mendoza, J.E.; Bobadilla, L.; Angulo, G.; Calderon, M.S. Phylogeny and species delimitations in the entomopathogenic genus Beauveria (Hypocreales, Ascomycota), including the description of B. peruviensis sp. nov. MycoKeys 2019, 58, 47–68. [Google Scholar] [CrossRef]
- Boluda, C.G.; Rico, V.J.; Divakar, P.K.; Nadyeina, O.; Myllys, L.; McMullin, R.T.; Zamora, J.C.; Scheidegger, C.; Hawksworth, D.L. Evaluating methodologies for species delimitation: The mismatch between phenotypes and genotypes in lichenized fungi (Bryoria sect. Implexae, Parmeliaceae). Persoonia 2019, 42, 75–100. [Google Scholar] [CrossRef]
- Boonmee, S.; Wanasinghe, D.N.; Calabon, M.S.; Huanraluek, N.; Chandrasiri, S.K.U.; Jones, G.E.B.; Rossi, W.; Leonardi, M.; Singh, S.K.; Rana, S.; et al. Fungal diversity notes 1387–1511: Taxonomic and phylogenetic contributions on genera and species of fungal taxa. Fungal Divers. 2021, 111, 1–335. [Google Scholar] [CrossRef]
- Pereira, D.S.; Phillips, A.J.L. Botryosphaeriaceae on palms—A new species of Neodeightonia, N. chamaeropicola, and new records from diseased foliage of ornamental palms in Portugal. Phytotaxa, 2023; submitted. [Google Scholar]
- Xu, G.; Qiu, F.; Li, X.; Zheng, F.-Q.; Zheng, L.; Miao, W.G.; Xie, C.P. Diaporthe limonicola causing leaf spot disease on Areca catechu in China. Plant Dis. 2020, 104, 1869. [Google Scholar] [CrossRef]
- Azuddin, N.F.; Mohd, M.H.; Rosely, N.F.N.; Mansor, A.; Zakaria, L. Molecular phylogeny of endophytic fungi from rattan (Calamus castaneus Griff.) spines and their antagonistic activities against plant pathogenic fungi. J. Fungi 2021, 7, 301. [Google Scholar] [CrossRef] [PubMed]
- Thompson, J.D.; Gibson, T.J.; Plewniak, F.; Jeanmougin, F.; Higgins, D.G. The ClustalX windows interface: Flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997, 25, 4876–4882. [Google Scholar] [CrossRef] [PubMed]
- Hall, T.A. BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Farris, J.S.; Kallersjo, M.; Kluge, A.G.; Bult, C. Testing significance of congruence. Cladistics 1994, 10, 315–319. [Google Scholar] [CrossRef]
- Swofford, D.L. PAUP*. Phylogenetic Analysis Using Parsimony (*and Other Methods); Version 4.0a165; Sinauer Associates: Sunderland, MA, USA, 2002. [Google Scholar]
- Miller, M.A.; Pfeiffer, W.; Schwartz, T. Creating the CIPRES Science Gateway for inference of large phylogenetic trees. In Proceedings of the 2010 Gateway Computing Environments Workshop (GCE), New Orleans, LA, USA, 14 November 2010; pp. 1–8. [Google Scholar]
- Kozlov, A.M.; Darriba, D.; Flouri, T.; Morel, B.; Stamatakis, A. RAxML-NG: A fast, scalable and user-friendly tool for maximum likelihood phylogenetic inference. Bioinformatics 2019, 35, 4453–4455. [Google Scholar] [CrossRef]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef]
- Rambaut, A. FigTree v1.4.4; 2018. Available online: http://tree.bio.ed.ac.uk/software/figtree/ (accessed on 15 June 2023).
- Xi, Z.; Liu, L.; Davis, C.C. The impact of missing data on species tree estimation. Mol. Biol. Evol. 2016, 33, 838–860. [Google Scholar] [CrossRef] [PubMed]
- Dettman, J.R.; Jacobson, D.J.; Taylor, J.W. A multilocus genealogical approach to phylogenetic species recognition in the model eukaryote Neurospora. Evolution 2003, 57, 2703–2720. [Google Scholar] [PubMed]
- Dettman, J.R.; Jacobson, D.J.; Taylor, J.W. Multilocus sequence data reveal extensive phylogenetic species diversity within the Neurospora discreta complex. Mycologia 2006, 98, 436–446. [Google Scholar] [CrossRef] [PubMed]
- Townsend, J.P. Profiling phylogenetic informativeness. Syst. Biol. 2007, 56, 222–231. [Google Scholar] [CrossRef] [PubMed]
- López-Giráldez, F.; Townsend, J.P. PhyDesign: An online application for profiling phylogenetic informativeness. BMC Evol. Biol. 2011, 11, 152. [Google Scholar] [CrossRef]
- Mello, B. Estimating TimeTrees with MEGA and the TimeTree Resource. Mol. Biol. Evol. 2018, 35, 2334–2342. [Google Scholar] [CrossRef]
- Tamura, K.; Battistuzzi, F.U.; Billing-Ross, P.; Murillo, O.; Filipski, A.; Kumar, S. Estimating divergence times in large molecular phylogenies. Proc. Natl. Acad. Sci. USA 2012, 109, 19333–19338. [Google Scholar] [CrossRef]
- Tamura, K.; Qiqing, T.; Kumar, S. Theoretical foundation of the RelTime method for estimating divergence times from variable evolutionary rates. Mol. Biol. Evol. 2018, 35, 1770–1782. [Google Scholar] [CrossRef]
- Pond, S.L.K.; Frost, S.D.W.; Muse, S.V. HyPhy: Hypothesis testing using phylogenies. Bioinformatics 2015, 21, 676–679. [Google Scholar] [CrossRef]
- Zhang, J.; Kapli, P.; Pavlidis, P.; Stamatakis, A. A general species delimitation method with applications to phylogenetic placements. Bioinformatics 2013, 29, 2869–2876. [Google Scholar] [CrossRef]
- Kapli, P.; Lutteropp, S.; Zhang, J.; Kobert, K.; Pavlidis, P.; Stamatakis, A.; Flouri, T. Multi-rate Poisson tree processes for single-locus species delimitation under maximum likelihood and Markov chain Monte Carlo. Bioinformatics 2017, 33, 1630–1638. [Google Scholar] [CrossRef] [PubMed]
- Nute, M.; Chou, J.; Molloy, E.K.; Warnow, T. The performance of coalescent-based species tree estimation methods under models of missing data. BMC Genom. 2018, 19, 286. [Google Scholar] [CrossRef]
- Bruen, T.C.; Philippe, H.; Bryant, D. A simple and robust statistical test for detecting the presence of recombination. Genetics 2006, 172, 2665–2681. [Google Scholar] [CrossRef] [PubMed]
- Huson, D.H.; Bryant, D. Application of phylogenetic networks in evolutionary studies. Mol. Biol. Evol. 2006, 23, 254–267. [Google Scholar] [CrossRef] [PubMed]
- Steel, M.A. Recovering a tree from the leaf colorations it generates under a Markov model. Appl. Math. Lett. 1994, 7, 19–24. [Google Scholar] [CrossRef]
- Bryant, D.; Moulton, V. Neighbor-Net: An agglomerative method for the construction of phylogenetic networks. Mol. Biol. Evol. 2004, 21, 255–265. [Google Scholar] [CrossRef]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef]
- Nei, M. Molecular Evolutionary Genetics; Columbia University Press: New York, NY, USA, 1987. [Google Scholar]
- Tajima, F. Evolutionary relationship of DNA sequences in finite populations. Genetics 1983, 105, 437–460. [Google Scholar] [CrossRef]
- Watterson, G.A. On the number of segregating sites in genetical models without recombination. Theor. Pop. Biol. 1975, 7, 256–276. [Google Scholar] [CrossRef]
- Tajima, F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 1989, 123, 585–595. [Google Scholar]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2021; Available online: https://www.R-project.org/ (accessed on 15 June 2023).
- Maechler, M.; Rousseeuw, P.; Struyf, A.; Hubert, M.; Hornik, K. Cluster: Cluster Analysis Basics and Extensions. R Package Version 2.1.4. 2022. Available online: https://CRAN.R-project.org/package=cluster (accessed on 15 June 2023).
- Kassambara, A.; Mundt, F. Factoextra: Extract and Visualize the Results of Multivariate Data Analyses. R Package Version 1.0.7. 2020. Available online: https://CRAN.R-project.org/package=factoextra (accessed on 15 June 2023).
- Galili, T. Dendextend: An R package for visualizing, adjusting, and comparing trees of hierarchical clustering. Bioinformatics 2015, 31, 3718–3720. [Google Scholar] [CrossRef]
- Charrad, M.; Ghazzali, N.; Boiteau, V.; Niknafs, A. NbClust: An r package for determining the relevant number of clusters in a data set. J. Stat. Softw. 2014, 61, 1–36. [Google Scholar] [CrossRef]
- Sokal, R.R.; Rohlf, F.J. The comparison of dendrograms by objective methods. Taxon 1962, 11, 33–40. [Google Scholar] [CrossRef]
- Guo, Y.S.; Crous, P.W.; Bai, Q.; Fu, M.; Yang, M.M.; Wang, X.H.; Du, Y.M.; Hong, N.; Xu, W.X.; Wang, G.P. High diversity of Diaporthe species associated with pear shoot canker in China. Persoonia 2020, 45, 132–162. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Hou, C.L. Three new species of Diaporthe from China based on morphological characters and DNA sequence data analyses. Phytotaxa 2019, 422, 157–174. [Google Scholar] [CrossRef]
- Chang, C.Q.; Cheng, Y.H.; Xiang, M.H.; Jiang, Z.D.; Chi, P.K. New species of Phomopsis on woody plants in Fujian province. Mycosystema 2005, 24, 6–11. [Google Scholar]
- Yang, Q.; Tang, J.; Zhou, G.Y. Characterization of Diaporthe species on Camellia oleifera in Hunan Province, with descriptions of two new species. MycoKeys 2021, 84, 15–33. [Google Scholar] [CrossRef]
- Crous, P.W.; Summerell, B.A.; Shivas, R.G.; Romberg, M.; Mel’nik, V.A.; Verkley, G.J.M.; Groenewald, J.Z. Fungal Planet description sheets: 92–106. Persoonia 2011, 27, 130–162. [Google Scholar] [CrossRef]
- Yang, Q.; Fan, X.L.; Guarnaccia, V.; Tian, C.M. High diversity of Diaporthe species associated with dieback diseases in China, with twelve new species described. MycoKeys 2018, 39, 97–149. [Google Scholar] [CrossRef]
- Huang, S.T.; Xia, J.W.; Zhang, X.G.; Sun, W.X. Morphological and phylogenetic analyses reveal three new species of Diaporthe from Yunnan, China. MycoKeys 2021, 78, 49–77. [Google Scholar] [CrossRef]
- Perera, R.H.; Hyde, K.D.; Maharachchikumbura, S.S.N.; Jones, E.B.G.; McKenzie, E.H.C.; Stadler, M.; Lee, H.B.; Samarakoon, M.C.; Ekanayaka, A.H.; Camporesi, E.; et al. Fungi on wild seeds and fruits. Mycosphere 2020, 11, 2108–2480. [Google Scholar] [CrossRef]
- Wrona, C.J.; Mohankumar, V.; Schoeman, M.H.; Tan, Y.P.; Shivas, R.G.; Jeff-Ego, O.S.; Akinsanmi, O.A. Phomopsis husk rot of macadamia in Australia and South Africa caused by novel Diaporthe species. Plant Pathol. 2020, 69, 911–921. [Google Scholar] [CrossRef]
- Dong, Z.; Manawasinghe, I.S.; Huang, Y.; Shu, Y.; Phillips, A.J.L.; Dissanayake, A.J.; Hyde, K.D.; Xiang, M.; Luo, M. Endophytic Diaporthe associated with Citrus grandis cv. Tomentosa in China. Front. Microbiol. 2021, 11, 1–20. [Google Scholar] [CrossRef]
- Dayarathne, M.C.; Jones, E.B.G.; Maharachchikumbura, S.S.N.; Devadatha, B.; Sarma, V.V.; Khongphinitbunjong, K.; Chomnunti, P.; Hyde, K.D. Morpho-molecular characterization of microfungi associated with marine based habitats. Mycosphere 2020, 11, 1–188. [Google Scholar] [CrossRef]
- Rossman, A.Y.; Allen, W.C.; Braun, U.; Castlebury, L.A.; Chaverri, P.; Crous, P.W.; Hawksworth, D.L.; Hyde, K.D.; Johnston, P.; Lombard, L.; et al. Overlooked competing asexual and sexually typified generic names of Ascomycota with recommendations for their use or protection. IMA Fungus 2016, 7, 289–308. [Google Scholar] [CrossRef]
- Chang, C.Q.; Xi, P.G.; Xiang, M.M.; Jiang, Z.D.; Chi, P.K. New species of Phomopsis on woody plants in Hunan province. Mycosystema 2005, 24, 145–154. [Google Scholar]
- Cao, L.; Luo, D.; Lin, W.; Yang, Q.; Deng, X. Four new species of Diaporthe (Diaporthaceae, Diaporthales) from forest plants in China. MycoKeys 2022, 91, 25–47. [Google Scholar] [CrossRef]
- Long, H.; Zhang, Q.; Hao, Y.Y.; Shao, X.Q.; Wei, X.X.; Hyde, K.D.; Wang, Y.; Zhao, D.G. Diaporthe species in south-western China. MycoKeys 2019, 57, 113–127. [Google Scholar] [CrossRef]
- Crous, P.W.; Groenewald, J.Z.; Shivas, R.G.; Edwards, J.; Seifert, K.A.; Alfenas, A.C.; Alfenas, R.F.; Burgess, T.I.; Carnegie, A.J.; Hardy, G.S.J.; et al. Fungal Planet description sheets: 69–91. Persoonia 2011, 26, 108–156. [Google Scholar] [CrossRef]
- Chen, K.-L.; Kirschner, R. Fungi from leaves of lotus (Nelumbo nucifera). Mycol. Prog. 2017, 17, 275–293. [Google Scholar] [CrossRef]
- Yang, Q.; Jiang, N.; Tian, C.M. New species and records of Diaporthe from Jiangxi Province, China. MycoKeys 2021, 77, 41–64. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Fan, X.L.; Du, Z.; Tian, C.M. Diaporthe species occurring on Senna bicapsularis in southern China, with descriptions of two new species. Phytotaxa 2017, 302, 145–155. [Google Scholar] [CrossRef]
- Ariyawansa, H.A.; Tsai, I.; Withee, P.; Tanjira, M.; Yen, C.Y.; Al-Rashed, S.; Elgorban, A.M.; Cheewangkoon, R. Diaporthe taiwanensis: A new taxon causing leaf spots and necrosis on Ixora chinensis in Taiwan. Phytotaxa 2020, 461, 155–165. [Google Scholar] [CrossRef]
- Abeywickrama, P.D.; Qian, N.; Jayawardena, R.S.; Li, Y.; Zhang, W.; Guo, K.; Zhang, L.; Zhang, G.Z.; Yan, J.; Li, X.; et al. Endophytic fungi in green manure crops; friends or foe? Mycosphere 2023, 14, 1–106. [Google Scholar] [CrossRef]
- Tan, Y.P.; Shivas, R.G. Nomenclatural novelties. Index Austral. Fungi 2022, 2, 1–12. [Google Scholar]
- Yang, E.F.; Karunarathna, S.C.; Dai, D.Q.; Stephenson, S.L.; Elgorban, A.M.; Al-Rejaie, S.; Xiong, Y.R.; Promputtha, I.; Samarakoon, M.C.; Tibpromma, S. Taxonomy and phylogeny of fungi associated with Mangifera indica from Yunnan, China. J. Fungi 2022, 8, 1249. [Google Scholar] [CrossRef]
- Tibpromma, S.; Hyde, K.D.; Bhat, J.D.; Mortimer, P.E.; Xu, J.C.; Promputtha, I.; Doilom, M.; Yang, J.B.; Tang, A.M.C.; Karunarathna, S.C. Identification of endophytic fungi from leaves of Pandanaceae based on their morphotypes and DNA sequence data from southern Thailand. MycoKeys 2018, 33, 25–67. [Google Scholar] [CrossRef]
- Farr, D.F.; Rossman, A.Y. Fungal Databases, US National Fungus Collections, ARS, USDA. 2023. Available online: https://nt.ars-grin.gov/fungaldatabases/ (accessed on 15 August 2023).
- Punithalingam, E. New species of Phomopsis. Trans. Br. Mycol. Soc. 1974, 63, 229–236. [Google Scholar] [CrossRef]
- Zerova, M.Y. Dekilyka novikh dlya SSSR vidiv Phomopsis. Bot. Zhurn. SSSR 1940, 1, 305–312. [Google Scholar]
- Taylor, J.W.; Turner, E.; Townsend, J.P.; Dettman, J.R.; Jacobson, D. Eukaryotic microbes, species recognition and the geographic limits of species: Examples from the kingdom Fungi. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2006, 361, 1947–1963. [Google Scholar] [CrossRef]
- Giraud, T.; Refrégier, G.; Le Gac, M.; de Vienne, D.M.; Hood, M.E. Speciation in fungi. Fungal Genet. Biol. 2008, 45, 791–802. [Google Scholar] [CrossRef]
- Cai, L.; Giraud, T.; Zhang, N.; Begerow, D.; Cai, G.; Shivas, R.G. The evolution of species concepts and species recognition criteria in plant pathogenic fungi. Fungal Divers. 2011, 50, 121–133. [Google Scholar] [CrossRef]
- Coetzee, M.P.A.; Wingfield, B.D.; Bloomer, P.; Wingfield, M.J. Phylogenetic analyses of DNA sequences reveal species partitions amongst isolates of Armillaria from Africa. Mycol. Res. 2005, 109, 1223–1234. [Google Scholar] [CrossRef] [PubMed]
- Aoki, T.; O’Donell, K.; Scandiani, M.M. Sudden death syndrome of soybean in South America is caused by four species of Fusarium: Fusarium brasiliense sp. nov., F. cuneirostrum sp. nov., F. tucumaniae, and F. virguliforme. Mycoscience 2005, 46, 162–183. [Google Scholar] [CrossRef]
- Laurence, M.H.; Summerell, B.A.; Burgess, L.W.; Liew, E.C.Y. Genealogical concordance phylogenetic species recognition in the Fusarium oxysporum species complex. Fungal Biol. 2014, 118, 374–384. [Google Scholar] [CrossRef] [PubMed]
- Mejia, L.C.; Castlebury, L.A.; Rossman, A.Y.; Sogonov, M.V.; White, J.F. A systematic account of the genus Plagiostoma (Gnomoniaceae, Diaporthales) based on morphology, host associations and a four gene phylogeny. Stud. Mycol. 2011, 68, 211–235. [Google Scholar] [CrossRef]
- Wang, X.H.; Chen, G.Q.; Huang, F.; Zhang, J.Z.; Hyde, K.D.; Li, H.G. Phyllosticta species associated with citrus diseases in China. Fungal Divers. 2011, 52, 209–224. [Google Scholar] [CrossRef]
- Cai, L.; Hyde, K.D.; Taylor, P.W.J.; Weir, B.S.; Waller, J.; Abang, M.M.; Zhang, J.Z.; Yang, Y.L.; Phoulivong, S.; Liu, Z.Y.; et al. A polyphasic approach for studying Colletotrichum. Fungal Divers. 2009, 39, 183–204. [Google Scholar]
- Crouch, J.A.; Clarke, B.B.; Hillman, B.I. What is the value of ITS sequence data in Colletotrichum systematics and species diagnosis? A case study using the falcate-spored graminicolous Colletotrichum group. Mycologia 2009, 101, 648–656. [Google Scholar] [CrossRef]
- Phoulivong, S.; Cai, L.; Chen, H.; McKenzie, E.H.C.; Abdelsalam, K.; Chukeatirote, E.; Hyde, K.D. Colletotrichum gloeosporioides is not a common pathogen on tropical fruits. Fungal Divers. 2010, 44, 33–43. [Google Scholar] [CrossRef]
- Lombard, L.; Crous, P.W.; Wingfield, B.D.; Wingfield, M.J. Phylogeny and systematics of the genus Calonectria. Stud. Mycol. 2010, 66, 31–69. [Google Scholar] [CrossRef] [PubMed]
- Hipp, A.L.; Hall, J.C.; Sytsma, K.J. Congruence versus phylogenetic accuracy: Revisiting the incongruence length difference test. Syst. Biol. 2004, 53, 81–89. [Google Scholar] [CrossRef]
- Milgroom, M.G. Recombination and the multilocus structure of fungal populations. Annu. Rev. Phytopathol. 1996, 34, 457–477. [Google Scholar] [CrossRef]
- Geiser, D.M.; Pitt, J.I.; Taylor, J.W. Cryptic speciation and recombination in the aflatoxin-producing fungus Aspergillus flavus. Proc. Natl. Acad. Sci. USA 1998, 95, 388–393. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, K.; Kistler, H.C.; Tacke, B.K.; Casper, H.C. Gene genealogies reveal global phylogeographic structure and reproductive isolation among lineages of Fusarium graminearum, the fungus causing wheat scab. Proc. Natl. Acad. Sci. USA 2000, 97, 7905–7910. [Google Scholar] [CrossRef]
- Matute, D.R.; McEwen, J.G.; Puccia, R.; Montes, B.A.; San-Blas, G.; Bagagli, E.; Taylor, J.W. Cryptic speciation and recombination in the fungus Paracoccidioides brasiliensis as revealed by gene genealogies. Mol. Biol. Evol. 2006, 23, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Mendez-Harclerode, F.M.; Strauss, R.E.; Fulhorst, C.F.; Milazzo, M.L.; Ruthven, D.C.; Bradley, R.D. Molecular evidence for high levels of intrapopulation genetic diversity in woodrats (Neotoma micropus). J. Mammal. 2007, 88, 360–370. [Google Scholar] [CrossRef]
- Grant, W.S.; Bowen, B.W. Shallow population histories in deep evolutionary lineages of marine fishes: Insights from sardines and anchovies and lessons for conservation. Genetics 1998, 89, 415–426. [Google Scholar] [CrossRef]
- Korneliussen, T.S.; Moltke, I.; Albrechtsen, A.; Nielsen, R. Calculation of Tajima’s D and other neutrality test statistics from low depth next-generation sequencing data. BMC Bioinf. 2013, 14, 289. [Google Scholar] [CrossRef]
- Carlson, C.S.; Thomas, D.J.; Eberle, M.; Livingston, R.; Rieder, M.; Nickerson, D.A. Genomic regions exhibiting positive selection identified from dense genotype data. Genome Res. 2005, 15, 1553–1565. [Google Scholar] [CrossRef]
- van Iersel, L.; Kelk, S.; Rupp, R.; Huson, D. Phylogenetic networks do not need to be complex: Using fewer reticulations to represent conflicting clusters. Bioinformatics 2010, 26, i124–i131. [Google Scholar] [CrossRef]
- Chen, S.; Kim, D.-K.; Chase, M.W.; Kim, J.-H. Networks in a large-scale phylogenetic analysis: Reconstructing evolutionary history of Asparagales (Lilianae) based on four plastid genes. PLoS ONE 2013, 8, e59472. [Google Scholar] [CrossRef]
- Linde, C.C. Population genetic analyses of plant pathogens: New challenges and opportunities. Australas. Plant Pathol. 2010, 39, 23e28. [Google Scholar] [CrossRef]
- Barton, N.H. Mutation and the evolution of recombination. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2010, 365, 1281–1294. [Google Scholar] [CrossRef]
- Croll, D.; Lendenmann, M.H.; Stewart, E.; McDonald, B.A. The impact of recombination hotspots on genome evolution of a fungal plant pathogen. Genetics 2015, 201, 1213–1228. [Google Scholar] [CrossRef] [PubMed]
- Matute, D.R.; Sepúlveda, V.E. Fungal species boundaries in the genomics era. Fungal Genet. Biol. 2019, 131, 103249. [Google Scholar] [CrossRef] [PubMed]
- Schoch, C.L.; Seifert, K.A.; Huhndorf, S.; Robert, V.; Spouge, J.L.; Levesque, C.A.; Chen, W.; Fungal Barcoding Consortium. Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for fungi. Proc. Natl. Acad. Sci. USA 2012, 109, 6241–6246. [Google Scholar] [CrossRef] [PubMed]
- Vu, D.; Groenewald, M.; de Vries, M.; Gehrmann, T.; Stielow, B.; Eberhardt, U.; Al-Hatmi, A.; Groenewald, J.Z.; Cardinali, G.; Houbraken, J.; et al. Large-scale generation and analysis of filamentous fungal DNA barcodes boosts coverage for kingdom fungi and reveals thresholds for fungal species and higher taxon delimitation. Stud. Mycol. 2019, 92, 135–154. [Google Scholar] [CrossRef]
- Gazis, R.; Rehner, S.; Chaverri, P. Species delimitation in fungal endophyte diversity studies and its implications in ecological and biogeographic inferences. Mol. Ecol. 2011, 20, 3001–3013. [Google Scholar] [CrossRef]
- Hibbett, D.S.; Ohman, A.; Glotzer, D.; Nuhn, M.; Kirk, P.; Nilsson, R.H. Progress in molecular and morphological taxon discovery in fungi and options for forma classification of environmental sequences. Fungal Biol. Rev. 2011, 25, 38–47. [Google Scholar] [CrossRef]
- Hughes, K.W.; Tulloss, R.H.; Petersen, R.H. Intragenomic nuclear RNA variation in a cryptic Amanita taxon. Mycologia 2018, 110, 93–103. [Google Scholar] [CrossRef]
- Piganeau, G.; Eyre-Walker, A. A reanalysis of the indirect evidence for recombination in human mitochondrial DNA. Heredity 2004, 92, 282–288. [Google Scholar] [CrossRef] [PubMed]
- Naranjo-Ortiz, M.A.; Gabaldón, T. Fungal evolution: Major ecological adaptations and evolutionary transitions. Biol. Rev. Camb. Philos. Soc. 2019, 94, 1443–1476. [Google Scholar] [CrossRef]
- Chethana, K.W.T.; Manawasinghe, I.S.; Hurdeal, V.G.; Bhunjun, C.S.; Appadoo, M.A.; Gentekaki, E.; Raspé, O.; Promputtha, I.; Hyde, K.D. What are fungal species and how to delineate them? Fungal Divers. 2021, 109, 1–25. [Google Scholar] [CrossRef]
- Stengel, A.; Stanke, K.M.; Quattrone, A.C.; Herr, J.R. Improving taxonomic delimitation of fungal species in the age of genomics and phenomics. Front. Microbiol. 2022, 13, 847067. [Google Scholar] [CrossRef]
- Harrington, T.C.; Rizzo, D.M. Defining species in the fungi. In Structure and Dynamics of Fungal Populations; Worrall, J.J., Ed.; Springer Science & Business Media: Dordrecht, The Netherlands, 1999; Volume 25, pp. 43–71. [Google Scholar]
- Steenkamp, E.T.; Wingfield, M.J.; McTaggart, A.R.; Wingfield, B.D. Fungal species and their boundaries matter e definitions, mechanisms and practical implications. Fung. Biol. Rev. 2018, 32, 104–116. [Google Scholar] [CrossRef]
- Brayford, D. Variation in Phomopsis isolates from Ulmus species in the British Isles and Italy. Mycol. Res. 1990, 94, 691–697. [Google Scholar] [CrossRef]
- Crous, P.W.; Groenewald, J.Z. Hosts, species and genotypes: Opinions versus data. Australas. Plant Pathol. 2005, 34, 463–470. [Google Scholar] [CrossRef]
- Hyde, K.D.; Jeewon, R.; Chen, Y.J.; Bhunjun, C.S.; Calabon, M.S.; Jiang, H.B.; Lin, C.-G.; Norphanphoun, C.; Sysouphathong, P.; Pem, D.; et al. The numbers of fungi: Is the descriptive curve flattening? Fungal Divers. 2020, 103, 219–271. [Google Scholar] [CrossRef]
- Hilário, S.; Gonçalves, M.F.M. Mechanisms underlying the pathogenic and endophytic lifestyles in Diaporthe: An omics-based approach. Horticulturae 2023, 9, 423. [Google Scholar] [CrossRef]
- McCartney, H.A.; Foster, S.J.; Fraaije, B.A.; Ward, E. Molecular diagnostics for fungal plant pathogens. Pest. Manag. Sci. 2003, 59, 129–142. [Google Scholar] [CrossRef] [PubMed]
- Anderson, P.K.; Cunningham, A.A.; Patel, N.G.; Morales, F.J.; Epstein, P.R.; Daszak, P. Emerging infectious diseases of plants: Pathogen pollution, climate change and agrotechnology drivers. Trends Ecol. Evol. 2004, 19, 535–544. [Google Scholar] [CrossRef] [PubMed]
- Desprez-Loustau, M.-L.; Marçais, B.; Nageleisen, L.-M.; Piou, D.; Vannini, A. Interactive effects of drought and pathogens in forest trees. Ann. For. Sci. 2006, 63, 597–612. [Google Scholar] [CrossRef]
- Desprez-Loustau, M.-L.; Robin, C.; Reynaud, G.; Déqué, M.; Badeau, V.; Piou, D.; Husson, C.; Marcais, B. Simulating the effects of a climate-change scenario on the geographical range and activity of forest-pathogenic fungi. Can. J. Plant Pathol. 2007, 29, 101–120. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Taxon 1 | Culture 2 and Status 3 | Host | Country | GenBank Accession Number 4 | ||||
|---|---|---|---|---|---|---|---|---|
| ITS | tef1 | tub2 | cal | his3 | ||||
| Diaporthe arecae | CBS 535.75 | Citrus sp. | Suriname | KC343033 | KC343759 | KC344001 | KC343275 | KC343517 |
| CBS 161.64IT | Areca catechu | India | KC343032 | KC343758 | KC344000 | KC343274 | KC343516 | |
| SM30 | Calamus castaneus | Malaysia | MN651492 | MT077090 | MT077061 | N/A | N/A | |
| D. arecae (“D. eugeniae”) | CBS 444.82 | Eugenia aromatica | Indonesia | KC343098 | KC343824 | KC344066 | KC343340 | KC343582 |
| D. arecae (“D. perseae”) | CBS 151.73 | Persea americana | Netherlands | KC343173 | KC343899 | KC344141 | KC343415 | KC343657 |
| D. arecae (syn. D. acuta) | CGMCC 3.19600T | Pyrus pyrifolia | China | MK626957 | MK654802 | MK691124 | MK691225 | MK726161 |
| PSCG 045 | Pyrus pyrifolia | China | MK626956 | MK654809 | MK691223 | MK691123 | MK726160 | |
| D. arecae (syn. D. anhuiensis) | CNUCC 201901T | Cunninghamia lanceolata | China | MN219718 | MN224668 | MN227008 | MN224549 | MN224556 |
| CNUCC 201902PT | Cunninghamia lanceolata | China | MN219727 | MN224669 | MN227009 | MN224550 | MN224557 | |
| D. arecae (syn. D. arengae) | CBS 114979T | Arenga engleri | Hong Kong | KC343034 | KC343760 | KC344002 | KC343276 | KC343518 |
| SM28 | Calamus castaneus | Malaysia | MN651480 | MT077093 | MT077062 | N/A | N/A | |
| SM29 | Calamus castaneus | Malaysia | MN651482 | MT077094 | MT077063 | N/A | N/A | |
| SM35 | Calamus castaneus | Malaysia | MN651483 | MT077099 | MT077068 | N/A | N/A | |
| SM38 | Calamus castaneus | Malaysia | MN651484 | MT077097 | MT077066 | N/A | N/A | |
| SM39 | Calamus castaneus | Malaysia | MN651485 | MT077098 | MT077067 | N/A | N/A | |
| SM41 | Calamus castaneus | Malaysia | MN651481 | MT077095 | MT077064 | N/A | N/A | |
| SM45 | Calamus castaneus | Malaysia | MN651486 | MT077096 | MT077065 | N/A | N/A | |
| SM49 | Calamus castaneus | Malaysia | MN651487 | MT077089 | MT077069 | N/A | N/A | |
| D. arecae (syn. D. averrhoae) | SCHM 3605H | Averrhoa carambola | China | AY618930 | N/A | N/A | N/A | N/A |
| D. arecae (syn. D. bounty) | BRIP 59361aH | Malus domestica | Australia | OM918690 | OM960599 | OM960617 | N/A | N/A |
| D. arecae (syn. D. camelliae-oleiferae) | HNZZ027T | Camellia oleifera | China | MZ509555 | MZ504707 | MZ504718 | MZ504685 | MZ504696 |
| HNZZ030 | Camellia oleifera | China | MZ509556 | MZ504708 | MZ504719 | MZ504686 | MZ504697 | |
| D. arecae (syn. D. ceratozamiae) | CBS 131306T | Ceratozamia robusta | Australia | JQ044420 | N/A | N/A | N/A | N/A |
| D. arecae (syn. D. cercidis) | CFCC 52565T | Cercis chinensis | China | MH121500 | MH121542 | MH121582 | MH121424 | MH121460 |
| CFCC 52566 | Cercis chinensis | China | MH121501 | MH121543 | MH121583 | MH121425 | MH121461 | |
| D. arecae (syn. D. chamaeropicola) | CDP 0460T | Chamaerops humilis | Portugal | MT022111 | MT011074 | MT011080 | MT011068 | N/A |
| D. arecae (syn. D. chrysalidocarpi) | SAUCC 194.33PT | N/A | China | MT822561 | MT855874 | MT855758 | MT855645 | MT855530 |
| SAUCC 194.35T | N/A | China | MT822563 | MT855876 | MT855760 | MT855646 | MT855532 | |
| D. arecae (syn. D. delonicis) | MFLU 16-1059H | Delonix regia | Thailand | MT215490 | N/A | MT212209 | N/A | N/A |
| D. arecae (syn. D. drenthii) | BRIP 66523 | Macadamia sp. | South Africa | MN708228 | MN696525 | MN696536 | N/A | N/A |
| BRIP 66524T | Macadamia sp. | South Africa | MN708229 | MN696526 | MN696537 | N/A | N/A | |
| D. arecae (syn. D. endocitricola) | ZHKUCC 20-0012T | Citrus grandis | China | MT355682 | MT409336 | MT409290 | MT409312 | N/A |
| ZHKUCC 20-0013PT | Citrus grandis | China | MT355683 | MT409337 | MT409291 | MT409313 | N/A | |
| D. arecae (syn. D. fraxini-angustifoliae) | BRIP 54781IT | Fraxinus angustifolia | Australia | JX862528 | JX862534 | KF170920 | N/A | N/A |
| MFLUCC 15-0748 | Vitis vinifera | China | KT459428 | KT459446 | KT459430 | KT459462 | N/A | |
| D. arecae (syn. D. fulvicolor) | CGMCC 3.19601T | Pyrus pyrifolia | China | MK626859 | MK654806 | MK691236 | MK691132 | MK726163 |
| PSCG 057 | Pyrus pyrifolia | China | MK626858 | MK654810 | MK691233 | MK691131 | MK726164 | |
| D. arecae (syn. D. gossiae) | BRIP 59730aH | Sesbania sp. | Australia | OM918693 | OM960602 | OM960620 | N/A | N/A |
| D. arecae (syn. D. guangxiensis) | JZB 320091 | Vitis vinifera | China | MK335769 | MK523564 | MK500165 | MK736724 | N/A |
| JZB 320094T | Vitis vinifera | China | MK335772 | MK523566 | MK500168 | MK736727 | N/A | |
| D. arecae (syn. D. hongheensis) | KUMCC 21-0457T | Mangifera indica | China | OM001331 | ON468649 | ON468658 | ON715010 | N/A |
| KUMCC 21-0458 | Mangifera indica | China | OM001330 | ON468650 | ON468659 | ON715009 | N/A | |
| D. arecae (syn. D. howardiae) | BRIP 59697aH | Agave sp. | Australia | OM918695 | OM960604 | OM960622 | N/A | N/A |
| D. arecae (syn. D. huangshanensis) | CNUCC 201903T | Camellia oleifera | China | MN219729 | MN224670 | MN227010 | N/A | MN224558 |
| CNUCC 201904PT | Camellia oleifera | China | MN219730 | MN224671 | MN227011 | N/A | MN224559 | |
| D. arecae (syn. D. hunanensis) | HNZZ023T | Camellia oleifera | China | MZ509550 | MZ504702 | MZ504713 | MZ504680 | MZ504691 |
| HNZZ025 | Camellia oleifera | China | MZ509551 | MZ504703 | MZ504714 | MZ504681 | MZ504692 | |
| D. arecae (syn. D. krabiensis) | MFLUCC 17-2481T | Submerged wood | Thailand | MN047101 | MN433215 | MN431495 | N/A | N/A |
| D. arecae (syn. D. limonicola) | CBS 142549T | Citrus limon | Malta | NR_154980 | MF418501 | MF418582 | MF418256 | MF418342 |
| CBS 142550 | Citrus limon | Malta | MF418423 | MF418502 | MF418583 | MF418257 | MF418343 | |
| CPC 27869 | Citrus limon | Malta | MF418419 | MF418498 | MF418579 | MF418253 | MF418339 | |
| HNHK02 | Areca catechu | China | MN424515 | MN424557 | MN424529 | MN424571 | MN424543 | |
| HNQH03 | Areca catechu | China | MN424526 | MN424568 | MN424540 | MN424582 | MN424554 | |
| HNQH02 | Areca catechu | China | MN424525 | MN424567 | MN424539 | MN424581 | MN424553 | |
| D. arecae (syn. D. liquidambaris) | SCHM 3621H | Liquidambar formosana | China | AY601919 | N/A | N/A | N/A | N/A |
| D. arecae (syn. D. litchiicola) | BRIP 54900T | Litchi chinensis | Australia | JX862533 | JX862539 | KF170925 | N/A | N/A |
| D. arecae (syn. D. loropetali) | SCHM 3615H | Loropetalum chinense | China | AY601917 | N/A | N/A | N/A | N/A |
| D. arecae (syn. D. meliae) | CFCC 53089T | Melia azedarach | China | MK432657 | ON081654 | MK578057 | N/A | ON081662 |
| CFCC 53090 | Melia azedarach | China | MK432658 | ON081655 | MK578058 | N/A | ON081663 | |
| D. arecae (syn. D. melitensis) | CBS 142551T | Citrus limon | Malta | MF418424 | MF418503 | MF418584 | MF418258 | MF418344 |
| CPC 27875 | Citrus limon | Malta | MF418425 | MF418504 | MF418585 | MF418259 | MF418345 | |
| D. arecae (syn. D. millettiae) | GUCC 9167T | Plant foliage | China | MK398674 | MK480609 | MK502089 | MK502086 | N/A |
| MFLUCC 20-0183 | Celtis formosana | China | MW114351 | MW192214 | MW148271 | MW151589 | N/A | |
| D. arecae (syn. D. musigena) | CBS 129519T | Musa sp. | Australia | KC343143 | KC343869 | KC344111 | KC343385 | KC343627 |
| D. arecae (syn. D. nelumbonis) | BCRC FU30382R | Nelumbo nucifera | China | KT821501 | N/A | LC069368 | N/A | N/A |
| D. arecae (syn. D. norfolkensis) | BRIP 59718aH | Mangifera indica | Australia | OM918699 | OM960608 | OM960626 | N/A | N/A |
| D. arecae (syn. D. oculi) | MAFF 246252T | Homo sapiens | Japan | LC373514 | LC373516 | LC373518 | N/A | N/A |
| D. arecae (syn. D. osmanthi) | GUCC 9165T | Camellia sinensis | China | MK398675 | MK480610 | MK502091 | MK502087 | N/A |
| SAUCC 194.21 | Camellia sinensis | China | MT822549 | MT855862 | MT855746 | MT855634 | MT855518 | |
| D. arecae (syn. D. pandanicola) | MFLUCC 17-0607T | Pandanus sp. | Thailand | MG646974 | N/A | MG646930 | N/A | N/A |
| SAUCC 194.82 | Milletia reticulata | China | MT822610 | MT855922 | MT855807 | MT855689 | MT855578 | |
| D. arecae (syn. D. pascoei) | BRIP 54847IT | Persea americana | Australia | JX862532 | JX862538 | KF170924 | N/A | N/A |
| D. arecae (syn. D. pescicola) | MFLUCC 16-0105T | Prunus persica | China | KU557555 | KU557623 | KU557579 | KU557603 | N/A |
| MFLUCC 16-0108 | Prunus persica | China | KU557558 | KU557626 | KU557582 | KU557606 | N/A | |
| PSCG 036 | Pyrus × bretschneideri | China | MK626855 | MK654796 | MK691226 | MK691116 | MK726159 | |
| PSCG 037 | Pyrus × bretschneideri | China | MK626857 | MK654799 | MK691230 | MK691130 | MK726157 | |
| D. arecae (syn. D. phyllanthicola) | SCHM 3680H | Phyllanthus emblicae | China | AY620819 | N/A | N/A | N/A | N/A |
| D. arecae (syn. D. podocarpi-macrophylli) | CGMCC 3.18281T | Podocarpus macrophyllus | Japan | KX986774 | KX999167 | KX999207 | KX999278 | KX999246 |
| LC 6229 | Olea europaea | Italy | KX986771 | KX999164 | KX999204 | KX999277 | KX999243 | |
| D. arecae (syn. D. pseudomangiferae) | CBS 101339T | Mangifera indica | Dominican Republic | KC343181 | KC343907 | KC344149 | KC343423 | KC343665 |
| CBS 388.89 | Mangifera indica | Mexico | KC343182 | KC343908 | KC344150 | KC343424 | KC343666 | |
| D. arecae (syn. D. pseudooculi) | MAFF 246452T | Homo sapiens | Japan | LC373515 | LC373517 | LC373519 | N/A | N/A |
| D. arecae (syn. D. pseudophoenicicola) | CBS 176.77 | Mangifera indica | Iraq | KC343183 | KC343909 | KC344151 | KC343425 | KC343667 |
| CBS 462.69T | Phoenix dactylifera | Spain | KC343184 | KC343910 | KC344152 | KC343426 | KC343668 | |
| CDP 0047 | Chamaerops humilis | Portugal | MT002357 | MT011069 | MT011075 | MT011065 | N/A | |
| CDP 0358 | Phoenix dactylifera | Portugal | MT004743 | MT011073 | MT011079 | MT011067 | N/A | |
| HNHK01 | Areca catechum | China | MN424514 | MN424556 | MN424528 | MN424570 | MN424542 | |
| HNHK03 | Areca catechum | China | MN424516 | MN424558 | MN424530 | MN424572 | MN424544 | |
| HNQZ01 | Areca catechum | China | MN424520 | MN424562 | MN424534 | MN424576 | MN424548 | |
| HNWC01 | Areca catechum | China | MN424517 | MN424559 | MN424531 | MN424573 | MN424545 | |
| HNWC02 | Areca catechum | China | MN424518 | MN424560 | MN424532 | MN424574 | MN424546 | |
| HNWN03 | Areca catechum | China | MN424524 | MN424566 | MN424538 | MN424580 | MN424552 | |
| LC 6150 | Phoenix canariensis | Uruguay | KY011891 | KY011902 | N/A | N/A | N/A | |
| LC 6151 | Phoenix canariensis | Uruguay | KY011892 | KY011903 | N/A | N/A | N/A | |
| D. arecae (syn. D. pterocarpicola) | MFLUCC 10-0580aT | Pterocarpus indicus | Thailand | JQ619887 | JX275403 | JX275441 | JX197433 | N/A |
| MFLUCC 10-0580bIT | Pterocarpus indicus | Thailand | JQ619888 | JX275404 | JX275442 | JX197434 | N/A | |
| D. arecae (syn. D. schimae) | CFCC 53103T | Schima superba | China | MK432640 | MK578116 | MK578043 | MK442962 | MK442987 |
| CFCC 53104 | Schima superba | China | MK432641 | MK578117 | MK578044 | MK442963 | MK442988 | |
| D. arecae (syn. D. searlei) | BRIP 66528T | Macadamia sp. | South Africa | MN708231 | N/A | MN696540 | N/A | N/A |
| D. arecae (syn. D. sennae) | CFCC 51636T | Cassia bicapsularis | China | KY203724 | KY228885 | KY228891 | KY228875 | N/A |
| CFCC 51637PT | Cassia bicapsularis | China | KY203725 | KY228886 | KY228892 | KY228876 | N/A | |
| D. arecae (syn. D. spinosa) | CGMCC 3.19602T | Pyrus pyrifolia | China | MK626849 | MK654811 | MK691234 | MK691129 | MK726156 |
| PSCG 388 | Pyrus pyrifolia | China | MK626860 | MK654798 | MK691229 | MK691128 | MK726171 | |
| D. arecae (syn. D. taiwanensis) | NTUCC 18-105-1T | Ixora sp. | China | MT241257 | MT251199 | MT251202 | MT251196 | N/A |
| NTUCC 18-105-2 | Ixora sp. | China | MT241258 | MT251200 | MT251203 | MT251197 | N/A | |
| D. arecae (syn. D. taoicola) | MFLUCC 16-0117T | Prunus persica | China | KU557567 | KU557635 | KU557591 | N/A | N/A |
| MFLUCC 16-0118 | Prunus persica | China | KU557568 | KU557636 | KU557592 | N/A | N/A | |
| PSCG 485 | Pyrus pyrifolia | China | MK626869 | MK654812 | MK691227 | MK691120 | MK726173 | |
| D. arecae (syn. D. viciae) | JZB 320179T | Vicia villosa | China | OP626092 | OP627280 | OP627281 | N/A | OP627279 |
| D. arecae (syn. D. viniferae) | JZB 320071T | Vitis vinifera | China | MK341550 | MK500107 | MK500112 | MK500119 | N/A |
| JZB 320072 | Vitis vinifera | China | MK341551 | MK500108 | MK500113 | MK500120 | N/A | |
| D. arecae (syn. D. annellsiae) | BRIP 59731aH | Mangifera indica | Australia | OM918687 | OM960596 | OM960614 | N/A | N/A |
| D. chiangmaiensis | MFLUCC 18-0544T | Magnolia liliifera | Thailand | OK393703 | OL439483 | N/A | N/A | N/A |
| MFLUCC 18-0935 | Magnolia liliifera | Thailand | OK393704 | OL439484 | N/A | N/A | N/A | |
| D. chiangmaiensis (“D. cf. heveae 2”) | CBS 681.84 | Hevea brasiliensis | India | KC343117 | KC343843 | KC344085 | KC343359 | KC343601 |
| D. chiangmaiensis (“D. cf. heveae”) | BR74 | Calamus castaneus | Malaysia | MN651490 | MT077091 | MT077079 | N/A | N/A |
| SM36 | Calamus castaneus | Malaysia | MN651489 | MT077092 | MT077080 | N/A | N/A | |
| D. citri | CBS 134239 | Citrus sinensis | USA | KC357553 | KC357522 | KC357456 | KC357488 | MF418280 |
| CBS 135422ET | Citrus sp. | USA | KC843311 | KC843071 | KC843187 | KC843157 | MF418281 | |
| D. corylicola | CFCC 53986T | Corylus heterophylla | China | MW839880 | MW815894 | MW883977 | MW836684 | MW836717 |
| CFCC 54696 | Corylus heterophylla | China | MW839881 | MW815907 | MW883990 | MW836697 | MW836730 | |
| D. longicolla | ATCC 60325T | Glycine max | USA | KJ590728 | KJ590767 | KJ610883 | KJ612124 | KJ659188 |
| CBS 116023 | Glycine max | USA | KC343198 | KC343924 | KC344166 | KC343440 | KC343682 | |
| D. sennicola | CFCC 51634T | Cassia bicapsularis | China | KY203722 | KY228883 | KY228889 | KY228873 | KY228879 |
| CFCC 51635 | Cassia bicapsularis | China | KY203723 | KY228884 | KY228890 | KY228874 | KY228880 | |
| D. smilacicola | CFCC 54582T | Smilax glabra | China | OP955933 | OP959770 | OP959776 | OP959779 | OP959788 |
| CFCC 58764 | Smilax glabra | China | OP955934 | OP959769 | OP959775 | OP959778 | OP959787 | |
| Analysis 1 | Characters Summary | 5-loci Dataset 2 | |||||
|---|---|---|---|---|---|---|---|
| ITS | tef1 | tub2 | cal | his3 | Combined | ||
| Number of strains/number of species | 60/29, including 8/4 as outgroup taxa | ||||||
| Total characters | 490 | 341 | 376 | 461 | 456 | 2124 | |
| Invariable characters (%) | 394 (80.4%) | 168 (49.3%) | 247 (65.7%) | 292 (63.3%) | 323 (70.8%) | 1424 (67.0%) | |
| MP | Parsimony-informative characters (%) | 85 (17.3%) | 165 (48.4%) | 115 (30.6%) | 155 (33.6%) | 116 (25.4%) | 636 (29.7%) |
| Parsimony-uninformative characters | 11 | 8 | 14 | 14 | 17 | 64 | |
| Tree length (TL) | 206 | 322 | 231 | 364 | 236 | 1655 | |
| Consistency index (CI) | 0.558 | 0.730 | 0.693 | 0.629 | 0.763 | 0.555 | |
| Homoplasy index (HI) | 0.442 | 0.270 | 0.307 | 0.371 | 0.237 | 0.445 | |
| Retention index (RI) | 0.875 | 0.885 | 0.872 | 0.821 | 0.893 | 0.778 | |
| Rescaled consistency index (RC) | 0.488 | 0.646 | 0.604 | 0.516 | 0.681 | 0.431 | |
| ML/BA | Unique alignment patterns/alignment sites (%) | 112/484 (23.1%) | 173/331 (52.3%) | 132/373 (35.4%) | 183/461 (39.7%) | 151/456 (33.1%) | 751/2105 (35.7%) |
| Invariant sites (%) | 80.2% | 47.7% | 65.4% | 63.3% | 70.8% | 66.5% | |
| Undetermined characters or gaps (%) | 7.8% | 8.6% | 7.9% | 8.4% | 7.4% | 8.0% | |
| Nucleotide substitution models * | TN93 +G+I | HKY +G | TN93 +G | GTR +G+I | GTR +G | Partitioned | |
| Analysis 1 | Characters summary | 4-loci dataset 2 | |||||
| ITS | tef1 | tub2 | cal | Combined | |||
| Number of strains/number of species | 83/39, including 8/4 as outgroup taxa | ||||||
| Total characters | 490 | 341 | 376 | 461 | 1668 | ||
| Invariable characters (%) | 383 (78.2%) | 167 (49.0%) | 241 (64.1%) | 269 (58.4%) | 1060 (63.5%) | ||
| MP | Parsimony-informative characters (%) | 94 (19.2%) | 169 (49.6%) | 119 (31.6%) | 159 (34.5%) | 541 (32.4%) | |
| Parsimony-uninformative characters | 13 | 5 | 16 | 33 | 67 | ||
| Tree length (TL) | 250 | 344 | 261 | 431 | 1642 | ||
| Consistency index (CI) | 0.532 | 0.692 | 0.644 | 0.608 | 0.488 | ||
| Homoplasy index (HI) | 0.468 | 0.308 | 0.356 | 0.392 | 0.512 | ||
| Retention index (RI) | 0.887 | 0.888 | 0.867 | 0.819 | 0.767 | ||
| Rescaled consistency index (RC) | 0.472 | 0.614 | 0.558 | 0.498 | 0.374 | ||
| ML/BA | Unique alignment patterns/alignment sites (%) | 130/488 (26.6%) | 182/331 (55.0%) | 145/374 (38.8%) | 201/461 (43.6%) | 658/1654 (39.78%) | |
| Invariant sites (%) | 78.1% | 47.4% | 63.9% | 58.4% | 63.3% | ||
| Undetermined characters or gaps (%) | 8.3% | 8.6% | 8.4% | 8.3% | 8.4% | ||
| Nucleotide substitution models * | TN93 +G+I | GTR +G | GTR +G+I | GTR +G+I | Partitioned | ||
| Analysis 1 | Characters summary | 3-loci dataset 2 | |||||
| ITS | tef1 | tub2 | Combined | ||||
| Number of strains/number of species | 114/53, including 8/4 as outgroup taxa | ||||||
| Total characters | 490 | 341 | 376 | 1207 | |||
| Invariable characters (%) | 374 (76.3%) | 156 (45.7%) | 224 (59.6%) | 754 (62.5%) | |||
| MP | Parsimony-informative characters (%) | 98 (20.0%) | 174 (51.0%) | 130 (34.6%) | 402 (33.3%) | ||
| Parsimony-uninformative characters | 18 | 11 | 22 | 51 | |||
| Tree length (TL) | 303 | 476 | 358 | 1518 | |||
| Consistency index (CI) | 0.488 | 0.592 | 0.567 | 0.417 | |||
| Homoplasy index (HI) | 0.512 | 0.408 | 0.433 | 0.583 | |||
| Retention index (RI) | 0.891 | 0.851 | 0.835 | 0.758 | |||
| Rescaled consistency index (RC) | 0.435 | 0.504 | 0.473 | 0.316 | |||
| ML/BA | Unique alignment patterns/alignment sites (%) | 143/490 (29.2%) | 206/341 (60.4%) | 167/376 (44.4%) | 516/1207 (42.8%) | ||
| Invariant sites (%) | 8.3% | 11.1% | 8.9% | 9.3% | |||
| Undetermined characters or gaps (%) | 76.3% | 45.8% | 59.6% | 62.5% | |||
| Nucleotide substitution models * | TN93 +G+I | GTR +G+I | GTR +G+I | Partitioned | |||
| Dataset Tested 1 | Φw-Statistic (p-Value) 2 | ||
|---|---|---|---|
| 5-loci | 4-loci | 3-loci | |
| ITS | 0.19 (4.34 × 10−4) * | 0.23 (0.01) * | 0.27 (0.02) * |
| tef1 | 0.11 (0.02) * | 0.13 (2.94 × 10−3) * | 0.17 (1.12 × 10−3) * |
| tub2 | 0.10 (0.07) | 0.12 (9.92 × 10−3) * | 0.14 (0.23) |
| cal | 0.20 (0.10) | 0.19 (0.99) | N/A |
| his3 | 0.13 (0.61) | N/A | N/A |
| Combined | 0.16 (0.00) * | 0.19 (0.00) * | 0.22 (0.00) * |
| Dataset Tested 1 | N 2 | Measures of Genetic Diversity 2 | Tajima’s D 3 | ||||||
|---|---|---|---|---|---|---|---|---|---|
| h | S | Hd ± SD | π ± SD | η | θ | ||||
| 5-loci | ITS | 52 | 31 | 51 | 0.977 ± 0.008 | 0.030 ± 0.002 | 57 | 0.031 | −0.09823 ns |
| tef1 | 52 | 31 | 72 | 0.961 ± 0.016 | 0.037 ± 0.003 | 75 | 0.060 | −1.31045 ns | |
| tub2 | 52 | 27 | 67 | 0.963 ± 0.012 | 0.033 ± 0.001 | 71 | 0.047 | −1.06861 ns | |
| cal | 52 | 33 | 73 | 0.965 ± 0.015 | 0.032 ± 0.002 | 91 | 0.057 | −1.52753 ns | |
| his3 | 52 | 27 | 58 | 0.963 ± 0.011 | 0.021 ± 0.003 | 62 | 0.035 | −1.43297 ns | |
| Combined | 52 | 42 | 321 | 0.992 ± 0.005 | 0.030 ± 0.002 | 356 | 0.045 | −1.18300 ns | |
| 4-loci | ITS | 75 | 40 | 52 | 0.980 ± 0.005 | 0.028 ± 0.001 | 58 | 0.029 | −0.09624 ns |
| tef1 | 75 | 38 | 63 | 0.969 ± 0.009 | 0.034 ± 0.002 | 67 | 0.054 | −1.19098 ns | |
| tub2 | 75 | 36 | 67 | 0.967 ± 0.009 | 0.028 ± 0.001 | 71 | 0.047 | −1.32256 ns | |
| cal | 75 | 43 | 99 | 0.975 ± 0.008 | 0.031 ± 0.002 | 122 | 0.071 | −1.92574 ss | |
| Combined | 75 | 57 | 281 | 0.993 ± 0.003 | 0.030 ± 0.001 | 318 | 0.049 | −1.33832 ns | |
| 3-loci | ITS | 106 | 60 | 59 | 0.987 ± 0.003 | 0.028 ± 0.001 | 65 | 0.031 | −0.25953 ns |
| tef1 | 106 | 52 | 85 | 0.974 ± 0.006 | 0.035 ± 0.002 | 105 | 0.081 | −1.86768 ss | |
| tub2 | 106 | 56 | 88 | 0.980 ± 0.005 | 0.029 ± 0.001 | 107 | 0.066 | −1.83873 ss | |
| Combined | 106 | 81 | 233 | 0.995 ± 0.002 | 0.030 ± 0.001 | 277 | 0.055 | −1.51365 ns | |
| Taxon 1 | Conidiomata | Conidiogenous Layer 2 | Conidia | Reference |
|---|---|---|---|---|
| Diaporthe arecae (H.C. Srivast., Zakia & Govindar.) R.R. Gomes, C. Glienke & Crous ≡ Subramanella arecae H.C. Srivast., Zakia & Govindar. (CBS H-7808IH) | Pycnosclerotium formed along the sclerotium cortex, lacking ostiole, exuding conidia through irregular openings, 160–360 × 240–860 μm | Conidiophores distinct, long, thin, hyaline, simple | Alpha conidia elliptic, hyaline, aseptate, 7.2–9.6 × 2.4 μm Beta-conidia needle-shaped, slightly curved, hyaline, aseptate, 14.4–24 × 1.2 μm Gamma conidia not observed | [24,38] |
| Diaporthe acuta Y.S. Guo & G.P. Wang * (CGMCC 3.19600T) | Pycnidia globose or irregular, dark brown to black, 230–544 μm diam. | N/A | Alpha conidia fusiform to oval, acutely rounded ends, hyaline, aseptate, bi- or multiguttulate, 6–9.5 × 2–3 μm ( = 7.8 × 2.6 μm, n = 50; L/W = 3) Beta and gamma conidia not observed | [104] |
| Diaporthe anhuiensis H. Zhou & C.L. Hou * (CNUCC 201901T) | Pycnidia globose, fuscous to black, exuding whitish to cream conidial droplets from ostiole, 250–340 μm diam. | Conidiophores cylindrical, tapering towards apex, hyaline, unbranched, 10.5–25.2 × 1.5–2.7 μm | Alpha conidia spindly or fusoid, hyaline, aseptate, bi-guttulate, rarely multiguttulate, 7.6–10.4 × 2.2–3.6 μm ( = 8.8 × 2.8 μm, n = 40) Beta and gamma conidia not observed | [105] |
| Diaporthe arengae R.R. Gomes, C. Glienke & Crous * (CBS 114979T) | Pycnidia subglobose, black, exuding cream conidial droplets through central ostiole, up to 250 μm diam. | Conidiophores cylindrical, straight to sinuous, hyaline apex, pale brown base, 0–6-septate smooth, branched, densely aggregated, 10–60 × 2.5–4 μm Conidiogenous cells cylindrical, terminal and lateral, slightly tapering towards apex (1–1.5 μm), phialidic (with periclinal thickening), with collarette not flared (up to 2 μm long), 8–15 × 1.5–2.5 μm | Alpha conidia fusoid-ellipsoid, tapering towards ends, subobtuse apex, flattened hilum at base, hyaline, aseptate, guttulate, (5–)6–7(–9) × (2–)2.5(–3) μm Beta conidia rarely observed, subcylindrical, bluntly rounded apex, truncate base, hyaline, aseptate, smooth, 20–25 × 1.5 μm Gamma conidia not observed | [24] |
| Diaporthe averrhoae (C.Q. Chang, Z.D. Jiang & P.K. Chi) Y.H. Gao & L. Cai * ≡ Phomopsis averrhoae C.Q. Chang, Z.D. Jiang & P.K. Chi (SCHM 3605H) | Pycnidia of eustroma, compressed triangle or triangle, unilocular, brown to dark brown, with thinner wall at the base, 188–388 × 83–175 μm | Conidiophores hyaline, septate, branched, 8.5–36 × 1.4–2.0 μm Conidiogenous cells hyaline, phialidic | Alpha conidia fusiform, hyaline, aseptate, biguttulate, 6.0–8.4 × 1.4–1.8 μm Beta conidia filiform, mostly hamate, hyaline, aseptate, 10–25.5 × 0.5–0.9 μm Gamma conidia not observed | [26,106] |
| Diaporthe camelliae-oleiferae Q. Yang * (HNZZ027T) | Pycnidia globose, dark brown to black, exuding pale-yellow conidial droplets from ostiole, 500–660 μm diam. | Conidiophores reduced to conidiogeneous cells. Conidiogenous cells cylindrical, tapering towards apex, straight, terminal, aseptate, densely aggregated, (7.5–)10–14(–15.5) × 1.5–2.3 μm (n = 30) | Alpha conidia ellipsoidal to fusiform, hyaline, aseptate, bi-guttulate, 5–6.5(–7.5) × 1.9–2.3 μm (n = 30) Beta conidia filiform, sinuous at one end, hyaline, aseptate, eguttulate, (26.5–)28.5–31(–33) × 0.8–1.2 μm (n = 30) Gamma conidia not observed | [107] |
| Diaporthe ceratozamiae Crous & R.G. Shivas * (CBS 131306T) | Pycnidia subglobose, black, exuding yellow conidial droplets from ostiole, up to 300 μm diam. | Conidiophores cylindrical, straight to sinuous, hyaline, 1–3-septate, smooth, branched, densely aggregated, 15–30 × 3–4 μm Conidiogenous cells cylindrical, terminal, and lateral, slightly tapering towards apex (1–1.5 μm), phialidic (with periclinal thickening), with collarette not flared (1 μm long) Paraphyses cylindrical, straight, flexuous, hyaline, usually 1–2-septate at base, smooth, wall thickened, unbranched or branched at base, extending above conidiophores, up to 60 μm long and 1.5–2.5 μm wide at base | Alpha conidia fusiform, tapering towards ends, acutely rounded apex, subtruncate base, hyaline, aseptate, (6.5–)8–9(–10) × 2–2.5(–3) μm Beta conidia and gamma conidia not observed | [108] |
| Diaporthe cercidis C.M. Tian & Q. Yang * (CFCC 52565T) | Pycnidia discoid (ectostromatic disc), with a solitary undivided circular locule, nearly flat, grey to brown, with one ostiole, 135–200 μm diam. | Conidiophores cylindrical, tapering towards apex, straight or slightly curved, unbranched, phialidic, 7–17 × 1.4–2.1 μm | Alpha conidia fusiform to oval, hyaline, aseptate, bi-guttulate, 6.5–10 × 3–3.5 μm ( = 8.6 × 3.3 μm, n = 30) Beta conidia filiform, straight or hamate, hyaline, aseptate, eguttulate, 20–28.5 × 1–1.3 μm ( = 25.5 × 1.2 μm, n = 30) Gamma conidia not observed | [109] |
| Diaporthe chamaeropicola D.S. Pereira & A.J.L. Phillips * (CDP 0460T) | Pycnidia subglobose, dark-brown to black, lacking an ostiole, exuding a creamy mucoid conidial mass through irregular fissures on pycnidial wall, up to 4 mm diam. | Conidiophores reduced to conidiogenous cells Conidiogenous cells cylindrical, occasionally ampulliform, tapering towards apex, straight, hyaline, aseptate or 1–3-septate, smooth, unbranched or branched, with collarette (up to 1 µm long), enteroblastic (with periclinal thickening and 1–2 annellations), dimorphic, short conidiogenous cells 4.9–19.4 × 0.9–2.6 µm ( = 13.66 × 1.75 µm), long conidiogenous cells 15.2–49.2 × 1.1–2.7 µm ( = 29.54 × 1.75 µm) Paraphyses cylindrical, straight, flexuous, tapering towards apex, hyaline, 1–2(–3)-septate at base, smooth, unbranched or branched at base, extending above conidiogeneous cells, 26.6–78.8 μm ( = 53.57 µm) long | Alpha conidia cylindrical to ellipsoidal, rounded apex, obtuse to truncate base, straight to slightly curved, hyaline, aseptate, smooth, biguttulate, 5.6–9.4 × 1.7–3 μm ( = 7.53 × 2.31 µm, L/W = 3.33) Beta and gamma conidia not observed | [64] |
| Diaporthe chrysalidocarpi S.T. Huang, J.W. Xia, W.X. Sun, & X.G. Zhang * (SAUCC 194.35T) | Pycnidia subglobose, black, exuding white or yellowish creamy conidial droplets from central ostiole | Conidiophores subcylindrical, swelling at base, straight or curved, hyaline, septate, smooth, branched, 27.5–35 × 1.4–2 μm Conidiogenous cells cylindrical, tapering towards apex, terminal, straight or sinuous, phialidic, 10.5–23 × 1.4–1.8 μm | Beta conidia filiform, subtruncate base, tapering towards base, straight or slightly curved, hyaline, aseptate, 28–32.5 × 1.2–1.6 μm ( = 30.3 × 1.3 μm, n = 20) Alpha and gamma conidia not observed | [110] |
| Diaporthe delonicis R.H. Perera, E.B.G. Jones & K.D. Hyde * (MFLU 16-1059H) | Pycnidia globose or near-globose, brown to dark brown, exuding white creamy conidial droplets, 78–190 μm ( = 120 μm) diam. | Conidiophores subcylindrical, hyaline, 6.4–15.2 × 1.4–2.2 μm ( = 11.6 × 1.9 μm) Conidiogenous cells cylindrical, tapering towards apex, with prominent collarette, phialidic, 5.3–10.5 × 1.3–2.5 μm ( = 7.9 × 1.9 μm) | Alpha conidia fusoid, obtuse ends, hyaline, aseptate, 4-guttulate, smooth, 4.4–9 × 1.3–2.2 μm ( = 7.7 × 1.8 μm) Beta conidia filiform, slightly curved at one end, rounded ends, hyaline, aseptate, smooth, 16–23 × 1–1.7 μm ( = 19.4 × 1.2 μm) Gamma conidia not observed | [111] |
| Diaporthe drenthii Y.P. Tan, Akinsanmi & R.G. Shivas * (BRIP 66524T) | Pycnidia globose or irregular, dark brown to black, up to 1 mm diam. | Conidiophores hyaline, smooth, densely aggregated, 15–25 μm long Conidiogeneous cells cylindrical, straight or flexuous, hyaline, phialidic, 10–20 × 1–2.5 μm | Alpha conidia fusiform, acute ends, hyaline, aseptate, 5.5–8.5 × 1.5–2 μm Beta conidia sparse, curved, 25–35 × 1 μm Gamma conidia not observed | [112] |
| Diaporthe endocitricola Z.Y. Dong, M. Luo, M.M. Xiang & K.D. Hyde * (ZHKUCC 20-0012T) | Pycnidia subglobose or lageniform, multilocular, exuding hyaline to dark black creamy conidial droplets from ostiole, 124–790 × 111–635 μm ( = 353 × 289 μm) | Conidiophores cylindrical, hyaline, 12–40 × 1–3 μm ( = 26 × 2 μm) | Alpha conidia cylindrical to ellipsoid, hyaline, aseptate, multi-guttulate, 6–8 × 2–3 μm ( = 7 × 3 μm) Beta conidia filiform, straight or slightly curved at one end, hyaline, aseptate, 12–30 × 1–2 μm ( = 19 × 2 μm) Gamma conidia fusiform, hyaline, multi-guttulate | [113] |
| Diaporthe fraxini-angustifoliae R.G. Shivas, J. Edwards & Y.P. Tan * (BRIP 54781IT) | Pycnidia subglobose, rarely with ostiolar beaks (up to 100 μm high), exuding tan to white conidial droplets from ostiole | Conidiophores reduced to conidiogenous cells or cylindrical to lageniform, straight to sinuous, hyaline to pale brown, 1-septate, 5–30 × 1.5–4 μm Conidiogenous cells cylindrical, hyaline, tapering towards apex, phialidic, 5–15 × 1–2 μm | Alpha conidia scarce, cylindrical to oval, attenuated ends, hyaline to subhyaline, (4–)5–8.5(–10) × 2–3 μm Beta conidia abundant, flexuous to lunate, mostly curved through 45°–180° in upper third, truncate base, narrowed towards acute apex, hyaline, aseptate, (16–)17–21(–22) × 1 μm Gamma conidia not observed | [39] |
| Diaporthe fulvicolor Y.S. Guo & G.P. Wang * (CGMCC 3.19601T) | Pycnidia globose or irregular, dark brown to black, 174–316 μm diam. | Conidiophores cylindrical, straight, hyaline, 1-septate, unbranched, smooth, densely aggregated, 5.5–8 × 2.5–3.5 μm Conidiogeneous cells ampulliform, terminal, tapering towards apex, hyaline, 6.5–10 × 1.5–2.5 μm | Alpha conidia fusiform to oval, acutely rounded ends, hyaline, aseptate, bi- or multi-guttulate, 7–9 × 2–3 μm ( = 7.8 × 2.5 μm, n = 50; L/W = 3.1) Beta and gamma conidia not observed | [104] |
| Diaporthe guangxiensis Dissanayake, X.H. Li & K.D. Hyde * (JZB 320094T) | Pycnidia globose, dark brown to black, 250–1550 μm ( = 1.1 mm, n = 20) diam. | Conidiophores cylindrical, straight or sinuous, slightly tapering towards apex, terminal, aseptate, densely aggregated, 21–35 × 1.5–2.5 μm ( = 27 × 2 μm) | Alpha conidia fusiform or oval, obtuse ends, hyaline, 5.3–7.8 × 1.5–3.2 μm ( = 6.8 × 2.5 μm, n = 40) Beta conidia filiform, hamate, tapering towards ends, hyaline, aseptate, guttulate, 20–32 × 1–1.5 μm ( = 27 × 1.5 μm, n = 20) Gamma conidia not observed | [21] |
| Diaporthe huangshanensis H. Zhou & C. L. Hou * (CNUCC 201903T) | Pycnidia globose, brown to black, exuding whitish translucent conidial droplets from apex, 210–270 μm diam. | Conidiophores cylindrical, straight to sinuous, hyaline, branched, 12.1–23.5 × 1.1–2.9 μm | Alpha conidia ellipsoidal to olivary body, hyaline, aseptate, bi-to multi-guttulate, 5.7–8.4 × 2.7–4.5 μm ( = 6.9 × 3.5 μm, n = 40) Beta conidia filiform, straight or hamate, partially guttulate, one end rounded and other acute and curved, 19.5–30 × 1.1–2.1 μm ( = 24.1 × 1.5 μm, n = 30) Gamma conidia not observed | [105] |
| Diaporthe hunanensis Q. Yang * (HNZZ023T) | Pycnidia globose, black, 180–300 μm diam. | Conidiophores reduced to conidiogeneous cells. Conidiogenous cells cylindrical, straight or slightly curved, aseptate, phialidic, (8–)9–15(–16.5) × 1.7–2.1 μm (n = 30) | Alpha conidia ellipsoidal, obtuse ends, hyaline, aseptate, bi-guttulate, 6.5–7.5(–8.5) × 2.4–2.9 μm (n = 30) Beta and gamma conidia not observed | [107] |
| Diaporthe krabiensis Dayarathne * (MFLUCC 17-2481T) | Pycnidia globose or irregular, uniloculate or multiloculate, black, 117–145 × 130–140 μm | Conidiophores cylindrical, straight to sinuous, 2–3-septate, branched, densely aggregated, rarely reduced to conidiogenous cells Conidiogenous cells subcylindrical, tapering towards apex, hyaline, phialidic (with periclinal thickening), with flared collarette, 15–32 × 0.9–1.4 μm ( = 28.5 × 1.2 μm, n = 20) | Beta conidia fusiform to hooked, hyaline, aseptate, smooth, 15–32 × 0.9–1.4 μm ( = 28.5 × 1.2 μm, n = 20) Alpha and gamma conidia not observed | [114] |
| Diaporthe limonicola Guarnaccia & Crous * (CBS 142549T) | Pycnidia dark brown to black, exuding whitish translucent to cream conidial droplets from ostiole, 250–670 μm diam. | Conidiophores cylindrical, straight, hyaline, 1-septate, smooth, densely aggregated, 5–20 × 1.5–4 μm Conidiogenous cells cylindrical, terminal, tapering towards apex, hyaline, phialidic, 5–12 × 1–2 μm Paraphyses hyaline, 1–3-septate, smooth, intermingled among conidiophores, up to 90 μm long and 1–2 μm diam. at apex | Alpha conidia fusiform, acute ends, hyaline, aseptate, mono- to biguttulate 5.5–8.5 × 1.5–2.5 μm ( = 6.8 × 2.1 μm, L/W = 2.8) Beta conidia filiform, curved, tapering towards ends, hyaline, aseptate, eguttulate, 15–26.5 × 1–2 μm ( = 22.7 × 1.4 μm, L/W = 16.2) Gamma conidia fusiform to subcylindrical, acute or rounded apex, hyaline, multiguttulate, 9–15.5 × 1–2 μm ( = 10.7 × 1.4 μm, L/W = 7.6) | [10] |
| Diaporthe liquidambaris (C.Q. Chang, Z.D. Jiang & P.K. Chi) Udayanga & Castl. * ≡ Phomopsis liquidambaris C.Q. Chang, Z.D. Jiang & P.K. Chi (SCHM 3621H) | Pycnidia of eustroma, tuberous or irregular, unilocular to multilocular, 143–350 × 88–250 μm | Conidiophores hyaline, septate, sympodially branched, 10–25 × 1.7–3.0 μm Conidiogenous cells hyaline, phialidic | Alpha conidia fusiform, acute ends, hyaline, aseptate, biguttulate, 6.5–8.1 × 1.7–2.2 μm Beta conidia filiform, hamate, hyaline, aseptate, 10.5–24.5 × 0.6–1 μm Gamma conidia not observed | [106,115] |
| Diaporthe litchiicola R.G. Shivas, Grice & Y.P. Tan [as “litchicola”] * (BRIP 54900T) | Pycnidia subglobose, with black cylindrical ostiolate neck (up to 1.5 mm), up to 400 μm diam. | Conidiophores reduced to conidiogeneous cells Conidiogenous cells cylindrical, straight to sinuous, hyaline, tapering towards apex, smooth, 20–45 × 1.5–2 μm | Alpha conidia fusiform to oval, tapered at ends, cylindrical to ellipsoidal, hyaline, smooth, guttulate, (5–)6.5–9.5(–10) × 1.5–2(–2.5) μm Beta conidia flexuous to lunate, (17–)20–32(–37) × 1–1.5 μm Gamma conidia not observed | [39] |
| Diaporthe loropetali (C.Q. Chang, Z.D. Jiang & P.K. Chi) Y.H. Gao & L. Cai * ≡ Phomopsis loropetali C.Q. Chang, Z.D. Jiang & P.K. Chi (SCHM 3615H) | Pycnidia of eustroma, ampullate or tuberous, unilocular, with darker and thicker wall near the ostiole, 163–338 × 88–218 μm | Conidiophores filiform, hyaline, septate, branched, 10–29 × 1.4–2.1 μm Conidiogenous cells hyaline, phialidic | Alpha conidia fusiform to lanceolate, acute apex, obtuse base, hyaline, aseptate, biguttulate, 6.2–8.4 × 1.5–1.9 μm Beta conidia filiform, straight or curved, hyaline, aseptate, 14–31 × 0.6–1.2 μm Gamma conidia not observed | [26,116] |
| Diaporthe meliae C.M. Tian & Qin Yang * (CFCC 53089T) | Pycnidia discoid (ectostromatic disc), with an undivided locule, dark brown, with one ostiole, (325–)135–200(–385) μm diam. (n = 30) | Conidiophores reduced to conidiogeneous cells. Conidiogenous cells cylindrical, tapering towards apex, straight or slightly curved, branched, hyaline, (13.5–)15–26.5(–28) × 1.3–2.1(–2.3) μm (n = 30) | Alpha conidia fusiform, hyaline, aseptate, multiguttulate, (6.7–)8–9.5(–10) × (2–)2.1–2.3 μm (L/W = 3.4–4.5, n = 30) Beta and gamma conidia not observed | [117] |
| Diaporthe melitensis Guarnaccia & Crous * (CBS 142551T) | Pycnidia dark brown to black, exuding whitish translucent to yellowish conidial droplets from ostiole, 250–650 μm diam. | Conidiophores cylindrical, straight, hyaline, 1-septate, smooth, densely aggregated, 5–15 × 1.5–5.5 μm Conidiogenous cells cylindrical, terminal, tapering towards apex, hyaline, phialidic, 6–12 × 1–3 μm | Alpha conidia fusiform, acute ends, hyaline, aseptate, 1–4-guttulate, 4.5–7 × 1.5–3 μm ( = 5.9 × 2.2 μm, L/W = 2.7) Beta and gamma conidia not observed | [10] |
| Diaporthe millettiae H. Long, K.D. Hyde & Yong Wang bis * (GUCC 9167T) | Pycnidia subglobose to irregular, with up to 1 mm necks when present, multilocular, ostiolate, 1.5–1.8 mm diam. | Conidiophores reduced to conidiogeneous cells or cylindrical, hyaline to pale yellowish-brown, 1-septate, 10–23 × 1–2.5 μm Conidiogenous cells cylindrical to flexuous, tapering towards apex, hyaline, 8–18 × 1.5–3 μm | Alpha conidia fusiform, narrowed towards ends, hyaline, mostly biguttulate, 4.5–9 × 2–3.5 μm Beta conidia scarce to abundant, flexuous to J-shaped, hyaline, 17.5–32 × 1–2 μm Gamma conidia not observed | [118] |
| Diaporthe musigena Crous & R.G. Shivas * (CBS 129519T) | Pycnidia subglobose, with elongated black necks, exuding yellow conidial droplets through ostiole, up to 250 μm diam. | Conidiophores cylindrical, straight to sinuous, hyaline, 1–3-septate, smooth, branched, densely aggregated, 15–40 × 1.5–2.5 μm Conidiogenous cells cylindrical, terminal and lateral, slightly tapering towards apex (0.5–1 μm), phialidic (with periclinal thickening), with collarette not flared (2–5 μm long) Paraphyses cylindrical, straight, flexuous, hyaline, septate, unbranched or branched, extending above conidiophores, up to 80 μm long and 2–2.5 μm wide at base | Alpha conidia fusiform, tapering towards ends, straight to slightly curved, acutely rounded apex, subobtuse base, hyaline, aseptate, smooth, guttulate, (7–)8–10(–12) × (2–)2.5(–3) μm Beta conidia observed in older cultures, spindle-shaped, acutely rounded apex, truncate base, tapering more prominently in upper third, straight to curve, hyaline, aseptate, smooth, (14–)19–22(–25) × (1.5–)2 μm Gamma conidia ellipsoid to fusoid, acutely rounded apex, subtruncate to acutely rounded base, hyaline, aseptate, smooth, 7–9 × 4–5 μm | [119] |
| Diaporthe nelumbonis Sawada ex R. Kirschner * ≡ Phyllosticta nelumbonis Sawada (BPI 352726H) (R. Kirschner 4114R) | Pycnidia slightly applanate, brown, ostiolate, 55–87 × 80–125 μm | Conidiophores reduced to conidiogenous cells or with a separate basal cell that often turns into an intercalary conidiogenous cell Conidiogenous cells pyriform to obclavate or lageniform, conspicuously narrowed apex, terminal or intercalary, with minute periclinal thickening, (3–)4.5–7.5(−9) × 2–3 μm (n = 30) in H, (6–)6.5–10(−11) × (1.5–)2–3 (n = 20) in R | Alpha conidia oblong-ellipsoidal, straight or slightly curved, rounded apex, attenuated towards base, hyaline, aseptate, mostly biguttulate, (6–)6.5–8(−9) × 2–2.5 μm (n = 30) in H, (5–)6–7 × (1.5–)2 μm (n = 30) in R Beta and gamma conidia not observed | [120] |
| Diaporthe oculi Mochiz. & Kaz. Tanaka * (MAFF 246252T) | Pycnidia globose to depressed globose, with cylindrical, central, dark brown ostiolar neck (150–480 × 80–140 μm diam.), exuding yellow to pink conidial mass, 90–250 × 110–310 μm diam. | Conidiophores reduced to conidiogenous cells Conidiogeneous cells cylindrical to lageniform, phialidic, 6–15 × 2–5 μm | Alpha conidia fusoid-ellipsoid, hyaline, aseptate, 5–8.5 × 2–3 μm ( = 6.7–2.4 μm, L/W = 2.3–3.2, n = 50), Beta and gamma conidia not observed | [42] |
| Diaporthe osmanthi H. Long, K.D. Hyde & Yong Wang bis * (GUCC 9165T) | Pycnidia globose, subglobose or irregular, with up to 1 mm necks when present, multilocular, ostiolate, up to 1–1.5 mm diam. | Conidiophores reduced to conidiogeneous cells or cylindrical, hyaline to pale yellowish-brown, 1-septate, 20.5–61 × 1–3 μm Conidiogenous cells cylindrical to flexuous, tapering towards apex, hyaline, 10–15 × 1.5–3 μm | Alpha conidia fusiform, narrowed towards ends, hyaline, biguttulate, 5.5–8.5 × 2–3 μm Beta conidia scarce to abundant, flexuous to J-shaped, hyaline, 20–31.5 × 1–2.5 μm Gamma conidia not observed | [118] |
| Diaporthe pascoei R.G. Shivas, J. Edwards & Y.P. Tan * (BRIP 54847IT) | Pycnidia with ostiolar beaks (mostly up to 1.5 mm high), exuding conidial droplets from ostiole | Conidiophores cylindrical, straight, hyaline, 1–2-septate near base, unbranched, 5–40 × 2–3 μm Conidiogenous cells cylindrical, terminal, hyaline, tapering towards apex, phialidic, 5–30 × 2–3 μm | Alpha conidia scarce, cylindrical, rounded apex, slightly attenuated base, hyaline, (3.5–)4–5 × 1–2 μm Beta conidia abundant, flexuous to lunate, often curved up to 90° at apex, truncated base, narrowed towards apex, hyaline, (15–)19–31(–39) × 1–1.5 μm Gamma conidia not observed | [39] |
| “Diaporthe perseae” (CBS 151.73) | Pycnidia globose, black, exuding cream conidial droplets through central ostiole, up to 400 μm diam. | Conidiophores cylindrical, straight to sinuous, hyaline, 1–3-septate, smooth, branched, densely aggregated, 15–35 × 3–4 μm Conidiogenous cells cylindrical, terminal and lateral, slightly tapering towards apex (1–1.5 μm), phialidic (with periclinal thickening), with prominent collarette (up to 5 μm long), 8–17 × 1.5–2.5 μm Paraphyses subcylindrical, obtuse ends, hyaline, 2–4-septate, smoooth, up to 60 μm long and 3 μm diam. | Alpha conidia fusoid to ellipsoid, tapering towards ends, straight, subobtuse apex, subtruncate base, hyaline, aseptate, smooth, guttulate, (6–)7–8(–9) × 2(–2.5) μm Beta conidia spindle-shaped, tapering from lower third towards apex, curved, acutely rounded apex, truncate base, hyaline, aseptate, smooth, (15–)22–25(–28) × 1.5(–2) μm Gamma conidia ellipsoid to fusoid, acutely rounded apex, subtruncate base, hyaline, aseptate, smooth, 9–14× 1.5–2 μm | [24] |
| Diaporthe pescicola Dissanayake, J.Y. Yan, X.H. Li & K.D. Hyde * (MFLUCC 16-0105T) | Pycnidia globose, dark brown to black, up to 300 μm diam. | Conidiophores cylindrical, straight or sinuous, terminal, slightly tapering towards apex, aseptate, densely aggregated, 21–35 × 1.5–2.5 μm ( = 27 × 2 μm) | Alpha conidia fusiform or oval, obtuse ends, hyaline, biguttulate, 6–8.5 × 2–3 μm ( = 8 × 3 μm) Beta conidia filiform, hamate, tapering towards ends, hyaline, aseptate, 18–37 × 1–1.5 μm ( = 27 × 1.5 μm) Gamma conidia not observed | [41] |
| Diaporthe phyllanthicola (C.Q. Chang, Z.D. Jiang & P.K. Chi) Y.H. Gao & L. Cai * ≡ Phomopsis phyllanthicola C.Q. Chang, Z.D. Jiang & P.K. Chi (SCHM 3680H) | Pycnidia of eustroma, triangle, tuberous or irregular, unilocular to multilocular, with darker and thicker wall at the base, 185–425 × 100–125 μm | Conidiophores hyaline, septate, branched, 12.5–29 × 1.7–2.6 μm Conidiogenous cells hyaline, phialidic | Alpha conidia fusiform, hyaline, aseptate, eguttulate or biguttulate, 6.6–8.2 × 1.5–1.8 μm Beta conidia filiform, curved or hamate, hyaline, aseptate, 13.5–26.5 × 0.6–0.9 μm Gamma conidia not observed | [26,106] |
| Diaporthe podocarpi- -macrophylli Y.H. Gao & L. Cai * (CGMCC 3.18281T) | Pycnidia subglobose, dark brown to black, exuding yellowish translucent conidial droplets from ostiole, 250–699 μm diam. | Alpha conidiophores cylindrical, straight to sinuous, sometimes inflated, hyaline, septate, branched, in dense clusters, 6–18 × 1.5–3 μm ( = 12.3 × 2.1 μm, n = 30) Beta conidiophores cylindrical to clavate, straight, hyaline, septate, branched, smooth, 10.5–27 × 1.5–2.5 μm ( = 15.3 × 2.1 μm, n = 30) | Alpha conidia fusiform, acute ends, hyaline, aseptate, biguttulate, 3.5–8.5 × 1–3 μm ( = 6.3 × 2.1 μm, n = 50) Beta conidia filiform, curved, tapering towards ends, truncate base, hyaline, aseptate, eguttulate, 8.5–31.5 × 0.5–2 μm ( = 19.5 × 1.1 μm, n = 30) Gamma conidia not observed | [26] |
| Diaporthe pseudomangiferae R.R. Gomes, Glienke & Crous * (CBS 101339T) | Pycnidia globose, with elongated necks with central ostioles, exuding yellow–orange to cream conidial droplets, up to 300 μm diam. | Conidiophores cylindrical, straight to sinuous, hyaline, 1–3-septate, smooth, branched, densely aggregated, 20–30 × 2–2.5 μm Conidiogenous cells cylindrical, terminal and lateral, slightly tapering towards apex, phialidic, with flared collarette (up to 3 μm long), 10–15 × 2–3 μm Paraphyses cylindrical, straight to flexuous, hyaline, septate, smooth, unbranched or branched at base, extending above conidiophores, up to 80 μm long and 2–3 μm wide at base | Alpha conidia fusiform, tapering towards ends, acutely rounded apex, truncate base, hyaline, aseptate, smooth, guttulate to granular, (6–)7–9(–10) × (2–)2.5(–3) μm Beta and gamma conidia not observed | [24] |
| Diaporthe pseudooculi Mochiz. and Kaz. Tanaka * (MAFF 246452T) | Pycnidia globose to depressed globose, with cylindrical to papillate, central ostiolar neck (100–220 × 45–130 μm diam.), exuding white to yellow conidial mass, 220–330 × 180–280 μm diam. | Conidiophores hyaline, 5–12 × 2–5 μm Conidiogeneous cells cylindrical, phialidic, 12–18 × 2 μm Paraphyses filamentous, 50–65 × 1.5–2.5 μm | Alpha conidia ellipsoid, hyaline, aseptate, 6–9 × 2–3.5 μm ( = 7.3–2.8 μm, L/W = 2.1–3.2, n = 50) Beta conidia sigmoid, hyaline, aseptate, 21.5–33.5 × 1.2–1.7 μm ( = 27–1.4 μm, n = 30) Gamma conidia not observed | [42] |
| Diaporthe pseudophoenicicola R.R. Gomes, C. Glienke & Crous * (CBS 462.69T) | Pycnidia globose, with neck, exuding yellow-orange conidial droplets through ostiole, up to 400 μm diam. | Conidiophores cylindrical, straight to curved, hyaline, 1–3-septate, smooth, branched, densely aggregated, 12–45 × 1.5–3 μm Conidiogenous cells cylindrical, terminal and lateral, slightly tapering towards apex, phialidic (with periclinal thickening), with collarette flared (2–5 μm long), 8–15 × 1.5–2.5 μm Paraphyses cylindrical, hyaline, 1–3-septate, smoooth, straigh to flexuous, extending above conidiophores, up to 100 μm long and 3 μm wide at base | Alpha conidia fusiform, tapering towards ends, straight, acutely rounded apex, truncate base, hyaline, aseptate, smooth, granular, (6–)7–8(–9) × (2–)2.5(–3) μm Beta and gamma conidia not observed | [24] |
| Diaporthe pterocarpicola Udayanga, Xing Z. Liu and K.D. Hyde * (MFLUCC 10-0580aT) | Pycnidia hemi-spherical, with slightly elongated black neck, exuding yellowish translucent conidial droplets from ostiole, up to 75 × 120 μm | Conidiophores subcylindrical to cylindrical, wide at base, straight to sinuous, hyaline, unbranched, densely aggregated, 7–18 × 1.5–3.5 μm, 2.5–3.5 wide at base Conidiogenous cells cylindrical, terminal, slightly tapering towards apex, phialidic (with periclinal thickening), 1–2 μm diam. Paraphyses occasionally present, cylindrical, straight to flexuous, hyaline, septate, smooth, unbranched, extending above conidiophores, up to 25 μm long and 1.5–2 μm wide at base | Alpha conidia ellipsoid or clavate, subtruncate base, hyaline, aseptate, multiguttulate, (5–)6–7(–8) × (2–)2.5(–3.5) μm Beta and gamma conidia not observed | [31] |
| Diaporthe schimae C.M. Tian and Q. Yang * (CFCC 53103T) | Pycnidia globose, exuding cream to yellowish translucent conidial droplets from ostiole, (150–)180–300(–373) μm diam. | Conidiophores reduced to conidiogeneous cells. Conidiogenous cells straight, slightly tapering towards apex, hyaline, septate, unbranched | Alpha conidia scarce, ellipsoidal to spindle-shaped, hyaline, aseptate, 4-guttulate, (7.5–)8–8.5(–9) × 2.5–3 μm Beta conidia filiform, straight to sinuous at one end, hyaline, aseptate, eguttulate, (25–)27.5–38.5(–40.5) × 1–1.5 µm Gamma conidia not observed | [121] |
| Diaporthe searlei R.G. Shivas, Akinsanmi & Y.P. Tan * (BRIP 66528T) | Pycnidia globose or irregular, dark brown to black, up to 1 mm diam. | Conidiophores hyaline, smooth, densely aggregated, 15–45 μm long Conidiogeneous cells cylindrical, straight or flexuous, hyaline, phialidic, 10–35 × 1–2.5 μm | Alpha conidia fusiform, acute ends, hyaline, aseptate, 5–9 × 1.5–2 μm Beta and gamma conidia not observed | [112] |
| Diaporthe sennae C.M. Tian and Qin Yang * (CFCC 51636T) | Pycnidia circular to ovoid, uniloculate and undivided, ectostromatic disc brown to dark, with one ostiole, (400–)500–600(–680) μm ( = 570 μm, n = 20) diam. | Conidiophores reduced to conidiogeneous cells. Conidiogenous cells straight or slightly curved, hyaline, phialidic | Alpha conidia ellipsoidal to oval, hyaline, aseptate, smooth, biguttulate, rarely 3-guttulate, (5–)5.5–6.3(–6.5) × 1.5–1.7(–1.8) μm ( = 6 × 1.6 μm, n = 50) Beta conidia straight to hamate, hyaline, aseptate, smooth, (17.3–)18.4–20(–23.3) × 0.9 μm ( = 19.1 × 0.9 μm, n = 50) Gamma conidia not observed | [122] |
| Diaporthe spinosa Y.S. Guo and G.P. Wang * (CGMCC 3.19602T) | Pycnidia globose, dark brown to black, 124–172 μm diam. | Conidiophores ampulliform, hyaline, 1-septate, smooth, unbranched, densely aggregated, 6–9 × 3–4.5 μm Conidiogeneous cells cylindrical, straight, terminal, tapering towards apex, hyaline, 8–29 × 1.5–2.5 μm | Alpha conidia fusiform to oval, acutely rounded ends, hyaline, aseptate, bi- or multi-guttulate, 5.5–8 × 2–3.5 μm ( = 7 × 2.6 μm, n = 50; L/W = 2.7) Beta conidia filiform, curved, tapering towards ends, multi-guttulate, 18.5–30.5 × 1–1.5 μm ( = 25.1 × 1.3 μm, n = 38; L/W = 19.3) Gamma conidia not observed | [104] |
| Diaporthe taiwanensis H.A. Ariyaw. and I. Tsai * (NTUCC 18-105-1T) | Pycnidia irregular, with hairy neck, exuding yellowish conidial droplets, up to 270 μm diam. | Conidiophores cylindrical, hyaline, septate, branched, 11–15 × 1–2.5 μm Conidiogenous cells subcylindrical, straight to curved, tapering towards apex, hyaline, 7–8.5 × 1–2.5 μm | Alpha conidia fusiform, acute ends, hyaline, aseptate, 1–3-guttulate, 7–9.5 × 2.5–3 μm Beta conidia acutely rounded and curved apex, hyaline, smooth, 24–30 × 1–2 μm Gamma conidia not observed | [123] |
| Diaporthe taoicola Dissanayake, X.H. Li & K.D. Hyde * (MFLUCC 16-0117T) | Pycnidia globose, black, multilocular, exuding cream conidial droplets from central ostiole, up to 300 μm diam. | Conidiophores cylindrical, straight to sinuous, hyaline, smooth, densely aggregated, 10–25 × 2–3 μm Conidiogenous cells cylindrical, terminal and lateral, slightly tapering towards apex, phialidic, 9–16 × 1.5–2 μm Paraphyses cylindrical, with obtuse ends, hyaline, 1–3-septate, smooth, extending above conidiophores | Alpha conidia fusoid to ellipsoid, subobtuse apex, bluntly rounded base with flattened hilum, tapering towards ends, straight, hyaline, smooth, guttulate, 7–9 × 2–3 μm ( = 8 × 3 μm) Beta conidia spindle-shaped, curved, tapering towards subacutely rounded apex, truncate base, hyaline, aseptate, 20–25 × 1.5–2 μm ( = 19 × 2 μm) Gamma conidia not observed | [41] |
| Diaporthe viciae W.S. Zhao, Q. Ning and J.Y. Yan * (JZB 320179T) | Pycnidia oval to round, black, 150–200 × 150–250 μm | Conidiophores cylindrical, aseptate, densely aggregated, 15–32.5 μm long Conidiogenous cells cylindrical, terminal and lateral, phialidic | Alpha conidia fusiform or oval, hyaline, 2–5-guttulate, 7–10 × 2–4 μm ( = 8.3 × 3 μm, n = 50) Beta and gamma conidia not observed | [124] |
| Diaporthe viniferae Dissanayake, X.H. Li & K.D. Hyde * (JZB 320071T) | Pycnidia globose, dark brown to black, 363–937 μm ( = 529 μm, n = 20) diam. | Conidiophores not observed Conidiogenous cells not observed | Alpha conidia fusiform or oval, obtuse ends, hyaline, bi-guttulate, 5–8.3 × 1.3–2.5 μm ( = 6.4 × 2.1 μm) Beta conidia filiform, hamate, tapering towards ends, hyaline, aseptate, 23–35 × 1–1.5 μm ( = 28 × 1.3 μm, n = 40) Gamma conidia not observed | [21] |
| Taxon 1 | Host | Country | Ecological Group 2 | Reference |
|---|---|---|---|---|
| Diaporthe arecae | Fruit of Areca catechu (Arecaceae) | India | Potential pathogen | [38] |
| Diaporthe acuta * | Diseased branches of Pyrus pyrifolia (Rosaceae) | China (Hubei) | Pathogen | [104] |
| Diaporthe annellsiae * | Fruit of Mangifera indica (Anacardiaceae) | Australia (Western Australia) | UN | [125] |
| Diaporthe anhuiensis * | Leaves of Cunninghamia lanceolata (Cupressaceae) | China (Anhui) | Endophyte | [105] |
| Diaporthe arengae * | Arenga engleri (Arecaceae) | China (Hong Kong) | UN | [24] |
| Diaporthe averrhoae * | Branches of Averrhoa carambola (Oxalidaceae) | China (Fujian) | UN | [106] |
| Diaporthe bounty * | Leaf spots of Malus domestica (Rosaceae) | Australia (Norfolk Island) | Potential pathogen | [125] |
| Diaporthe camelliae-oleiferae * | Leaf spots of Camellia oleifera (Theaceae) | China (Hunan) | Potential pathogen | [107] |
| Diaporthe ceratozamiae * | Leaf spots of Ceratozamia robusta (Zamiaceae) | Australia (Queensland) | Potential pathogen | [108] |
| Diaporthe cercidis * | Twigs and branches of Cercis chinensis (Fabaceae) | China (Jiangsu) | UN | [109] |
| Diaporthe chamaeropicola * | Leaf spots of Chamaerops humilis (Arecaceae) | Portugal (Lisbon) | Potential pathogen | [64] |
| Diaporthe chrysalidocarpi * | Leaf spots of Chrysalidocarpus lutescens (Arecaceae) | China (Yunnan) | Potential pathogen | [110] |
| Diaporthe delonicis * | Seed pods of Delonix regia (Fabaceae) | Thailand (Chiang Rai) | Saprophyte | [111] |
| Diaporthe drenthii * | Rotten husk of Macadamia sp. (Proteaceae) | South Africa (KwaZulu-Natal) | Pathogen | [112] |
| Diaporthe endocitricola * | Fruits of Citrus grandis (Rutaceae) | China (Guangdong) | Endophyte | [113] |
| Diaporthe fraxini-angustifoliae * | Diseased stems of Fraxinus angustifolia (Oleaceae) | Australia (Victoria) | Potential pathogen | [39] |
| Diaporthe fulvicolor * | Diseased branches of Pyrus pyrifolia (Rosaceae) | China (Hubei) | Pathogen | [104] |
| Diaporthe gossiae * | Stem of Sesbania sp. (Fabaceae) | Australia (Western Australia) | UN | [125] |
| Diaporthe guangxiensis * | Diseased trunk of Vitis vinifera (Vitaceae) | China (Guangxi) | Pathogen | [21] |
| Diaporthe hongheensis * | Branch of Mangifera indica (Anacardiaceae) | China (Yunnan) | Saprophyte | [126] |
| Diaporthe howardiae * | Leaf spots of Agave sp. (Asparagaceae) | Australia (Norfolk Island) | Potential pathogen | [125] |
| Diaporthe huangshanensis * | Leaves of Camellia oleifera (Theaceae) | China (Anhui) | Endophyte | [105] |
| Diaporthe hunanensis * | Leaf spots of Camellia oleifera (Theaceae) | China (Hunan) | Potential pathogen | [107] |
| Diaporthe krabiensis * | Submerged wood of Bruguiera sp. (Rhizophoraceae) | Thailand (Krabi) | Saprophyte | [114] |
| Diaporthe limonicola * | Branch canker of Citrus limon (Rutaceae) | Malta (Gozo) | Pathogen | [10] |
| Diaporthe liquidambaris * | Branches of Liquidambar formosana (Altingiaceae) | China (Fujian) | UN | [106] |
| Diaporthe litchiicola * | Diseased Litchi chinensis (Sapindaceae) | Australia (Queensland) | Potential pathogen | [39] |
| Diaporthe loropetali * | Branches of Loropetalum chinense (Hamamelidaceae) | China (Hunan) | UN | [116] |
| Diaporthe meliae * | Branche canker of Melia azedarach (Meliaceae) | China (Shandong) | Potential pathogen | [117] |
| Diaporthe melitensis * | Branch canker of Citrus limon (Rutaceae) | Malta (Gozo) | Pathogen | [10] |
| Diaporthe millettiae * | Leaves of Millettia reticulata (Fabaceae) | China (Guangxi) | UN | [118] |
| Diaporthe musigena * | Necrotic leaves of Musa sp. (Musaceae) | Australia (Queensland) | Potential pathogen | [119] |
| Diaporthe nelumbonis * | Leaf spots of Nelumbo nucifera (Nelumbonaceae) | China (Taiwan, Taipei) | Potential pathogen | [120] |
| Diaporthe norfolkensis * | Panicle of Mangifera indica (Anacardiaceae) | Australia (Norfolk Island) | UN | [125] |
| Diaporthe oculi * | Diseased human eye | Japan (Gifu) | Pathogen | [42] |
| Diaporthe osmanthi * | Leaves of Osmanthus fragrans (Oleaceae) | China (Guangxi) | UN | [118] |
| Diaporthe pandanicola * | Leaves of Pandanus sp. (Pandanaceae) | Thailand (Chumphon) | Endophyte | [127] |
| Diaporthe pascoei * | Roten fruit of Persea Americana (Lauraceae) | Australia (Victoria) | Potential pathogen | [39] |
| Diaporthe pescicola * | Shoots of Prunus persica (Rosaceae) | China (Hubei) | Pathogen | [41] |
| Diaporthe phyllanthicola * | Branches of Phyllanthus emblica (Phyllanthaceae) | China (Fujian) | UN | [106] |
| Diaporthe podocarpi-macrophylli * | Leaves of Podocarpus macrophyllus (Podocarpaceae) | Japan | UN | [26] |
| Diaporthe pseudomangiferae * | Mangifera indica (Anacardiaceae) | Dominican Republic | UN | [24] |
| Diaporthe pseudooculi * | Diseased human eye | Japan (Gifu) | Pathogen | [42] |
| Diaporthe pseudophoenicicola * | Dead tops of green leaves on Phoenix dactylifera (Arecaceae) | Spain (Mallorca) | UN | [24] |
| Diaporthe pterocarpicola * | Leaf spot of Pterocarpus indicus (Fabaceae) | Thailand (Chiang Rai) | Potential pathogen | [31] |
| Diaporthe schimae * | Leaf spots of Schima superba (Theaceae) | China (Jiangxi) | Potential pathogen | [121] |
| Diaporthe searlei * | Rotten husk of Macadamia sp. (Proteaceae) | South Africa (Mpumalanga) | Pathogen | [112] |
| Diaporthe sennae * | Diseased twigs and branches of Senna bicapsularis (Fabaceae) | China (Guangxi) | Potential pathogen | [122] |
| Diaporthe spinosa * | Diseased branches of Pyrus pyrifolia (Rosaceae) | China (Jiangsu) | Pathogen | [104] |
| Diaporthe taiwanensis * | Leaf spots of Ixora chinensis (Rubiaceae) | China (Taiwan, Taoyuan) | Pathogen | [123] |
| Diaporthe taoicola * | Shoots of Prunus persica (Rosaceae) | China (Hubei) | Pathogen | [41] |
| Diaporthe viciae * | Stems of Vicia villosa (Fabaceae) | China (Guangxi) | Endophyte | [124] |
| Diaporthe viniferae * | Diseased trunk of Vitis vinifera (Vitaceae) | China (Guangxi) | Pathogen | [21] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pereira, D.S.; Hilário, S.; Gonçalves, M.F.M.; Phillips, A.J.L. Diaporthe Species on Palms: Molecular Re-Assessment and Species Boundaries Delimitation in the D. arecae Species Complex. Microorganisms 2023, 11, 2717. https://doi.org/10.3390/microorganisms11112717
Pereira DS, Hilário S, Gonçalves MFM, Phillips AJL. Diaporthe Species on Palms: Molecular Re-Assessment and Species Boundaries Delimitation in the D. arecae Species Complex. Microorganisms. 2023; 11(11):2717. https://doi.org/10.3390/microorganisms11112717
Chicago/Turabian StylePereira, Diana S., Sandra Hilário, Micael F. M. Gonçalves, and Alan J. L. Phillips. 2023. "Diaporthe Species on Palms: Molecular Re-Assessment and Species Boundaries Delimitation in the D. arecae Species Complex" Microorganisms 11, no. 11: 2717. https://doi.org/10.3390/microorganisms11112717
APA StylePereira, D. S., Hilário, S., Gonçalves, M. F. M., & Phillips, A. J. L. (2023). Diaporthe Species on Palms: Molecular Re-Assessment and Species Boundaries Delimitation in the D. arecae Species Complex. Microorganisms, 11(11), 2717. https://doi.org/10.3390/microorganisms11112717