
Features of Mycobacterium bovis Complete Genomes Belonging to 5 Different Lineages

 , , , and
, , , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Mycobacterium bovis Strains
2.2. Additional Genomes
2.3. DNA Extraction
2.4. MinION Sequencing
2.5. Illumina Sequencing
2.6. Genome Assembly Method
2.7. Genome Comparison
2.8. Pangenomic Analysis
2.9. Whole Genome SNP Identification and Selection
2.10. Phylogeny Based on SNP
3. Results
3.1. Complete Genomes Features
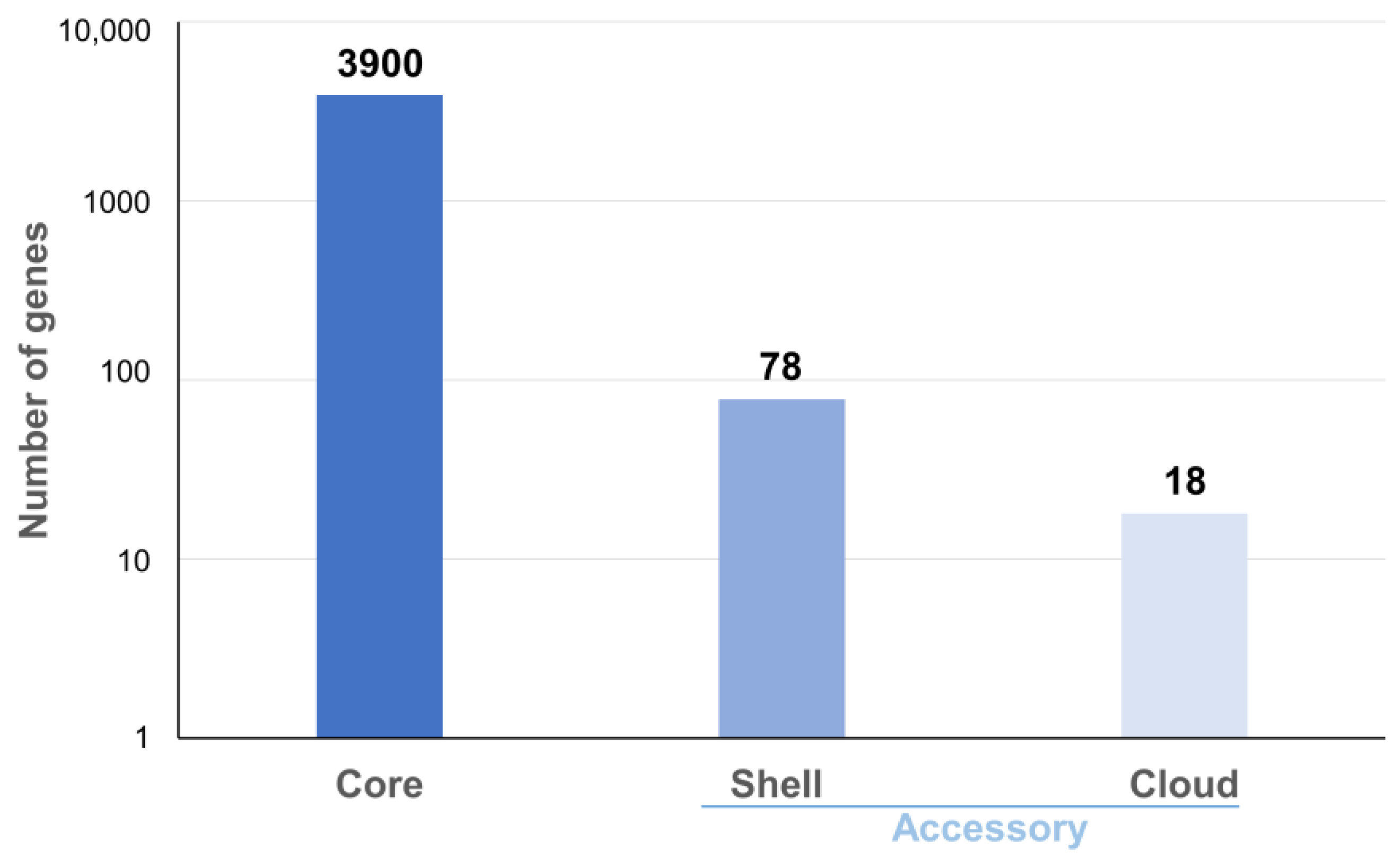
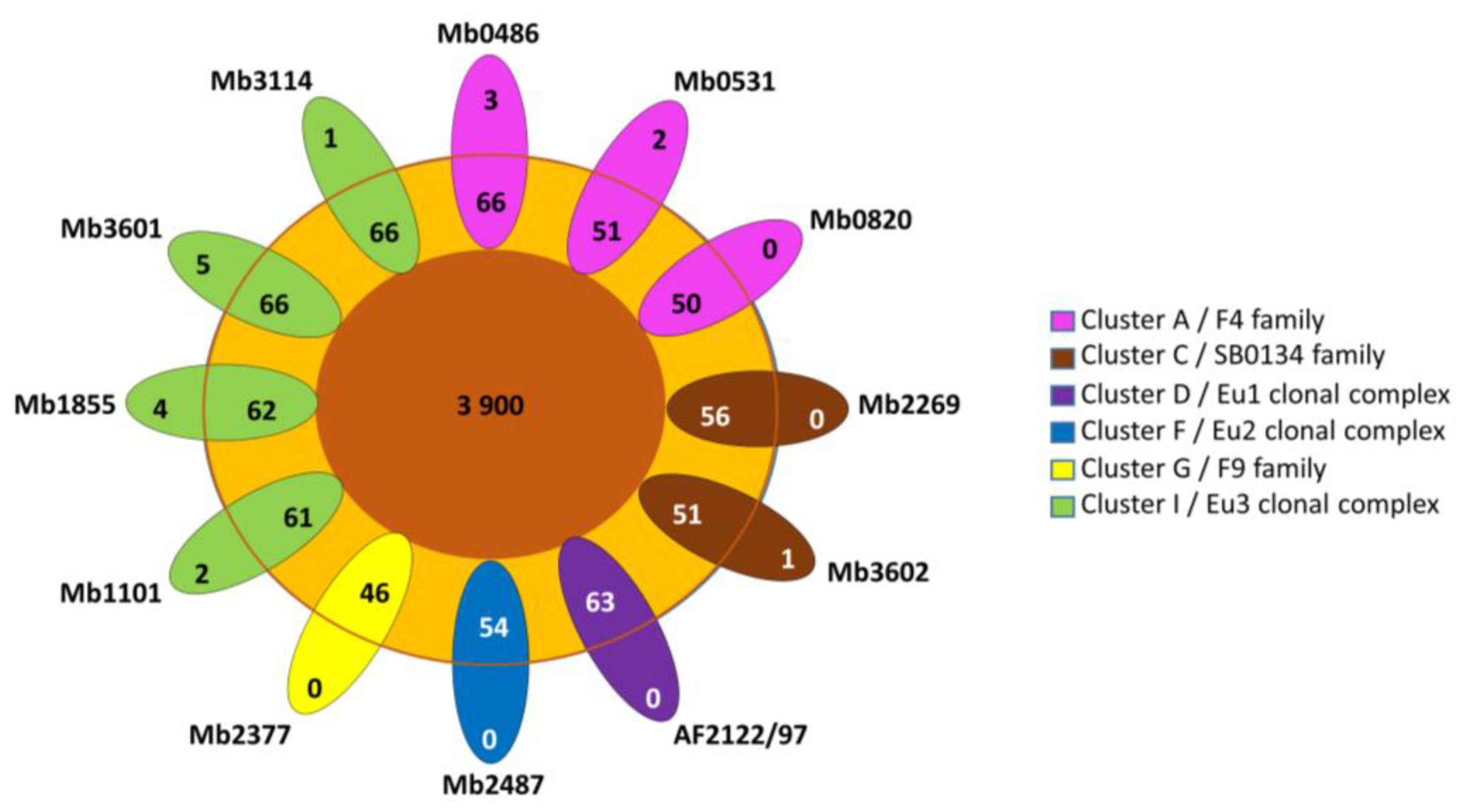
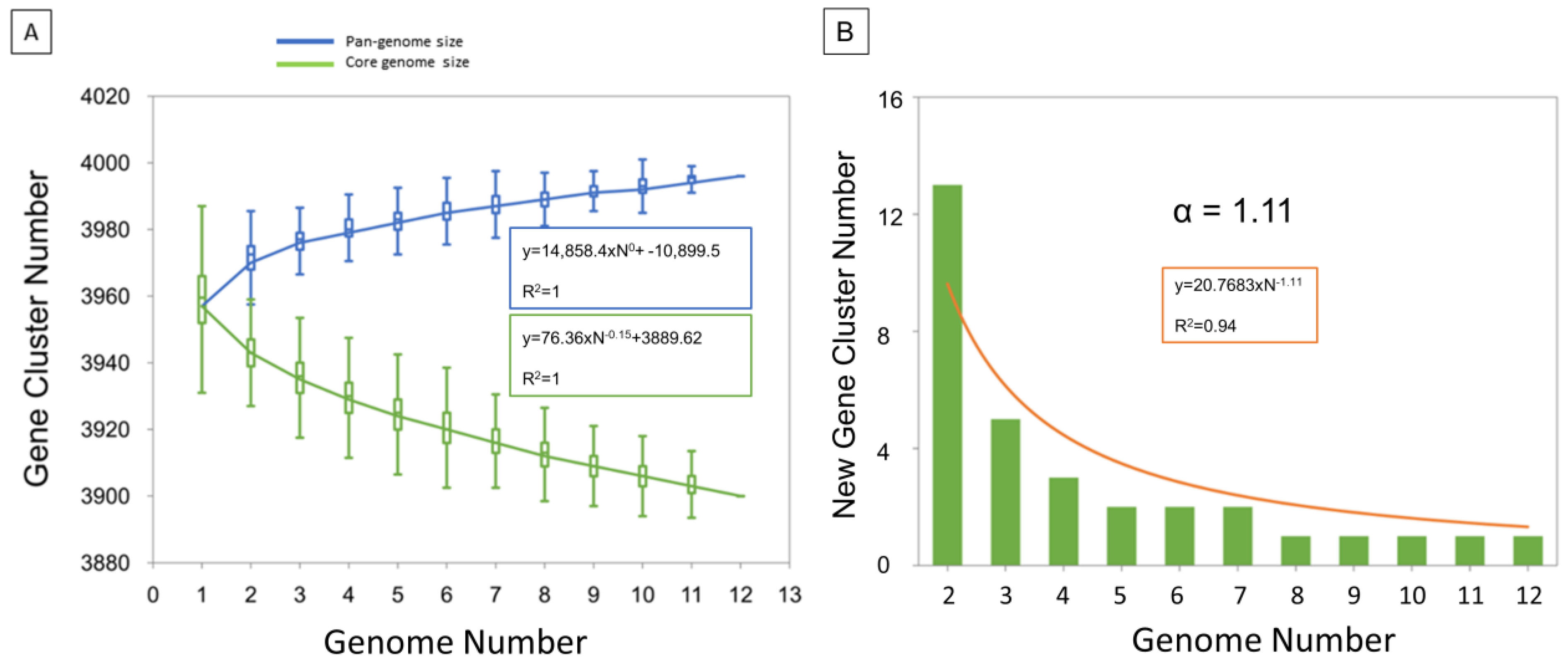
3.2. Pangenome Analysis and Gene Content Variation
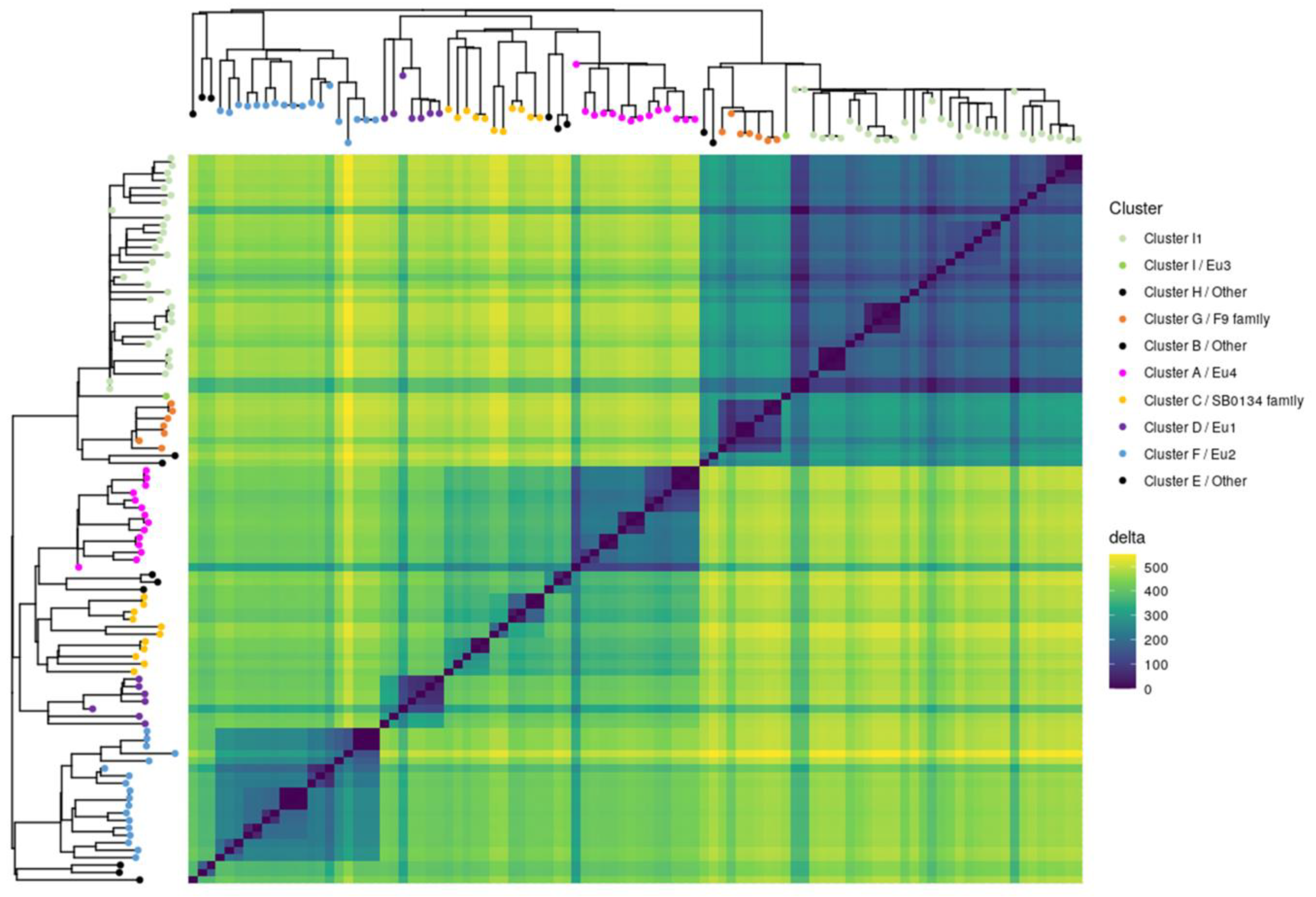
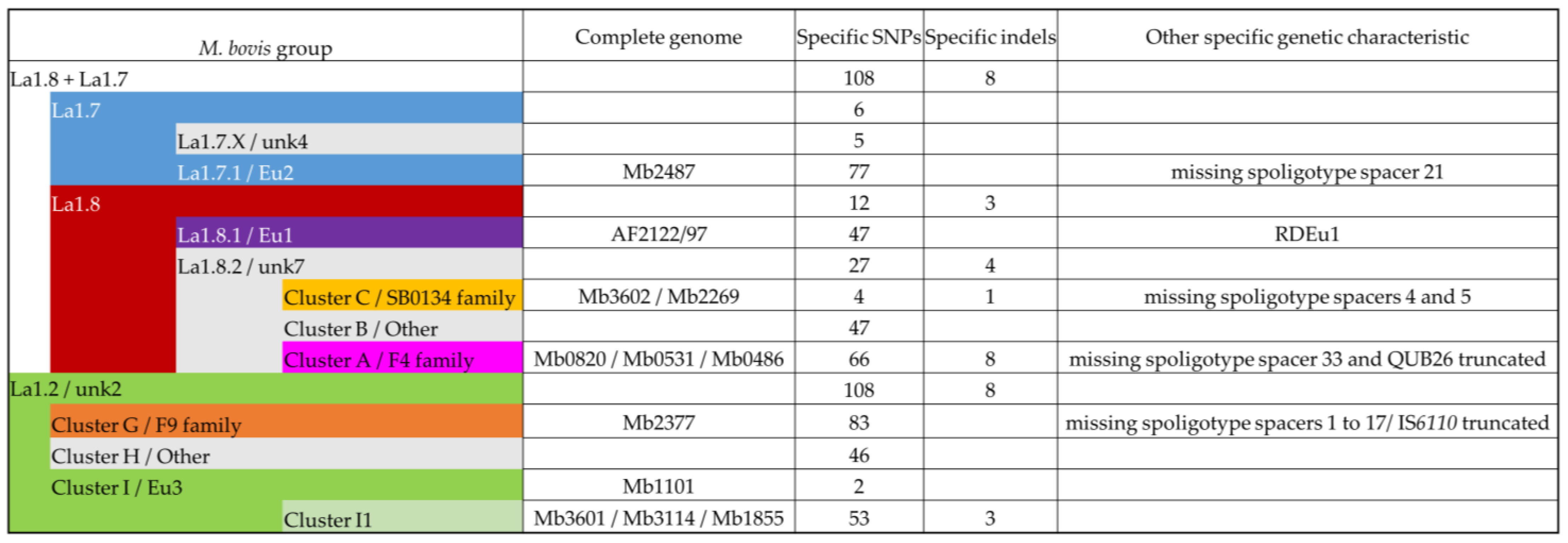
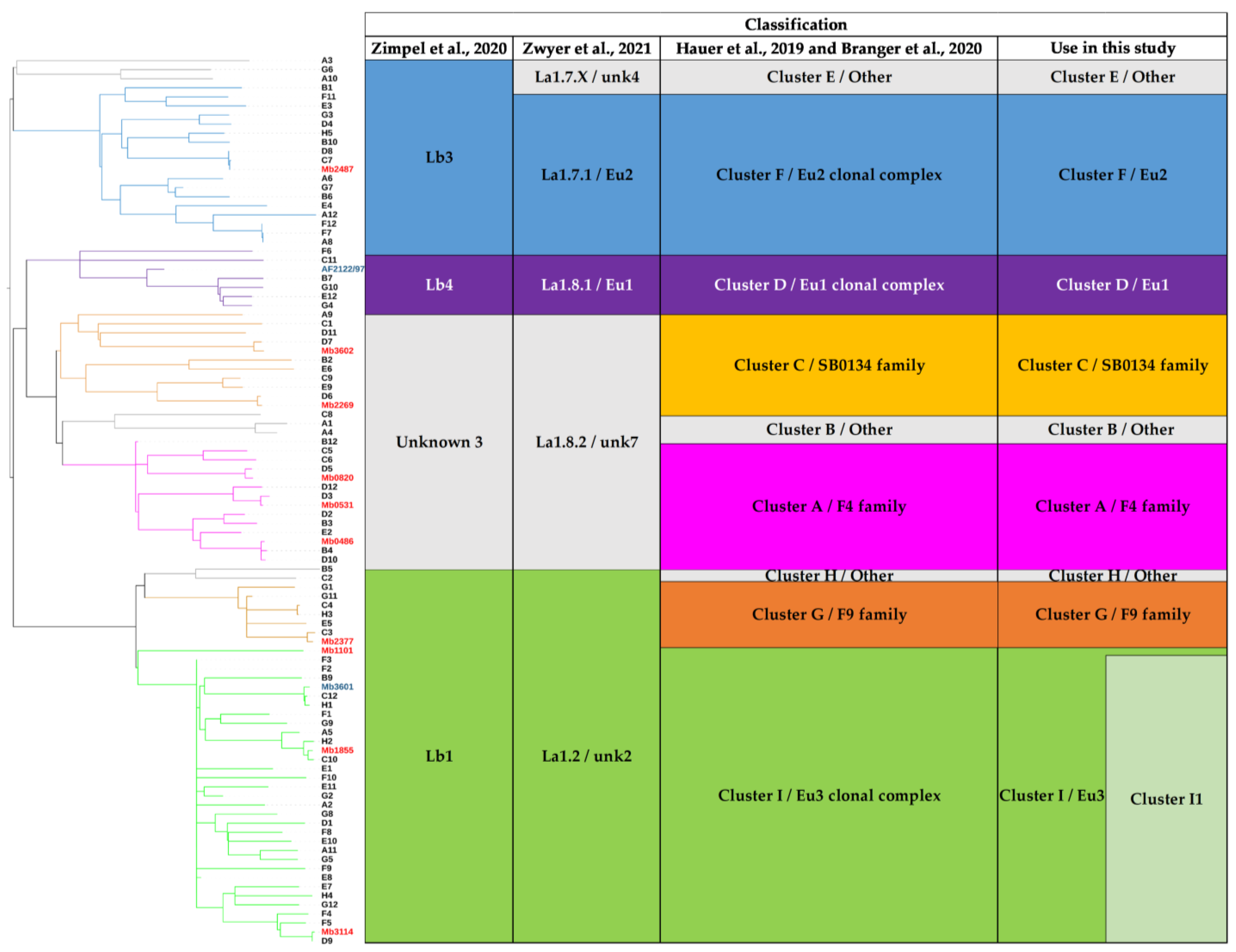
3.3. Contribution of the Complete Genome to M. bovis Lineages Definition
3.3.1. Cluster A/F4 Family
3.3.2. Cluster C/SB0134 Family
3.3.3. Cluster F/Eu2
3.3.4. Lineages 1.7 and 1.8
3.3.5. Cluster G/F9 Family
3.3.6. Cluster I/Eu3
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Berg, S.; Garcia-Pelayo, M.C.; Müller, B.; Hailu, E.; Asiimwe, B.; Kremer, K.; Dale, J.; Boniotti, M.B.; Rodriguez, S.; Hilty, M.; et al. African 2, a Clonal Complex of Mycobacterium bovis Epidemiologically Important in East Africa. J. Bacteriol. 2011, 193, 670–678. [Google Scholar] [CrossRef]
- Muller, B.; Hilty, M.; Berg, S.; Garcia-Pelayo, M.C.; Dale, J.; Boschiroli, M.L.; Cadmus, S.; Ngandolo, B.N.R.; Godreuil, S.; Diguimbaye-Djaibé, C.; et al. African 1, an Epidemiologically Important Clonal Complex of Mycobacterium bovis Dominant in Mali, Nigeria, Cameroon, and Chad. J. Bacteriol. 2009, 191, 1951–1960. [Google Scholar] [CrossRef]
- Rodriguez-Campos, S.; Schürch, A.C.; Dale, J.; Lohan, A.J.; Cunha, M.V.; Botelho, A.; De Cruz, K.; Boschiroli, M.L.; Boniotti, M.B.; Pacciarini, M.; et al. European 2—A clonal complex of Mycobacterium bovis dominant in the Iberian Peninsula. Infect. Genet. Evol. 2012, 12, 866–872. [Google Scholar] [CrossRef]
- Smith, N.H.; Berg, S.; Dale, J.; Allen, A.; Rodriguez, S.; Romero, B.; Matos, F.; Ghebremichael, S.; Karoui, C.; Donati, C.; et al. European 1: A globally important clonal complex of Mycobacterium bovis. Infect. Genet. Evol. 2011, 11, 1340–1351. [Google Scholar] [CrossRef]
- Almaw, G.; Mekonnen, G.A.; Mihret, A.; Aseffa, A.; Taye, H.; Conlan, A.J.K.; Gumi, B.; Zewude, A.; Aliy, A.; Tamiru, M.; et al. Population structure and transmission of Mycobacterium bovis in Ethiopia. Microb. Genom. 2021, 7, 000539. [Google Scholar] [CrossRef]
- da Conceição, M.L.; Conceição, E.C.; Furlaneto, I.P.; Da Silva, S.P.; Guimarães, A.E.D.S.; Gomes, P.; Boschiroli, M.L.; Michelet, L.; Kohl, T.A.; Kranzer, K.; et al. Phylogenomic Perspective on a Unique Mycobacterium bovis Clade Dominating Bovine Tuberculosis Infections among Cattle and Buffalos in Northern Brazil. Sci. Rep. 2020, 10, 1747. [Google Scholar] [CrossRef]
- Hauer, A.; Michelet, L.; Cochard, T.; Branger, M.; Nunez, J.; Boschiroli, M.-L.; Biet, F.; Hauer, A.; Michelet, L.; Cochard, T.; et al. Accurate Phylogenetic Relationships among Mycobacterium bovis Strains Circulating in France Based on Whole Genome Sequencing and Single Nucleotide Polymorphism Analysis. Front. Microbiol. 2019, 10, 955. [Google Scholar] [CrossRef]
- Kohl, T.A.; Kranzer, K.; Andres, S.; Wirth, T.; Niemann, S.; Moser, I. Population Structure of Mycobacterium bovis in Germany: A Long-Term Study Using Whole-Genome Sequencing Combined with Conventional Molecular Typing Methods. J. Clin. Microbiol. 2020, 58, e01573-20. [Google Scholar] [CrossRef]
- Loiseau, C.; Menardo, F.; Aseffa, A.; Hailu, E.; Gumi, B.; Ameni, G.; Berg, S.; Rigouts, L.; Robbe-Austerman, S.; Zinsstag, J.; et al. An African origin for Mycobacterium bovis. Evol. Med. Public Health 2020, 2020, 49–59. [Google Scholar] [CrossRef]
- Zimpel, C.K.; Patané, J.S.L.; Guedes, A.C.P.; de Souza, R.F.; Silva-Pereira, T.T.; Camargo, N.C.S.; Filho, A.F.D.S.; Ikuta, C.Y.; Neto, J.S.F.; Setubal, J.C.; et al. Global Distribution and Evolution of Mycobacterium bovis Lineages. Front. Microbiol. 2020, 11, 843. [Google Scholar] [CrossRef]
- Zwyer, M.; Çavusoglu, C.; Ghielmetti, G.; Pacciarini, M.L.; Scaltriti, E.; Van Soolingen, D.; Dötsch, A.; Reinhard, M.; Gagneux, S.; Brites, D. A new nomenclature for the livestock-associated Mycobacterium tuberculosis complex based on phylogenomics. Open Res. Eur. 2021, 1, 100. [Google Scholar] [CrossRef]
- Boschiroli, M.; Michelet, L.; Hauer, A.; De Cruz, K.; Courcoul, A.; Hénault, S.; Palisson, A.; Karoui, C.; Biet, F.; Zanella, G. Tuberculose bovine en France: Cartographie des souches de Mycobacterium bovis entre 2000–2013. Bull. Épidémiologique 2015, 70, 2–8. [Google Scholar]
- Hauer, A.; De Cruz, K.; Cochard, T.; Godreuil, S.; Karoui, C.; Henault, S.; Bulach, T.; Bañuls, A.-L.; Biet, F.; Boschiroli, M. Genetic evolution of Mycobacterium bovis causing tuberculosis in livestock and wildlife in France since. PLoS ONE 2015, 10, e0117103. [Google Scholar] [CrossRef]
- Michelet, L.; Durand, B.; Boschiroli, M.-L. Tuberculose bovine: Bilan génotypique de M. bovis à l’origine des foyers bovins entre 2015 et 2017 en France Métropolitaine. Bull. Epidemiol. 2020, 91, 13. [Google Scholar]
- Ceres, K.M.; Stanhope, M.J.; Gröhn, Y.T. A critical evaluation of Mycobacterium bovis pangenomics, with reference to its utility in outbreak investigation. Microb. Genom. 2022, 8, 000839. [Google Scholar] [CrossRef]
- Guimaraes, A.M.S.; Zimpel, C.K. Mycobacterium bovis: From Genotyping to Genome Sequencing. Microorganisms 2020, 8, 667. [Google Scholar] [CrossRef]
- Farrell, D.; Crispell, J.; Gordon, S.V. Updated functional annotation of the Mycobacterium bovis AF2122/97 reference genome. Access Microbiol. 2020, 2, e000129. [Google Scholar] [CrossRef]
- Garnier, T.; Eiglmeier, K.; Camus, J.-C.; Medina, N.; Mansoor, H.; Pryor, M.; Duthoy, S.; Grondin, S.; Lacroix, C.; Monsempe, C.; et al. The complete genome sequence of Mycobacterium bovis. Proc. Natl. Acad. Sci. USA 2003, 100, 7877–7882. [Google Scholar] [CrossRef]
- Crispell, J.; Cassidy, S.; Kenny, K.; McGrath, G.; Warde, S.; Cameron, H.; Rossi, G.; MacWhite, T.; White, P.C.L.; Lycett, S.; et al. Mycobacterium bovis genomics reveals transmission of infection between cattle and deer in Ireland. Microb. Genom. 2020, 6, e000388. [Google Scholar] [CrossRef]
- Crispell, J.; Zadoks, R.N.; Harris, S.R.; Paterson, B.; Collins, D.M.; De-Lisle, G.W.; Livingstone, P.; Neill, M.A.; Biek, R.; Lycett, S.J.; et al. Using whole genome sequencing to investigate transmission in a multi-host system: Bovine tuberculosis in New Zealand. BMC Genom. 2017, 18, 180. [Google Scholar] [CrossRef]
- Ortiz, A.P.; Perea, C.; Davalos, E.; Velázquez, E.F.; González, K.S.; Camacho, E.R.; Latorre, E.A.G.; Lara, C.S.; Salazar, R.M.; Bravo, D.M.; et al. Whole Genome Sequencing Links Mycobacterium bovis from Cattle, Cheese and Humans in Baja California, Mexico. Front. Veter-Sci. 2021, 8, 674307. [Google Scholar] [CrossRef]
- Price-Carter, M.; Brauning, R.; De Lisle, G.W.; Livingstone, P.; Neill, M.; Sinclair, J.; Paterson, B.; Atkinson, G.; Knowles, G.; Crews, K.; et al. Whole Genome Sequencing for Determining the Source of Mycobacterium bovis Infections in Livestock Herds and Wildlife in New Zealand. Front. Veter-Sci. 2018, 5, 272. [Google Scholar] [CrossRef]
- Salvador, L.C.M.; O’Brien, D.J.; Cosgrove, M.K.; Stuber, T.P.; Schooley, A.M.; Crispell, J.; Church, S.V.; Gröhn, Y.T.; Robbe-Austerman, S.; Kao, R.R. Disease management at the wildlife-livestock interface: Using whole-genome sequencing to study the role of elk in Mycobacterium bovis transmission in Michigan, USA. Mol. Ecol. 2019, 28, 2192–2205. [Google Scholar] [CrossRef]
- Branger, M.; Loux, V.; Cochard, T.; Boschiroli, M.L.; Biet, F.; Michelet, L. The complete genome sequence of Mycobacterium bovis Mb3601, a SB0120 spoligotype strain representative of a new clonal group. Infect. Genet. Evol. 2020, 82, 104309. [Google Scholar] [CrossRef]
- Delavenne, C.; Pandofi, F.; Girard, S.; Réveillaud, É.; Boschiroli, M.-L.; Dommergues, L.; Garapin, F.; Keck, N.; Martin, F.; Moussu, M.; et al. Tuberculose bovine: Bilan et évolution de la situation épidémiologique entre 2015 et 2017 en France Métropolitaine. Bull Epidemiol 91:12 (in French). Bull. Epidemiol. 2020, 91, 12. [Google Scholar]
- Biek, R.; O’Hare, A.; Wright, D.; Mallon, T.; McCormick, C.; Orton, R.J.; McDowell, S.; Trewby, H.; Skuce, R.A.; Kao, R.R. Whole Genome Sequencing Reveals Local Transmission Patterns of Mycobacterium bovis in Sympatric Cattle and Badger Populations. PLOS Pathog. 2012, 8, e1003008. [Google Scholar] [CrossRef]
- Imai, T.; Ohta, K.; Kigawa, H.; Kanoh, H.; Taniguchi, T.; Tobari, J. Preparation of High-Molecular-Weight DNA: Application to Mycobacterial Cells. Anal. Biochem. 1994, 222, 479–482. [Google Scholar] [CrossRef]
- Wingett, S.W.; Andrews, S. FastQ Screen: A tool for multi-genome mapping and quality control. F1000Research 2018, 7, 1338. [Google Scholar] [CrossRef]
- Xia, E.; Teo, Y.-Y.; Ong, R.T.-H. SpoTyping: Fast and accurate in silico Mycobacterium spoligotyping from sequence reads. Genome Med. 2016, 8, 1–9. [Google Scholar] [CrossRef]
- De Coster, W.; D’Hert, S.; Schultz, D.T.; Cruts, M.; Van Broeckhoven, C. NanoPack: Visualizing and processing long-read sequencing data. Bioinformatics 2018, 34, 2666–2669. [Google Scholar] [CrossRef]
- Wick, R.R.; Judd, L.M.; Cerdeira, L.T.; Hawkey, J.; Méric, G.; Ben Vezina, B.; Wyres, K.L.; Holt, K.E. Trycycler: Consensus long-read assemblies for bacterial genomes. Genome Biol. 2021, 22, 266. [Google Scholar] [CrossRef]
- Kolmogorov, M.; Yuan, J.; Lin, Y.; Pevzner, P.A. Assembly of long, error-prone reads using repeat graphs. Nat. Biotechnol. 2019, 37, 540–546. [Google Scholar] [CrossRef]
- Vaser, R.; Šikić, M. Raven: A de novo genome assembler for long reads. bioRxiv 2021. [Google Scholar] [CrossRef]
- Wick, R.R.; Judd, L.M.; Gorrie, C.L.; Holt, K.E. Unicycler: Resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput. Biol. 2017, 13, e1005595. [Google Scholar] [CrossRef]
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.A.; Zeng, Q.; Wortman, J.; Young, S.K.; et al. Pilon: An Integrated Tool for Comprehensive Microbial Variant Detection and Genome Assembly Improvement. PLoS ONE 2014, 9, e112963. [Google Scholar] [CrossRef]
- Hunt, M.; De Silva, N.; Otto, T.D.; Parkhill, J.; Keane, J.A.; Harris, S.R. Circlator: Automated circularization of genome assemblies using long sequencing reads. Genome Biol. 2015, 16, 294. [Google Scholar] [CrossRef]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef]
- Darling, A.E.; Tritt, A.; Eisen, J.A.; Facciotti, M.T. Mauve Assembly Metrics. Bioinformatics 2011, 27, 2756–2757. [Google Scholar] [CrossRef]
- Bespiatykh, D.; Bespyatykh, J.; Mokrousov, I.; Shitikov, E. A Comprehensive Map of Mycobacterium tuberculosis Complex Regions of Difference. Msphere 2021, 6, e0053521. [Google Scholar] [CrossRef]
- Seemann, T. Prokka: Rapid Prokaryotic Genome Annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef]
- Hyatt, D.; Chen, G.-L.; Locascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 2010, 11, 119. [Google Scholar] [CrossRef]
- Tonkin-Hill, G.; MacAlasdair, N.; Ruis, C.; Weimann, A.; Horesh, G.; Lees, J.A.; Gladstone, R.A.; Lo, S.; Beaudoin, C.; Floto, R.A.; et al. Producing polished prokaryotic pangenomes with the Panaroo pipeline. Genome Biol. 2020, 21, 180. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Jia, X.; Yang, J.; Ling, Y.; Zhang, Z.; Yu, J.; Wu, J.; Xiao, J. PanGP: A tool for quickly analyzing bacterial pan-genome profile. Bioinformatics 2014, 30, 1297–1299. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows—Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve years of SAMtools and BCFtools. GigaScience 2021, 10, giab008. [Google Scholar] [CrossRef]
- Marin, M.; Vargas, R.; Harris, M.; Jeffrey, B.; Epperson, L.E.; Durbin, D.; Strong, M.; Salfinger, M.; Iqbal, Z.; Akhundova, I.; et al. Genomic sequence characteristics and the empiric accuracy of short-read sequencing. bioRxiv 2021. [Google Scholar] [CrossRef]
- Lorente-Leal, V.; Farrell, D.; Romero, B.; Álvarez, J.; de Juan, L.; Gordon, S.V. Performance and Agreement Between WGS Variant Calling Pipelines Used for Bovine Tuberculosis Control: Toward International Standardization. Front. Veter-Sci. 2021, 8, 780018. [Google Scholar] [CrossRef]
- Meehan, C.J.; Goig, G.; Kohl, T.A.; Verboven, L.; Dippenaar, A.; Ezewudo, M.; Farhat, M.R.; Guthrie, J.L.; Laukens, K.; Miotto, P.; et al. Whole genome sequencing of Mycobacterium tuberculosis: Current standards and open issues. Nat. Rev. Genet. 2019, 17, 533–545. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef] [PubMed]
- Charles, C.; Conde, C.; Biet, F.; Boschiroli, M.L.; Michelet, L. IS6110 Copy Number in Multi-Host Mycobacterium bovis Strains Circulating in Bovine Tuberculosis Endemic French Regions. Front. Microbiol. 2022, 13, 891902. [Google Scholar] [CrossRef] [PubMed]
- Dale, J.W. Mobile genetic elements in mycobacteria. Eur. Respir. J. Suppl. 1995, 20, 633s–648s. [Google Scholar]
- Mendiola, M.; Martin, C.; Otal, I.; Gicquel, B. Analysis of the regions responsible for IS6110 RFLP in a single Mycobacterium tuberculosis strain. Res. Microbiol. 1992, 143, 767–772. [Google Scholar] [CrossRef]
- Gupta, A.; Alland, D. Reversible gene silencing through frameshift indels and frameshift scars provide adaptive plasticity for Mycobacterium tuberculosis. Nat. Commun. 2021, 12, 4702. [Google Scholar] [CrossRef]
- Galagan, J.E. Genomic insights into tuberculosis. Nat. Rev. Genet. 2014, 15, 307–320. [Google Scholar] [CrossRef]
- Contreras-Moreira, B.; Vinuesa, P. GET_HOMOLOGUES, a Versatile Software Package for Scalable and Robust Microbial Pangenome Analysis. Appl. Environ. Microbiol. 2013, 79, 7696–7701. [Google Scholar] [CrossRef]
- Reis, A.C.; Cunha, M.V. The open pan-genome architecture and virulence landscape of Mycobacterium bovis. Microb. Genom. 2021, 7, 000664. [Google Scholar] [CrossRef]
- Richard, G. Eukaryotic Pangenomes. In The Pangenome: Diversity, Dynamics and Evolution of Genomes; Tettelin, H., Medini, D., Eds.; Springer: Cham, Switzerland, 2020. [Google Scholar] [CrossRef]
- Zimpel, C.K.; Brandão, P.E.; de Souza Filho, A.F.; de Souza, R.F.; Ikuta, C.Y.; Ferreira Neto, J.S.; Camargo, N.C.S.; Heinemann, M.B.; Guimarães, A.M.S. Complete Genome Sequencing of Mycobacterium bovis SP38 and Comparative Genomics of Mycobacterium bovis and M. tuberculosis Strains. Front. Microbiol. 2017, 8, 2389. [Google Scholar] [CrossRef]
- Baumler, A.; Fang, F.C. Host Specificity of Bacterial Pathogens. Cold Spring Harb. Perspect. Med. 2013, 3, a010041. [Google Scholar] [CrossRef]
- Bolotin, E.; Hershberg, R. Gene Loss Dominates As a Source of Genetic Variation within Clonal Pathogenic Bacterial Species. Genome Biol. Evol. 2015, 7, 2173–2187. [Google Scholar] [CrossRef]
- Gonzalo-Asensio, J.; Pérez, I.; Aguilo, N.; Uranga, S.; Picó, A.; Lampreave, C.; Cebollada, A.; Otal, I.; Samper, S.; Martín, C. New insights into the transposition mechanisms of IS6110 and its dynamic distribution between Mycobacterium tuberculosis Complex lineages. PLOS Genet. 2018, 14, e1007282. [Google Scholar] [CrossRef]
- Refrégier, G.; Sola, C.; Guyeux, C. Unexpected diversity of CRISPR unveils some evolutionary patterns of repeated sequences in Mycobacterium tuberculosis. BMC Genom. 2020, 21, 1–12. [Google Scholar] [CrossRef]
- Soto, C.Y.; Menéndez, M.C.; Pérez, E.; Samper, S.; Gómez, A.B.; García, M.J.; Martín, C. IS6110 Mediates Increased Transcription of the phoP Virulence Gene in a Multidrug-Resistant Clinical Isolate Responsible for Tuberculosis Outbreaks. J. Clin. Microbiol. 2004, 42, 212–219. [Google Scholar] [CrossRef]
- Fan, X.; Alla, A.A.E.A.; Xie, J. Distribution and function of prophage phiRv1 and phiRv2 among Mycobacterium tuberculosis complex. J. Biomol. Struct. Dyn. 2015, 34, 233–238. [Google Scholar] [CrossRef]
- Mahairas, G.G.; Sabo, P.J.; Hickey, M.J.; Singh, D.C.; Stover, C. Molecular analysis of genetic differences between Mycobacterium bovis BCG and virulent M. bovis. J. Bacteriol. 1996, 178, 1274–1282. [Google Scholar] [CrossRef]
- Lombardi, G.; Botti, I.; Pacciarini, M.L.; Boniotti, M.B.; Roncarati, G.; Monte, P.D. Five-year surveillance of human tuberculosis caused by Mycobacterium bovis in Bologna, Italy: An underestimated problem. Epidemiology Infect. 2017, 145, 3035–3039. [Google Scholar] [CrossRef]
- Driscoll, J.R. Spoligotyping for Molecular Epidemiology of the Mycobacterium tuberculosis Complex. Methods Mol. Biol. 2009, 551, 117–128. [Google Scholar] [CrossRef]
- Kamerbeek, J.; Schouls, L.; Kolk, A.; van Agterveld, M.; van Soolingen, D.; Kuijper, S.; Bunschoten, A.; Molhuizen, H.; Shaw, R.; Goyal, M.; et al. Simultaneous detection and strain differentiation of Mycobacterium tuberculosis for diagnosis and epidemiology. J. Clin. Microbiol. 1997, 35, 907–914. [Google Scholar] [CrossRef]
- Supply, P.; Allix, C.; Lesjean, S.; Cardoso-Oelemann, M.; Rüsch-Gerdes, S.; Willery, E.; Savine, E.; de Haas, P.; van Deutekom, H.; Roring, S.; et al. Proposal for Standardization of Optimized Mycobacterial Interspersed Repetitive Unit-Variable-Number Tandem Repeat Typing of Mycobacterium tuberculosis. J. Clin. Microbiol. 2006, 44, 4498–4510. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Mb2487 | Mb3602 | Mb2269 | Mb0820 | Mb0531 | Mb0486 | Mb2377 | Mb1101 | Mb1855 | Mb3114 |
|---|---|---|---|---|---|---|---|---|---|---|
| Accesion Number | CP096839 | CP096843 | CP096840 | CP096841 | CP096847 | CP096848 | CP096846 | CP096845 | CP096844 | CP096842 |
| Host species | Cattle | Deer | Cattle | Cattle | Cattle | Cattle | Cattle | Cattle | Cattle | Cattle |
| Spoligotype ID | SB0999 | SB0134 | SB0134 | SB0840 | SB0826 | SB0821 | SB0853 | SB0120 | SB0120 | SB0120 |
| MLVA profile * | 6 4 5 2 8 2 4 7 | 7 4 5 3 10 4 5 10 | 6 5 5 3 6 4 5 6 | 7 5 5 3 8 2 5 s 4 | 6 7 3 3 10 2 5 s 8 | 6 5 5 3 11 2 5 s 4 | 3 6 5 2 9 3 4 6 | 5 2 3 3 10 3 3 10 | 5 3 5 3 9 4 5 6 | 5 5 5 3 11 3 5 4 |
| Cluster | F | C | C | A | A | A | G | I | I | I |
| Alias | Eu2 CC | SB0134 family | SB0134 family | F4 family | F4 family | F4 family | F9 family | Eu3 CC | Eu3 CC | Eu3 CC |
| Lenght (bp) | 4,344,516 | 4,343,218 | 4,351,057 | 4,344,564 | 4,342,977 | 4,340,629 | 4,338,946 | 4,343,846 | 4,362,894 | 4,353,147 |
| GC (%) | 65.62 | 65.65 | 65.64 | 65.64 | 65.64 | 65.65 | 65.65 | 65.64 | 65.64 | 64.65 |
| CDS | 4012 | 3999 | 4014 | 4006 | 4005 | 3991 | 3986 | 4014 | 4034 | 4015 |
| rRNA | 3 | 3 | 3 | 3 | 3 | 3 | 3 | 3 | 3 | 3 |
| tRNA | 52 | 52 | 52 | 52 | 52 | 52 | 52 | 52 | 52 | 51 |
| tmRNA | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| IS6110 Nb | 3 | 1 | 3 | 2 | 4 | 3 | 1 truncated | 1 | 12 | 1 |
| IS1561 Nb | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| IS1081 Nb | 5 + 1 truncated | 5 + 1 truncated | 5 + 1 truncated | 5 + 1 truncated | 5 + 1 truncated | 5 + 1 truncated | 5 + 1 truncated | 5 + 1 truncated | 5 + 1 truncated | 5 + 1 truncated |
| Nomenclature | Length (in bp) | Number of Locus Tags Associated | Genome |
|---|---|---|---|
| Indel-Mb0531-33 | 2384 | 5 | Mb0531 |
| Indel-Mb0486-6 | 3148 | 4 | Mb0486 |
| Indel-Mb0486-11 | 3634 | 4 | Mb0486 |
| Indel-Mb3602-33 | 2150 | 2 | Mb3602 |
| Indel-Mb2269-1 | 2122 | 4 | Mb2269 |
| Indel-Mb2269-24 | 2368 | 6 | Mb2269 |
| Indel-Mb2487-5 | 2691 | 3 | Mb2487 |
| Indel-Mb2487-36 | 2387 | 6 | Mb2487 |
| Indel-Mb2487-50/RDBovis | 2409 | 3 | Mb2487 |
| Indel-Mb2377-27 | 5539 | 6 | Mb2377 |
| Indel-Mb1101-1 | 2966 | 2 | Mb1101 |
| Indel-Mb1101-8 | 4384 | 2 | Mb1101 |
| Indel-Mb1101-21 | 1160 | 1 | Mb1101 |
| Indel-Mb1855-26 | 1730 | 1 | Mb1855 |
| Indel-Mb1855-29 | 3058 | 3 | Mb1855 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Charles, C.; Conde, C.; Vorimore, F.; Cochard, T.; Michelet, L.; Boschiroli, M.L.; Biet, F. Features of Mycobacterium bovis Complete Genomes Belonging to 5 Different Lineages. Microorganisms 2023, 11, 177. https://doi.org/10.3390/microorganisms11010177
Charles C, Conde C, Vorimore F, Cochard T, Michelet L, Boschiroli ML, Biet F. Features of Mycobacterium bovis Complete Genomes Belonging to 5 Different Lineages. Microorganisms. 2023; 11(1):177. https://doi.org/10.3390/microorganisms11010177
Chicago/Turabian StyleCharles, Ciriac, Cyril Conde, Fabien Vorimore, Thierry Cochard, Lorraine Michelet, Maria Laura Boschiroli, and Franck Biet. 2023. "Features of Mycobacterium bovis Complete Genomes Belonging to 5 Different Lineages" Microorganisms 11, no. 1: 177. https://doi.org/10.3390/microorganisms11010177
APA StyleCharles, C., Conde, C., Vorimore, F., Cochard, T., Michelet, L., Boschiroli, M. L., & Biet, F. (2023). Features of Mycobacterium bovis Complete Genomes Belonging to 5 Different Lineages. Microorganisms, 11(1), 177. https://doi.org/10.3390/microorganisms11010177