
Genomic Diversity of the Rarely Observed Genotype of the Mycobacterium tuberculosis Central Asian (CAS) Lineage 3 from North Brazil

 , , , , ,
, , , , ,  ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Population
2.2. Genotyping Based on MIRU-VNTR
2.3. Whole-Genome Sequencing
2.4. Genome Analysis and Phylogenetic Reconstruction
2.5. Ethical Statement
3. Results
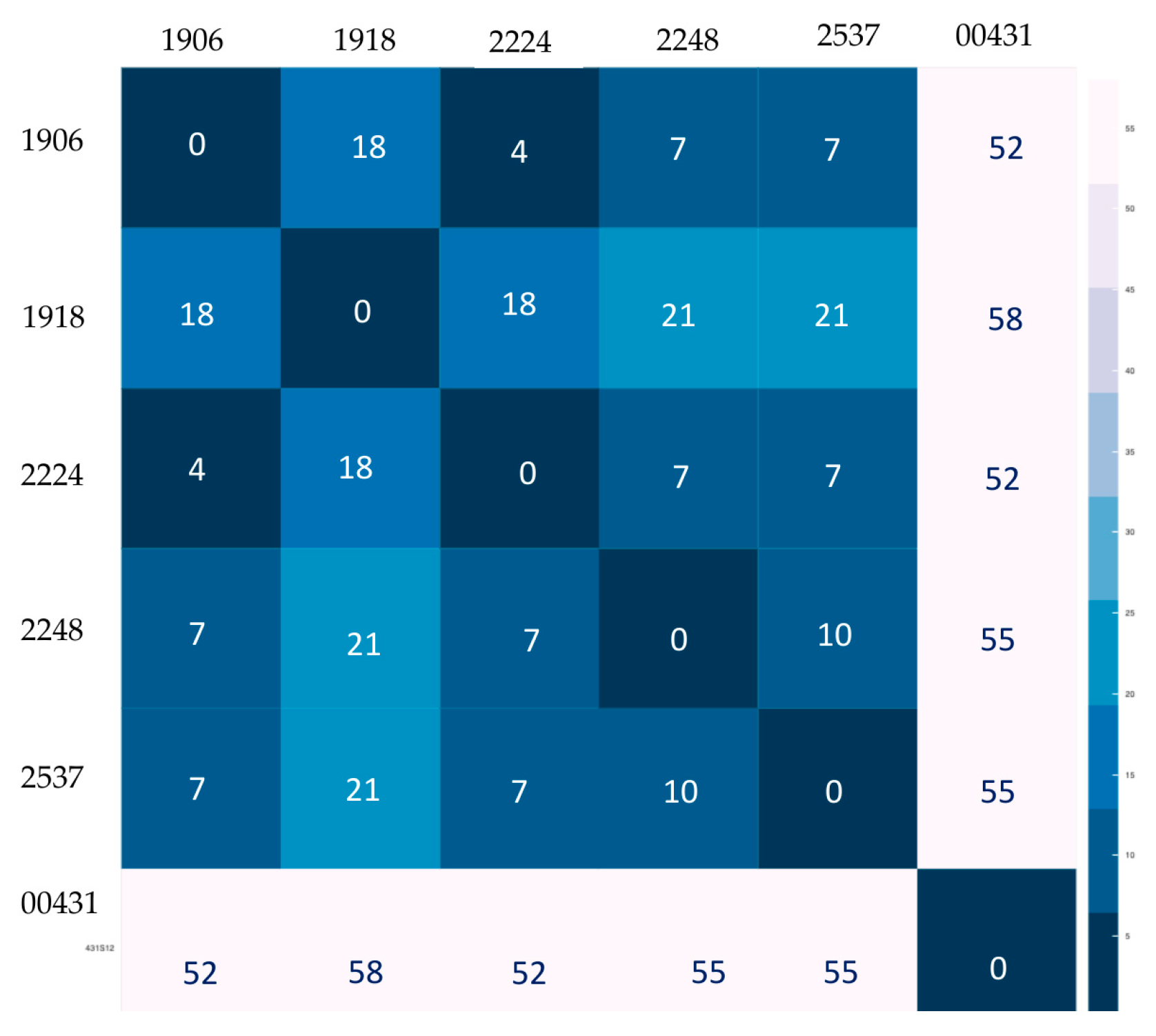
3.1. MIRU-VNTR Result
3.2. Whole Genome Sequencing Analysis and Phylogenomic Assessment
3.3. In Silico Genomic Analyses
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Couvin, D.; Reynaud, Y.; Rastogi, N. Two tales: Worldwide distribution of Central Asian (CAS) versus ancestral East-African Indian (EAI) lineages of Mycobacterium tuberculosis underlines a remarkable cleavage for phylogeographical, epidemiological and demographical characteristics. PLoS ONE 2019, 14, e0219706. [Google Scholar] [CrossRef] [PubMed]
- Menardo, F.; Rutaihwa, L.K.; Zwyer, M.; Borrell, S.; Comas, I.; Conceição, E.C.; Coscolla, M.; Cox, H.; Joloba, M.; Dou, H.-Y.; et al. Local adaptation in populations of Mycobacterium tuberculosis endemic to the Indian Ocean Rim. F1000Research 2021, 10, 60. [Google Scholar] [CrossRef] [PubMed]
- Coscolla, M.; Gagneux, S. Consequences of genomic diversity in Mycobacterium tuberculosis. Semin. Immunol. 2014, 26, 431–444. [Google Scholar] [CrossRef] [PubMed]
- Yimer, S.A.; Kalayou, S.; Homberset, H.; Birhanu, A.G.; Riaz, T.; Zegeye, E.D.; Lutter, T.; Abebe, M.; Holm-Hansen, C.; Aseffa, A.; et al. Lineage-Specific Proteomic Signatures in the Mycobacterium tuberculosis Complex Reveal Differential Abundance of Proteins Involved in Virulence, DNA Repair, CRISPR-Cas, Bioenergetics and Lipid Metabolism. Front. Microbiol. 2020, 11, 550760. [Google Scholar] [CrossRef] [PubMed]
- Conceição, E.C.; Salvato, R.S.; Gomes, K.M.; Guimarães, A.E.D.S.; da Conceição, M.L.; Guimarães, R.J.D.P.S.E.; Sharma, A.; Furlaneto, I.P.; Barcellos, R.B.; Bollela, V.R.; et al. Molecular epidemiology of Mycobacterium tuberculosis in Brazil before the whole genome sequencing era: A literature review. Memórias Do Inst. Oswaldo Cruz 2021, 116, e200517. [Google Scholar] [CrossRef]
- Conceição, E.C.; Rastogi, N.; Couvin, D.; Lopes, M.L.; Furlaneto, I.P.; Gomes, H.M.; Vasconcellos, S.E.G.; Suffys, P.N.; Schneider, M.P.C.; de Sousa, M.S.; et al. Genetic diversity of Mycobacterium tuberculosis from Pará, Brazil, reveals a higher frequency of ancestral strains than previously reported in South America. Infect. Genet. Evol. 2017, 56, 62–72. [Google Scholar] [CrossRef]
- Conceição, E.C.; Guimarães, A.E.D.S.; Lopes, M.L.; Furlaneto, I.P.; Rodrigues, Y.C.; Da Conceição, M.L.; Barros, W.A.; Cardoso, N.C.; Sharma, A.; Lima, L.N.G.C.; et al. Analysis of potential household transmission events of tuberculosis in the city of Belem, Brazil. Tuberculosis 2018, 113, 125–129. [Google Scholar] [CrossRef]
- Guimarães, A.E.D.S.; Sharma, A.; Furlaneto, I.P.; Rutaihwa, L.; Cardoso, J.F.; da Conceição, M.L.; Spinassé, L.B.; Machado, E.; Lopes, M.L.; Duarte, R.S.; et al. Evaluation of drug susceptibility profile of Mycobacterium tuberculosis Lineage 1 from Brazil based on whole genome sequencing and phenotypic methods. Memórias Do Inst. Oswaldo Cruz 2020, 115, e200520. [Google Scholar] [CrossRef]
- Reyes, J.F.; Chan, C.H.; Tanaka, M.M. Impact of homoplasy on variable numbers of tandem repeats and spoligotypes in Mycobacterium tuberculosis. Infect. Genet. Evol. 2012, 12, 811–818. [Google Scholar] [CrossRef]
- Yasmin, M.; Le Moullec, S.; Siddiqui, R.T.; De Beer, J.; Sola, C.; Refrégier, G. Quick and cheap MIRU-VNTR typing of Mycobacterium tuberculosis species complex using duplex PCR. Tuberculosis 2016, 101, 160–163. [Google Scholar] [CrossRef]
- Weniger, T.; Krawczyk, J.; Supply, P.; Niemann, S.; Harmsen, D. MIRU-VNTRplus: A web tool for polyphasic genotyping of Mycobacterium tuberculosis complex bacteria. Nucleic Acids Res. 2010, 38, W326–W331. [Google Scholar] [CrossRef][Green Version]
- Wingett, S.W.; Andrews, S. FastQ Screen: A tool for multi-genome mapping and quality control. F1000Res 2018, 7, 1338. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- García-Alcalde, F.; Okonechnikov, K.; Carbonell, J.; Cruz, L.; Götz, S.; Tarazona, S.; Dopazo, J.; Meyer, T.; Conesa, A. Qualimap: Evaluating Next-Generation Sequencing Alignment Data—PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/22914218/ (accessed on 30 November 2022).
- Tarasov, A.; Vilella, A.J.; Cuppen, E.; Nijman, I.J.; Prins, P. Genome Analysis Sambamba: Fast Processing of NGS Alignment Formats. Bioinformatics 2015, 31, 2032–2034. [Google Scholar] [CrossRef]
- Grüning, B.; Dale, R.; Sjödin, A.; Chapman, B.A.; Rowe, J.; Tomkins-Tinch, C.H.; Valieris, R.; Köster, J.; Bioconda Team. Bioconda: Sustainable and comprehensive software distribution for the life sciences. Nat. Methods 2018, 15, 475–476. [Google Scholar] [CrossRef]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef]
- Phelan, J.E.; O’Sullivan, D.M.; Machado, D.; Ramos, J.; Oppong, Y.E.A.; Campino, S.; O’Grady, J.; McNerney, R.; Hibberd, M.L.; Viveiros, M.; et al. Integrating informatics tools and portable sequencing technology for rapid detection of resistance to anti-tuberculous drugs. Genome Med. 2019, 11, 41. [Google Scholar] [CrossRef]
- Xia, E.; Teo, Y.-Y.; Ong, R.T.-H. SpoTyping: Fast and accurate in silico Mycobacterium spoligotyping from sequence reads. Genome Med. 2016, 8, 19. [Google Scholar] [CrossRef]
- Faksri, K.; Xia, E.; Tan, J.H.; Teo, Y.-Y.; Ong, R.T.-H. In silico region of difference (RD) analysis of Mycobacterium tuberculosis complex from sequence reads using RD-Analyzer. BMC Genom. 2016, 17, 847. [Google Scholar] [CrossRef]
- Seemann, T. Prokka: Rapid Prokaryotic Genome Annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef]
- Gomes, H.M.; Elias, A.R.; Oelemann, M.A.C.; Pereira, M.A.D.S.; Montes, F.F.O.; Marsico, A.G.; Kritski, A.L.; Filho, L.D.A.; Caldas, P.C.; Possuelo, L.G.; et al. Spoligotypes of Mycobacterium tuberculosis complex isolates from patients residents of 11 states of Brazil. Infect. Genet. Evol. 2012, 12, 649–656. [Google Scholar] [CrossRef]
- De Almeida, I.N.; Vasconcellos, S.E.G.; Figueredo, L.J.D.A.; Dantas, N.G.T.; Augusto, C.J.; Hadaad, J.P.A.; Suffys, P.N.; Carvalho, W.D.S.; De Miranda, S.S. Frequency of the Mycobacterium tuberculosis RDRio genotype and its association with multidrug-resistant tuberculosis. BMC Infect. Dis. 2019, 19, 556. [Google Scholar] [CrossRef]
- Garzon-Chavez, D.; Garcia-Bereguiain, M.A.; Mora-Pinargote, C.; Granda-Pardo, J.C.; Leon-Benitez, M.; Franco-Sotomayor, G.; Trueba, G.; de Waard, J.H. Population structure and genetic diversity of Mycobacterium tuberculosis in Ecuador. Sci. Rep. 2020, 10, 6237. [Google Scholar] [CrossRef]
- Hadifar, S.; Kamakoli, M.; Fateh, A.; Siadat, S.D.; Vaziri, F. Enhancing the Differentiation of Specific Genotypes in Mycobacterium Tuberculosis Population—PubMed. Available online: https://pubmed.ncbi.nlm.nih.gov/31784605/ (accessed on 30 November 2022).
- Merker, M.; Kohl, T.A.; Niemann, S.; Supply, P. The Evolution of Strain Typing in the Mycobacterium tuberculosis Complex. Strain Var. Mycobacterium Tuberc. Complex Its Role Biol. Epidemiol. Control 2017, 1019, 43–78. [Google Scholar] [CrossRef]
- da Conceição, M.L.; Conceição, E.C.; Furlaneto, I.P.; Da Silva, S.P.; Guimarães, A.E.D.S.; Gomes, P.; Boschiroli, M.L.; Michelet, L.; Kohl, T.A.; Kranzer, K.; et al. Phylogenomic Perspective on a Unique Mycobacterium bovis Clade Dominating Bovine Tuberculosis Infections among Cattle and Buffalos in Northern Brazil. Sci. Rep. 2020, 10, 1747. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Strain | Year | DST 1 | Sex | Age | MIRU-VNTR 2 | WGS Cluster 3 |
|---|---|---|---|---|---|---|
| 431 | 2001 | MDR | F | 34 | 242335542224434138443843 | Non clustered |
| 1906 | 2005 | Susc | M | 29 | 242335542124424178343843 | Cluster 1 |
| 1918 | 2010 | Susc | F | 61 | 242335542124424178343843 | Non clustered |
| 2224 | 2006 | Susc | M | 26 | 242334542124424178343843 | Cluster 1 |
| 2248 | 2006 | Susc | M | - | 242335542124424178343843 | Cluster 1 |
| 2537 | 2008 | Susc | F | 38 | 242335542124424178343843 | Cluster 1 |
| Strain | Mean Coverage | Mapping 1 | Contigs | Largest Contig | N50 | N75 | Insertion 1 | Deletion 1 | SNPs 1 |
|---|---|---|---|---|---|---|---|---|---|
| 431 | 261.13 | 99.26% | 165 | 173,577 | 61,250 | 39,904 | 109 | 162 | 1395 |
| 1906 | 277.38 | 99.23% | 167 | 155,704 | 60,542 | 37,278 | 118 | 130 | 1381 |
| 1918 | 281.59 | 99.29% | 162 | 161,736 | 62,665 | 37,278 | 139 | 120 | 1402 |
| 2224 | 267.62 | 99.32% | 158 | 158,538 | 55,650 | 35,572 | 131 | 135 | 1387 |
| 2248 | 187.94 | 99.30% | 178 | 214,707 | 52,142 | 33,124 | 135 | 121 | 1369 |
| 2537 | 251.62 | 99.33% | 164 | 179,030 | 56,137 | 35,288 | 127 | 120 | 1386 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Conceição, E.C.; da Conceição, M.L.; Marcon, D.J.; Loubser, J.; Andrade, G.L.; Silva, S.P.d.; Cruz, A.C.R.; Sharma, A.; Suffys, P.; Lima, K.V.B. Genomic Diversity of the Rarely Observed Genotype of the Mycobacterium tuberculosis Central Asian (CAS) Lineage 3 from North Brazil. Microorganisms 2023, 11, 132. https://doi.org/10.3390/microorganisms11010132
Conceição EC, da Conceição ML, Marcon DJ, Loubser J, Andrade GL, Silva SPd, Cruz ACR, Sharma A, Suffys P, Lima KVB. Genomic Diversity of the Rarely Observed Genotype of the Mycobacterium tuberculosis Central Asian (CAS) Lineage 3 from North Brazil. Microorganisms. 2023; 11(1):132. https://doi.org/10.3390/microorganisms11010132
Chicago/Turabian StyleConceição, Emilyn Costa, Marília Lima da Conceição, Davi Josué Marcon, Johannes Loubser, Gabrielly Leite Andrade, Sandro Patroca da Silva, Ana Cecília Ribeiro Cruz, Abhinav Sharma, Philip Suffys, and Karla Valéria Batista Lima. 2023. "Genomic Diversity of the Rarely Observed Genotype of the Mycobacterium tuberculosis Central Asian (CAS) Lineage 3 from North Brazil" Microorganisms 11, no. 1: 132. https://doi.org/10.3390/microorganisms11010132
APA StyleConceição, E. C., da Conceição, M. L., Marcon, D. J., Loubser, J., Andrade, G. L., Silva, S. P. d., Cruz, A. C. R., Sharma, A., Suffys, P., & Lima, K. V. B. (2023). Genomic Diversity of the Rarely Observed Genotype of the Mycobacterium tuberculosis Central Asian (CAS) Lineage 3 from North Brazil. Microorganisms, 11(1), 132. https://doi.org/10.3390/microorganisms11010132