Abstract
Bats have been identified as reservoirs of zoonotic and potentially zoonotic pathogens. Significant progress was made in the field of molecular biology with regard to infectious diseases, especially those that infect more than one species. Molecular methods, sequencing and bioinformatics have recently become irreplaceable tools in emerging infectious diseases research and even outbreak prediction. Modern methods in the molecular biology field have shed more light on the unique relationship between bats and viruses. Here we provide readers with a concise summary of the potential and limitations of molecular methods for studying the ecology of bats and bat-related pathogens and microorganisms.
1. Introduction
Bats (order Chiroptera) are a group of mammals with unique anatomy and physiology, which predispose them to fulfill a variety of ecological functions [1]. Known as the only mammals capable of sustained flight [1], bats occur ubiquitously except in Antarctica [2].
The impact of bats as reservoirs of zoonotic viruses has been defined in numerous studies [3,4,5,6]. Infectious pathogens, especially those that infect more than one species, are a complex subject that has recently captured the attention of many researchers. This impact is underlined by the fact that about 75% of zoonoses are spread to humans from wildlife [7,8,9,10,11,12,13]. One of the most important bat-borne zoonoses is bat rabies. Human–bat interactions are now the source of the majority of locally acquired human lyssavirus infections in many high-income countries without hematophagous or ‘vampire’ bat species [14]. On the other hand, experimental European bat lyssavirus (EBLV) infections in other mammals have revealed only low susceptibility of foxes by the intramuscular route [15]; an experimental EBLV infection of sheep resulted only in a peripheral abortive infection without neurological signs [16]. As for known or supposed natural infections, evidence suggests that Hendra virus and Nipah virus may have been transmitted from bats to other mammals beside humans [17,18,19]. Hendra virus was not highly contagious in experimental conditions [20].
Besides their role in emerging infectious diseases (EID), bats also fulfil indispensable roles in the functioning of the ecosystem [1,21]. While nectarivorous species are important pollinators of fruit-bearing trees and frugivorous bats play a role in the process of reforestation [22,23,24], insectivorous species are significant predators of insects [1,21,25]. Whenever bat populations experience a higher mortality rate, the decline in numbers may affect the whole ecosystem, even economics [26]. The guano of bats is rich in nitrogen and is used as fertilizer [27]. In several countries, bats are listed as species protected by local laws. Therefore, research of bat-related EID is ruled by ethical committees; specific bat biological sample availability is limited in some cases, as non-invasive sampling methods must often be preferred in these species [28].
A definitive reservoir species for a pathogen, according to Mlera et al. [29], may be defined in two ways: a species from which it is possible to isolate infectious pathogens, or a species that demonstrates high positivity for the said pathogen in surveillance studies [29]. Potential reservoir species may be proven to be seropositive and/or viral RNA may be present in their tissues [29]. On the other hand, Villarreal [30] states that the term persistence (including latent and chronic infection) better describes some instances of “symbiotic virus–host relationships” that are different from a simple reservoir of viruses. Since the invention of the first molecular methods, a reservoir status has been described in bats from which pathogens may be transmitted by many different routes (Figure 1) [4,6]; some viruses seem to persist in a few bat species as well; cases of possible viral elements inserted in bat genome have been identified [31,32]. Reservoir species are responsible for maintaining a pathogen in a region in the long term and for transmitting it to other species of concern [33]. To elucidate complex patterns of infection, long-term serological data with advanced analytical tools have proven necessary [34].
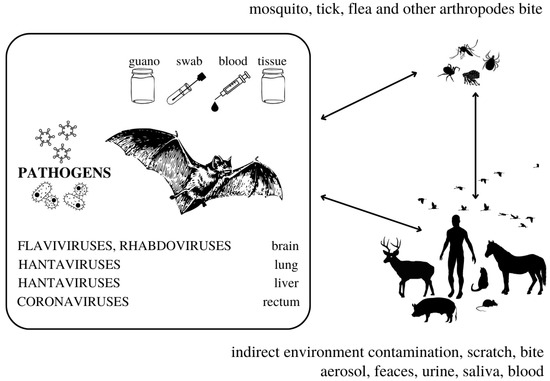
Figure 1.
The complexity of bat-borne pathogens transmission. The information in this figure was compiled using studies cited in this review.
Bats have been identified as an important reservoir species for many viral and a few bacterial pathogens [27]. Numerous studies have linked bats as potential reservoir species of lyssaviruses, henipaviruses, hantaviruses and coronaviruses [2,12,35,36,37,38,39,40,41,42,43,44,45,46,47]. The authors Cadar et al. [48], Pilipski et al. [49] and Kading et al. [50] report flavivirus detection in bats. Other viruses, such as adenoviruses, circoviruses, picornaviruses, papillomaviruses, herpesviruses, etc., were also detected in bats [12,51,52]; however, the role of bats as reservoirs in these cases still remains to be elucidated.
Since these EIDs have been linked to bats, and the most recent SARS-CoV-2 pandemic seems to also involve bat SARS-related coronaviruses [47,53,54]; therefore, the bats serve as a focal point of our mini-review. We address qualitative rather than quantitative aspects of the research subject. Our mini-review highlights the works that demonstrate the potential, as well as limitations, of molecular methods for studying the ecology of bats and bat-related microorganisms.
2. Molecular Methods in Bat EID Research
Molecular methods, sequencing and bioinformatics have recently become irreplaceable tools in EID research and even outbreak prediction. As the number of available studies implies, bats seem to be linked more to research concerning RNA viruses than DNA viruses [55,56]. Although several DNA viruses were detected in bats [12,57,58], these do not seem to have a significant impact on bat populations; neither do these viruses seem to be the etiological agent of a zoonotic disease outbreak.
RNA isolation, transcription and a variation of PCR [59] are therefore used quite often as first-choice methods in virus detection in bats, and sequences of the most common viruses are available in databases like GenBank.
2.1. PCR Leading to Discoveries
A standard PCR protocol followed by sequencing of purified amplicon is a versatile way of both identifying well-known viruses and finding new viruses. Examples of the relevance of this approach are studies similar to Straková et al. [44], who identified a novel hantavirus in bat liver tissue, performing a total RNA extraction (QIAamp viral RNA Mini Kit or Qiazol/triazol method) from internal organ tissues collected from dead individuals and proceeding to screen tissue samples for hantavirus RNA presence and sequencing of PCR amplicons (from a broad-spectrum RT-PCR). To determine the entire genomic sequence of this novel virus, IonTorrent HTS analysis was performed.
Cadar et al. [48] identified Usutu virus RNA in bat brain tissue by performing both total DNA and RNA extraction from tissues, then subjecting extracted nucleic acids to RT-PCR. Amplicons were sequenced directly and these sequences were used for phylogenetic analysis. Similar to Straková et al. [44], a complete genome sequence of the bat Usutu virus was then obtained from bat brain tissue using a set of primers designed from multiple comparisons of Usutu virus genomes available in databases [48].
A novel bat-borne hantavirus was detected in samples of bat lung tissue by Arai et al. [60], using a heminested L-segment primer set and a nested S-segment primer set. Their results might be indicative of a host-switching event [60], which might help understand the ecology of these viruses.
Luo et al. [61] collected 1044 bat brains and 3532 saliva swab samples to search for molecular evidence of rhabdoviruses. For initial screening, they used a previously described combined real-time reverse transcription PCR (RT-qPCR) that includes a probe-based RT-qPCR for pan-rabies virus detection and another pan-lyssavirus RT-qPCR [61]. Their effort led to the discovery of six new rhabdoviruses, the sequences of which were determined by next-generation sequencing and confirmed by Sanger sequencing. One of the tentative rhabdovirus species identified in this study clustered with two insect-related viruses; the authors did not exclude a possible role for arthropods in the life cycle of the identified bat viruses [61]. Although these rhabdoviruses were considered unlikely to present a high risk of spillover events, further information about transmission and shedding of these viruses in bats is needed to determine their zoonotic potential [61].
A wide range of rhabdoviruses was discovered in bats and bat parasites by Aznar-Lopez et al. [62], who performed a nested RT-PCR on 1488 oropharyngeal swabs from bats.
2.2. Coronaviruses Are Found Abundantly Using Non-Invasive Methods
Bat guano was successfully used as a sample for coronavirus detection in a long-term study performed by Lo et al. [63]. The authors obtained 512 fecal samples over the course of 4 years. RNA was isolated from these samples and carried out a nested PCR for coronavirus detection. Analysis of the sequences obtained in their study revealed that the detected coronaviruses belonged to the genera Alphacoronavirus and Betacoronavirus, some of which were grouped with the SARS-like coronavirus clade [63]. Using non-invasive sampling methods, such as guano collection, can lead to significant discoveries in bat-borne virus research; identification of non-invasive or less invasive samples such as saliva, urine or feces is also a key element in terms of bat conservation [61].
Anthony et al. [64] screened 606 bats for coronaviruses, collecting blood, oral and rectal swabs. Their method of choice was PCR using broadly reactive consensus primers.
2.3. Modern Sequencing Methods and Viral Diversity
Sanger’s sequencing method [65] and its more modern modifications belong to the first generation of sequencing methods. This method has been reported to still be the most accurate. It is widely accepted that NGS variants need to be validated with the gold standard Sanger sequencing technique prior to reporting, even though both the costs and turnaround time of this approach are considerable [66]. Another question needing an answer is how much can be concluded about the reservoir status of the bat even after successfully confirming the presence of a pathogen nucleic acid fragment in the samples. To fully confirm the reservoir status of a species, much more data is required; however, depending on the location of the detected pathogen fragment in the body of the host, the direction of further research into the virus–host relationship can be determined.
It was pointed out that traditional Sanger sequencing can only be applied to individual samples (or a low number thereof), which makes the method too painstaking for processing complex samples, especially for large-scale studies [67].
In the literature, the term next-generation sequencing (NGS) is often used to describe sequencing platforms other than those based on the Sanger method (pyrosequencing, sequencing by synthesis, ligation and two-base coding) [68]. NGS, also known as massively parallel or deep sequencing, is characterized by the ability to sequence millions of short DNA fragments in parallel [69]. NGS has proved to be a very efficient method to determine the virome of mammals, including bats, such as the extensive and highly efficient study performed by Wu et al. [12] in which a broad range of viruses, most of them novel, were identified in swab samples from 4440 bats. Metagenomics, defined as the direct genetic analysis of genomes contained with an environmental sample [70], has led to important findings, some of which encompass novel and/or potentially zoonotic viruses in bats [71,72,73,74,75]. Library preparation, being an important part of metagenomics, has also been pivotal in the research of full-genome sequencing of novel bat-associated viruses [51,52]. Wu et al. [12] used a series of sequence-independent RT-PCR, sequence-based PCR and specific nested PCR amplification methods, along with viral library construction and NGS, to analyze the viral community in the sampled bat species. Wu et al. [12] stated that the purpose of their study was to survey the ecological and biological diversities of viruses residing in these bat species, to investigate the presence of potential bat-borne zoonotic viruses and to evaluate the impacts of these viruses on public health. Recently, metagenomics has found a potential use in diagnostics [76] and surveillance [77].
Broad-spectrum studies, like Wu et al. from 2016 and 2018 [12,78], provide molecular proof for viral diversity in mammals. As the authors Wu et al. [78] conclude, combined with previous bat virome data, such studies greatly increase our knowledge of the viral community in wildlife in a densely populated country in an EID hotspot and continued efforts in viral discovery in these and other mammalian hosts may reveal greater diversity of viral lineages.
2.4. In Silico Analyses May Reveal Bases for Further Research
While it has long been known that the eukaryotic genome contains endogenous retroviruses, it was surprising to discover that sequences of RNA viruses that do not make a DNA intermediate and do not usually enter the nucleus are also present in eukaryotic genomes [79,80]. A useful collective term reflecting their fragmentary nature, EVE (endogenous viral elements), has been coined by Katzourakis and Gifford [24]; Holmes [80] uses this term to refer to all endogenous viruses regardless of taxonomy.
Using an initial PCR screening and phylogenetic analyses, Horie et al. [81] demonstrated that bats of the genus Eptesicus carry an inheritable endogenous bornavirus-like L (EBLL) element in their genome. Representatives of the genus Eptesicus occur in a wide geographical area including the northern hemisphere within Europe, Asia and the Americas [82]. These findings provide novel insights into the co-evolution of RNA viruses and mammalian species [81].
Taylor et al. [32] performed first an in silico screening of NIRV (non-retroviral integrated RNA viruses) in bat sequences; they further tested the presence of integrated copies of DNA-based filovirus sequences in the two species with the highest copy number, the wallaby species Macropus eugenii and the bat species Myotis lucifugus. They designed PCR primers from the mammalian genomic sequence belonging to the longer identical sequences and performed amplification of these segments in individuals of these species other than those used in existing genomic projects. The sequence from Macropus eugenii had only one mutation compared to the sequence from the mentioned genome project [32]. Taylor et al. [32] subsequently tested samples of bats of Myotis lucifugus and Eptesicus fuscus for the presence of these sequences. In all cases, the similarity of the new sequences was consistent with the hypothesis of integration of a filovirus-like copy of DNA into mammalian genomes. This laboratory finding supported their hypothesis expressed after an in silico examination of the genomic database; the phylogenetic analysis and sequencing performed in this work is consistent with the hypothesis of integration of filoviral elements into mammalian genomes [32]. Phylogenetic evidence suggests that the direction of transfer was from viral to mammalian genome [32].
Integrated viral elements have been found not only in bats but also in the genome of arthropod vectors [31,32]. Katzourakis and Gifford [31] reported that when these nucleotide sequences are fixed in the genome of a mammalian species, they can provide phylogenetic benefits to the species. These authors are of the opinion that the low frequency of recording events of the integration of the viral genome into the host genome is due to the low number of sequenced genomes of species and individuals and not to the fact that they are not retroviruses or viruses replicating in the nucleus of cells. Integrated viral elements can cause the host immune system to recognize these viral transcripts as “self” if they co-evolve with a mammalian host [31,83].
A more recent study by Skirmuntt et al. [84], which focuses heavily on bat immunology as a factor influencing EIDs, corroborates the conclusions of the aforementioned authors; the in silico method seems to continue gaining importance within the molecular biology field.
3. Discussion
To determine a working solution to the threat posed by EID, it seems to be important to consider what a reservoir is and also to consider the evolutionary history of reservoir species. Several authors have compiled different EID management frameworks over the course of the past years [85,86,87]. It is necessary to include bat immunology [84], biological, social and environmental science and even mathematics [86] in a ‘one health’, multidisciplinary approach [85] to EID research. Deeply understudied social and environmental questions should also be taken into account when dealing with EIDs [86]. Holmes [80] describes in some cases the relationships between a macro-organism and a virus as an intense “arms race”. The author concludes his work by stating that the discovery of EVE has raised some questions, including whether these can be beneficial to the host cells. Thorough, systematic, long-term studies comparing bat viromes to the viromes of other reservoir species, including research in EVEs and phylogenetic studies, could provide an insight into the true “molecular” nature of their reservoir/viral persistence status. Modern metagenomics and high-throughput sequencing methods, combined with PCR screening and phylogenetic analysis, have proven their use in providing an insight into the EIDsʼ true identity by identifying their molecular nature. These methods could, when combined, shed light on the relationships between viral hosts, reservoirs and viruses—in other words, to the ecology of viruses. Some zoonotic viruses are part of the ecosystem within the phenomenon of natural outbreaks; they are well adapted to reservoir macro-organisms. Since the most effective place to address such zoonotic threats is at the wildlife–human interface [86], public health infrastructure will also remain one of the most important factors involved in prevention and control of EIDs, regardless of future progress in laboratory diagnostics.
Studying bat ecology and evolution will also provide a better understanding of zoonotic virus–host dynamics, as well as the long-term co-evolutionary process between hosts and some viruses [84]. Viruses and parasites in general are a part of the ecosystem, especially those that display the phenomenon of natural outbreaks. While straightforward pathogen detection methods (sample collection, PCR, sequencing and phylogenetic analysis) are indispensable; bioinformatics hand-in-hand with ecology are necessary to analyze the “bigger picture”. Even hyperparasitism has been described in bats and their parasites, suggesting a potential for new discoveries in this field [88].
PCR and sequencing belong to the most relevant techniques in identifying EID outbreaks. In silico methods, as demonstrated by cited authors, have recently become equally relevant in EID management, as they can shed more light onto the underlying reasons behind the origin of zoonotic diseases. Input is needed from both human and veterinary medicine, along with deeper immunological, microbiological and bioinformatic insights.
Metagenomic analyses of non-invasive samples can provide information useful for surveillance [71,76] and early detection of viral zoonoses [71]. However, the nature of the sample affects its informative value. The mere fact that the nucleic acid of a virus is detected in, for instance, bat feces may indicate bat involvement in the ecology of the virus and may therefore point the direction for further research but will not inform exhaustively about their reservoir status. This is supported by the fact that various insect virus nucleic acids were detected in insectivorous bat guano [74]. Insectivorous bats have been proven to have a far more abundant virome than frugivorous bats [12]. In their study, Vicente-Santos et al. [89] conclude that bats serve only as dead-end hosts for Dengue virus, the presence of this virus being the result of ingestion of a positive mosquito vector. Several other studies suggested that bat diet and ectoparasite load may be related to the detection of arthropode viruses in samples from bats. A broad range of “dietary viruses” [57,90] have been discovered in bat guano [57], which are the result of passive transport of insect virome, including mosquito virome, through the gastrointestinal tract, while until now it remains unclear if these viruses can cross the intestinal wall and infect bats [90]. This arthropod–virus–bat relationship has been described both in frugivorous [90] and insectivorous bats [57]. Vicente-Santos et al. [89] combined PCR and serology from both bats and mosquitoes to reach their conclusion.
Worldwide, the access to different sample types is unbalanced, mostly because bats are protected by law in most European countries. To ensure proper bat conservation, non-invasive samples should be preferred. In future, bat-related EID research will benefit from finding ways to enhance the informative value of non-invasive samples, for example a systematic approach to metagenomics analysis of multiple non-invasive sample types collected from the same individual, combined with the research of viromes of bat-related arthropods and their immediate environment (Table 1). Collecting samples from the environment and other mammals may provide further information about bats as reservoirs [91].

Table 1.
Summary of use of the molecular methods in research of bats. The table was composed using the studies cited in this review.
Author Contributions
S.Z.: conceptualization and administration of the project, addition of important data, writing—manuscript; Ľ.K. and J.T.: manuscript writing—revision and editing; M.P. and M.D.: analysis of the literature, editing and review of the manuscript; Ł.M., P.T., G.N. and A.M.: revision of the manuscript; A.O.: supervision, revision and editing of the manuscript, raising funds. All authors have read and agreed to the published version of the manuscript.
Funding
This publication is the result of the project implementation: “Open scientific community for modern interdisciplinary research in medicine (OPENMED)”, ITMS2014+: 313011V455 supported by the Operational Programme Integrated Infrastructure, funded by the ERDF; this work was supported by the project KEGA No. 005UVLF-4/2022 granted by The Ministry of Education, Science, Research and Sport of the Slovak Republic.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
The authors would like to thank David Idris Lewis for the English language corrections.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study, in the collection, analyses, or interpretation of data, in the writing of the manuscript, or in the decision to publish the results.
References
- Kunz, T.H.; Braun de Torrez, E.; Bauer, D.; Lobova, T.; Fleming, T.H. Ecosystem services provided by bats. Ann. N. Y. Acad. Sci. 2011, 1223, 1–38. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, A.; Kulcsar, K.; Misra, V.; Frieman, M.; Mossman, K. Bats and Coronaviruses. Viruses 2019, 11, 41. [Google Scholar] [CrossRef] [PubMed]
- Dobson, A.P. What links bats to emerging infectious diseases? Science 2005, 310, 628–629. [Google Scholar] [CrossRef] [PubMed]
- Calisher, C.H.; Childs, J.E.; Field, H.E.; Holmes, K.V.; Schountz, T. Bats: Important reservoir hosts of emerging viruses. Clin. Microbiol. Rev. 2006, 19, 531–545. [Google Scholar] [CrossRef]
- Han, H.J.; Wen, H.L.; Zhou, C.M.; Chen, F.F.; Luo, L.M.; Liu, J.W.; Yu, X.J. Bats as reservoirs of severe emerging infectious diseases. Virus Res. 2015, 205, 1–6. [Google Scholar] [CrossRef]
- Kohl, C.; Nitsche, A.; Kurth, A. Update on Potentially Zoonotic Viruses of European Bats. Vaccines 2021, 9, 690. [Google Scholar] [CrossRef]
- Wolfe, N.D.; Dunavan, C.P.; Diamond, J. Origins of major human infectious diseases. Nature 2007, 447, 279–283. [Google Scholar] [CrossRef]
- Jones, K.E.; Patel, N.G.; Levy, M.A.; Storeygard, A.; Balk, D.; Gittleman, J.L.; Daszak, P. Global trends in emerging infectious diseases. Nature 2008, 451, 990–993. [Google Scholar] [CrossRef]
- Lloyd-Smith, J.O.; George, D.; Pepin, K.M.; Pitzer, V.E.; Pulliam, J.R.; Dobson, A.P.; Hudson, P.J.; Grenfell, B.T. Epidemic dynamics at the human-animal interface. Science 2009, 326, 1362–1367. [Google Scholar] [CrossRef]
- Marsh, G.A.; Wang, L.F. Hendra and Nipah viruses: Why are they so deadly? Curr. Opin. Virol. 2012, 2, 242–247. [Google Scholar] [CrossRef]
- Smith, I.; Wang, L.F. Bats and their virome: An important source of emerging viruses capable of infecting humans. Curr. Opin. Virol. 2013, 3, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Yang, L.; Ren, X.; He, G.; Zhang, J.; Yang, J.; Qian, Z.; Dong, J.; Sun, L.; Zhu, Y.; et al. Deciphering the bat virome catalog to better understand the ecological diversity of bat viruses and the bat origin of emerging infectious diseases. ISME J. 2016, 10, 609–620. [Google Scholar] [CrossRef] [PubMed]
- Madrières, S.; Castel, G.; Murri, S.; Vulin, J.; Marianneau, P.; Charbonnel, N. The Needs for Developing Experiments on Reservoirs in Hantavirus Research: Accomplishments, Challenges and Promises for the Future. Viruses 2019, 11, 664. [Google Scholar] [CrossRef] [PubMed]
- Wright, E.; Anuradha, S.; Richards, R.; Reid, S. A review of the circumstances and health-seeking behaviours associated with bat exposures in high-income countries. Zoonoses Public Health 2022, 69, 593–605. [Google Scholar] [CrossRef]
- Cliquet, F.; Picard-Meyer, E.; Barrat, J.; Brookes, S.M.; Healy, D.M.; Wasniewski, M.; Litaize, E.; Biarnais, M.; Johnson, L.; Fooks, A.R. Experimental infection of foxes with European Bat Lyssaviruses type-1 and 2. BMC Vet.-Res. 2009, 5, 19. [Google Scholar] [CrossRef]
- Tjørnehøj, K.; Fooks, A.R.; Agerholm, J.S.; Rønsholt, L. Natural and experimental infection of sheep with European bat lyssavirus type-1 of Danish bat origin. J. Comp. Pathol. 2006, 134, 190–201. [Google Scholar] [CrossRef]
- Young, P.L.; Halpin, K.; Selleck, P.W.; Field, H.; Gravel, J.L.; Kelly, M.A.; Mackenzie, J.S. Serologic evidence for the presence in Pteropus bats of a paramyxovirus related to equine morbillivirus. Emerg. Infect. Dis. 1996, 2, 239–240. [Google Scholar] [CrossRef]
- Halpin, K.; Young, P.L.; Field, H.E.; Mackenzie, J.S. Isolation of Hendra virus from pteropid bats: A natural reservoir of Hendra virus. J. Gen. Virol. 2000, 81 Pt 8, 1927–1932. [Google Scholar] [CrossRef]
- Chua, K.B.; Koh, C.L.; Hooi, P.S.; Wee, K.F.; Khong, J.H.; Chua, B.H.; Chan, Y.P.; Lim, M.E.; Lam, S.K. Isolation of Nipah virus from Malaysian Island flying-foxes. Microbes Infect. 2002, 4, 145–151. [Google Scholar] [CrossRef]
- Williamson, M.M.; Hooper, P.T.; Selleck, P.W.; Gleeson, L.J.; Daniels, P.W.; Westbury, H.A.; Murray, P.K. Transmission studies of Hendra virus (equine morbillivirus) in fruit bats, horses and cats. Aust. Veter. J. 1998, 76, 813–818. [Google Scholar] [CrossRef]
- Ramírez-Fráncel, L.A.; García-Herrera, L.V.; Losada-Prado, S.; Reinoso-Flórez, G.; Sánchez-Hernández, A.; Estrada-Villegas, S.; Lim, B.K.; Guevara, G. Bats and their vital ecosystem services: A global review. Integr. Zool. 2022, 17, 2–23. [Google Scholar] [CrossRef] [PubMed]
- Gonzales, R.S.; Ingle, N.R.; Lagunzad, D.A.; Nakashizuka, T. Seed Dispersal by Birds and Bats in Lowland Philippine Forest Successional Area. Biotropica 2009, 41, 452–458. [Google Scholar] [CrossRef]
- Sheherazade; Yasman; Pradana, D.H.; Tsang, L.M. The role of fruit bats in plant community changes in an urban forest in Indonesia. Raffles Bull. Zool. 2017, 65, 497–505. [Google Scholar]
- Fidelino, J.S.; Duya, M.R.M.; Duya, M.V.; Ong, P.S. Fruit bat diversity patterns for assessing restoration success in reforestation areas in the Philippines. Acta Oecologica 2020, 108, 103637. [Google Scholar] [CrossRef]
- Williams-Guillén, K.; Olimpi, E.; Maas, B.; Taylor, P.J.; Arlettaz, R. Bats in the Anthropogenic Matrix: Challenges and Opportunities for the Conservation of Chiroptera and Their Ecosystem Services in Agricultural Landscapes. In Bats in the Anthropocene: Conservation of Bats in a Changing World; Voigt, C., Kingston, T., Eds.; Springer: Berlin, Germany, 2016; pp. 151–186, (In Cham). [Google Scholar] [CrossRef]
- Boyles, J.G.; Cryan, P.M.; McCracken, G.F.; Kunz, T.H. Economic importance of bats in agriculture. Science 2011, 332, 41–42. [Google Scholar] [CrossRef]
- Allocati, N.; Petrucci, A.G.; Di Giovanni, P.; Masulli, M.; Di Illio, C.; De Laurenzi, V. Bat-man disease transmission: Zoonotic pathogens from wildlife reservoirs to human populations. Cell Death Discov. 2016, 2, 16048. [Google Scholar] [CrossRef]
- Hornok, S.; Estók, P.; Kováts, D.; Flaisz, B.; Takács, N.; Szőke, K.; Krawczyk, A.; Kontschán, J.; Gyuranecz, M.; Fedák, A.; et al. Screening of bat faeces for arthropod-borne apicomplexan protozoa: Babesia canis and Besnoitia besnoiti-like sequences from Chiroptera. Parasites Vectors 2015, 8, 441. [Google Scholar] [CrossRef]
- Mlera, L.; Melik, W.; Bloom, M.E. The role of viral persistence in flavivirus biology. Pathog. Dis. 2014, 71, 137–163. [Google Scholar] [CrossRef]
- Villarreal, L.P. The Widespread Evolutionary Significance of Viruses. In Origin and Evolution of Viruses; Domingo, E., Parrish, C.R., Holland, J.J., Eds.; Academic Press: Cambridge, MA, USA, 2008; pp. 477–516. [Google Scholar] [CrossRef]
- Katzourakis, A.; Gifford, R.J. Endogenous viral elements in animal genomes. PLoS Genet. 2010, 6, e1001191. [Google Scholar] [CrossRef]
- Taylor, D.J.; Leach, R.W.; Bruenn, J. Filoviruses are ancient and integrated into mammalian genomes. BMC Evol. Biol. 2010, 10, 193. [Google Scholar] [CrossRef]
- Haydon, D.T.; Cleaveland, S.; Taylor, L.H.; Laurenson, M.K. Identifying reservoirs of infection: A conceptual and practical challenge. Emerg. Infect. Dis. 2002, 8, 1468–1473. [Google Scholar] [CrossRef] [PubMed]
- Viana, M.; Cleaveland, S.; Matthiopoulos, J.; Halliday, J.; Packer, C.; Craft, M.E.; Hampson, K.; Czupryna, A.; Dobson, A.P.; Dubovi, E.J.; et al. Dynamics of a morbillivirus at the domestic-wildlife interface: Canine distemper virus in domestic dogs and lions. Proc. Natl. Acad. Sci. USA 2015, 112, 1464–1469. [Google Scholar] [CrossRef] [PubMed]
- Hooper, P.; Zaki, S.; Daniels, P.; Middleton, D. Comparative pathology of the diseases caused by Hendra and Nipah viruses. Microbes Infect. 2001, 3, 315–322. [Google Scholar] [CrossRef]
- Field, H.E.; Breed, A.C.; Shield, J.; Hedlefs, R.M.; Pittard, K.; Pott, B.; Summers, P.M. Epidemiological perspectives on Hendra virus infection in horses and flying foxes. Aust. Veter. J. 2007, 85, 268–270. [Google Scholar] [CrossRef]
- Kuzmin, I.V.; Bozick, B.; Guagliardo, S.A.; Kunkel, R.; Shak, J.R.; Tong, S.; Rupprecht, C.E. Bats, emerging infectious diseases, and the rabies paradigm revisited. Emerg. Health Threat. J. 2011, 4, 7159. [Google Scholar] [CrossRef]
- Rupprecht, C.E.; Turmelle, A.; Kuzmin, I.V. A perspective on lyssavirus emergence and perpetuation. Curr. Opin. Virol. 2011, 1, 662–670. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.U.; Gurley, E.S.; Hossain, M.J.; Nahar, N.; Sharker, M.A.; Luby, S.P. A Randomized Controlled Trial of Interventions to Impede Date Palm Sap Contamination by Bats to Prevent Nipah Virus Transmission in Bangladesh. PLoS ONE 2012, 7, e42689. [Google Scholar] [CrossRef] [PubMed]
- Banyard, A.C.; Evans, J.S.; Luo, T.R.; Fooks, A.R. Lyssaviruses and bats: Emergence and zoonotic threat. Viruses 2014, 6, 2974–2990. [Google Scholar] [CrossRef]
- Hayman, D.T.; Fooks, A.R.; Marston, D.A.; Garcia-R, J.C. The Global phylogeography of lyssaviruses—Challenging the ‘out of Africa’ hypothesis. PLoS Negl. Trop. Dis. 2016, 10, e0005266. [Google Scholar] [CrossRef]
- Field, H.E. Hendra virus ecology and transmission. Curr. Opin. Virol. 2016, 16, 120–125. [Google Scholar] [CrossRef]
- Goldstein, S.A.; Weiss, S.R. Origins and pathogenesis of Middle East respiratory syndrome-associated coronavirus: Recent advances. F1000Reserch 2017, 6, 1628. [Google Scholar] [CrossRef] [PubMed]
- Straková, P.; Dufkova, L.; Širmarová, J.; Salát, J.; Bartonička, T.; Klempa, B.; Pfaff, F.; Höper, D.; Hoffmann, B.; Ulrich, R.G. Novel hantavirus identified in European bat species Nyctalus noctula. Infect. Genet. Evol. 2017, 48, 127–130. [Google Scholar] [CrossRef] [PubMed]
- Velasco-Villa, A.; Mauldin, M.R.; Shi, M.; Escobar, L.E.; Gallardo-Romero, N.F.; Damon, I.; Olson, V.A.; Streicker, D.G.; Emerson, G. The history of rabies in the Western Hemisphere. Antivir. Res. 2017, 146, 221–232. [Google Scholar] [CrossRef] [PubMed]
- Andersen, K.G.; Rambaut, A.; Lipkin, W.I.; Holmes, E.C.; Garry, R.F. The proximal origin of SARS-CoV-2. Nat. Med. 2020, 26, 450–452. [Google Scholar] [CrossRef]
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef]
- Cadar, D.; Becker, N.; de Mendonca Campos, R.; Börstler, J.; Jöst, H.; Schmidt-Chanasit, J. Usutu virus in bats, Germany, 2013. Emerg. Infect. Dis. 2014, 20, 1771–1773. [Google Scholar] [CrossRef]
- Pilipski, J.D.; Pilipski, L.M.; Risley, L.S. West Nile Virus Antibodies in Bats from New Jersey and New York. J. Wildl. Dis. 2004, 40, 335–337. [Google Scholar] [CrossRef]
- Kading, R.C.; Kityo, R.; Nakayiki, T.; Ledermann, J.; Crabtree, M.B.; Lutwama, J.; Miller, B.R. Detection of Entebbe Bat Virus After 54 Years. Am. J. Trop. Med. Hyg. 2015, 93, 475–477. [Google Scholar] [CrossRef]
- Yinda, C.K.; Rector, A.; Zeller, M.; Conceição-Neto, N.; Heylen, E.; Maes, P.; Ghogomu, S.M.; Van Ranst, M.; Matthijnssens, J. A single bat species in Cameroon harbors multiple highly divergent papillomaviruses in stool identified by metagenomics analysi. Virol. Rep. 2016, 6, 74–80. [Google Scholar] [CrossRef][Green Version]
- Lazov, C.M.; Belsham, G.J.; Bøtner, A.; Rasmussen, T.B. Full-Genome Sequences of Alphacoronaviruses and Astroviruses from Myotis and Pipistrelle Bats in Denmark. Viruses 2021, 13, 1073. [Google Scholar] [CrossRef]
- Zhou, H.; Chen, X.; Hu, T.; Li, J.; Song, H.; Liu, Y.; Wang, P.; Liu, D.; Yang, J.; Holmes, E.C.; et al. A novel bat coronavirus closely related to SARS-CoV-2 contains natural insertions at the S1/S2 cleavage site of the spike protein. Curr. Biol. 2020, 30, 2196–2203. [Google Scholar] [CrossRef]
- Cerri, J.; Mori, E.; Ancillotto, L.; Russo, D.; Bertolino, S. COVID-19, media coverage of bats and related Web searches: A turning point for bat conservation? Mamm. Rev. 2022, 52, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.; Lau, S.; Woo, P.; Yuen, K.-Y. Bats as a continuing source of emerging infections in humans. Rev. Med. Virol. 2007, 17, 67–91. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.-F.; Anderson, D.E. Viruses in bats and potential spillover to animals and humans. Curr. Opin. Virol. 2019, 34, 79–89. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Ge, X.; Hon, C.C.; Zhang, H.; Zhou, P.; Zhang, Y.; Wu, Y.; Wang, L.F.; Shi, Z. Prevalence and genetic diversity of adeno-associated viruses in bats from China. J. Gen. Virol. 2010, 91, 2601–2609. [Google Scholar] [CrossRef]
- Donaldson, E.F.; Haskew, A.N.; Gates, J.E.; Huynh, J.; Moore, C.J.; Frieman, M.B. Metagenomic analysis of the viromes of three North American bat species: Viral diversity among different bat species that share a common habitat. J. Virol. 2010, 84, 13004–13018. [Google Scholar] [CrossRef]
- Mullis, K.; Faloona, F.; Scharf, S.; Saiki, R.; Horn, G.; Erlich, H. Specific enzymatic amplification of DNA in vitro: The polymerase chain reaction. Cold Spring Harb. Symp. Quant. Biol. 1986, 51, 263–273. [Google Scholar] [CrossRef]
- Arai, S.; Nguyen, S.T.; Boldgiv, B.; Fukui, D.; Araki, K.; Dang, C.N.; Ohdachi, S.D.; Nguyen, N.X.; Pham, T.D.; Boldbaatar, B.; et al. Novel bat-borne hantavirus, Vietnam. Emerg. Infect. Dis. 2013, 19, 1159–1161. [Google Scholar] [CrossRef]
- Luo, D.-S.; Li, B.; Shen, X.-R.; Jiang, R.-D.; Zhu, Y.; Wu, J.; Fan, Y.; Bourhy, H.; Hu, B.; Ge, X.-Y.; et al. Characterization of Novel Rhabdoviruses in Chinese Bats. Viruses 2021, 13, 64. [Google Scholar] [CrossRef]
- Aznar-Lopez, C.; Vázquez-Morón, S.; Marston, D.; Juste, J.; Ibanez, C.; Berciano, J.M.; Salsamendi, E.; Aihartza, J.; Banyard, A.; McElhinney, L.; et al. Detection of rhabdovirus viral RNA in oropharyngeal swabs and ectoparasites of Spanish bats. J. Gen. Virol. 2013, 94 Pt 1, 69–75. [Google Scholar] [CrossRef]
- Lo, V.T.; Yoon, S.-W.; Noh, J.Y.; Kim, Y.; Choi, Y.G.; Jeong, D.G.; Kim, H.K. Long-term surveillance of bat coronaviruses in Korea: Diversity and distribution pattern. Transbound. Emerg. Dis. 2020, 67, 2839–2848. [Google Scholar] [CrossRef] [PubMed]
- Anthony, S.J.; Ojeda-Flores, R.; Rico, O.; Navarrete-Macias, I.; Zambrana-Torrelio, C.M.; Rostal, M.K.; Epstein, J.H.; Tipps, T.; Liang, E.; Sanchez-Leon, M.; et al. Coronaviruses in bats from Mexico. J. Gen. Virol. 2013, 94 Pt 5, 1028–1038. [Google Scholar] [CrossRef] [PubMed]
- Sanger, F.; Nicklen, S.; Coulson, A.R. DNA sequencing with chain-terminating inhibitors. Proc. Natl. Acad. Sci. USA 1977, 74, 5463–5467. [Google Scholar] [CrossRef] [PubMed]
- Arteche-López, A.; Ávila-Fernández, A.; Romero, R.; Riveiro-Álvarez, R.; López-Martínez, M.A.; Giménez-Pardo, A.; Vélez-Monsalve, C.; Gallego-Merlo, J.; García-Vara, I.; Almoguera, B.; et al. Sanger sequencing is no longer always necessary based on a single-center validation of 1109 NGS variants in 825 clinical exomes. Sci. Rep. 2021, 11, 5697. [Google Scholar] [CrossRef] [PubMed]
- Shokralla, S.; Spall, J.L.; Gibson, J.F.; Hajibabaei, M. Next-generation sequencing technologies for environmental DNA research. Mol. Ecol. 2012, 21, 1794–1805. [Google Scholar] [CrossRef]
- Liu, L.; Li, Y.; Li, S.; Hu, N.; He, Y.; Pong, R.; Lin, D.; Lu, L.; Law, M. Comparison of Next-Generation Sequencing Systems. J. Biomed. Biotechnol. 2012, 2012, 251364. [Google Scholar] [CrossRef]
- Behjati, S.; Tarpey, P.S. What is next generation sequencing? Arch. Dis. Child. Educ. Pract. Ed. 2013, 98, 236–238. [Google Scholar] [CrossRef]
- Thomas, T.; Gilbert, J.; Meyer, F. Metagenomics—A guide from sampling to data analysis. Microb. Inform. Exp. 2012, 2, 3. [Google Scholar] [CrossRef]
- Hardmeier, I.; Aeberhard, N.; Qi, W.; Schoenbaechler, K.; Kraettli, H.; Hatt, J.-M.; Fraefel, C.; Kubacki, J. Metagenomic analysis of fecal and tissue samples from 18 endemic bat species in Switzerland revealed a diverse virus composition including potentially zoonotic viruses. PLoS ONE 2021, 16, e0252534. [Google Scholar] [CrossRef]
- Kohl, C.; Brinkmann, A.; Radonić, A.; Dabrowski, P.W.; Nitsche, A.; Mühldorfer, K.; Wibbelt, G.; Kurth, A. Zwiesel bat banyangvirus, a potentially zoonotic Huaiyangshan banyangvirus (Formerly known as SFTS)-like banyangvirus in Northern bats from Germany. Sci. Rep. 2020, J10, 1370. [Google Scholar] [CrossRef]
- Bolatti, E.M.; Viarengo, G.; Zorec, T.M.; Cerri, A.; Montani, M.E.; Hosnjak, L.; Casal, P.E.; Bortolotto, E.; Di Domenica, V.; Chouhy, D.; et al. Viral Metagenomic Data Analyses of Five NewWorld Bat Species from Argentina: Identification of 35 Novel DNA Viruses. Microorganisms 2022, 10, 266. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Altan, E.; Reyes, G.; Halstead, B.; Deng, X.; Delwart, E. Virome of bat guano from nine Northern California roosts. J. Virol. 2021, 95, e01713–e01720. [Google Scholar] [CrossRef] [PubMed]
- He, B.; Huang, X.; Zhang, F.; Tan, W.; Matthijnssens, J.; Qin, S.; Xu, L.; Zhao, Z.; Yang, L.; Wang, Q.; et al. Group A rotaviruses in Chinese bats: Genetic composition, serology, and evidence for bat-to-human transmission and reassortment. J. Virol. 2017, 91, e02493-16. [Google Scholar] [CrossRef] [PubMed]
- Höper, D.; Wylezich, C.; Beer, M. Loeffler 4.0: Diagnostic Metagenomics. Adv. Virus Res. 2017, 99, 17–37. [Google Scholar] [CrossRef]
- Mohsin, H.; Asif, A.; Fatima, M.; Rehman, Y. Potential role of viral metagenomics as a surveillance tool for the early detection of emerging novel pathogens. Arch. Microbiol. 2021, 203, 865–872. [Google Scholar] [CrossRef]
- Wu, Z.; Lu, L.; Du, J.; Yang, L.; Ren, X.; Liu, B.; Jiang, J.; Yang, J.; Dong, J.; Sun, L.; et al. Comparative analysis of rodent and small mammal viromes to better understand the wildlife origin of emerging infectious diseases. Microbiome 2018, 6, 178. [Google Scholar] [CrossRef]
- Crochu, S.; Cook, S.; Attoui, H.; Charrel, R.N.; De Chesse, R.; Belhouchet, M.; Lemasson, J.J.; de Micco, P.; de Lamballerie, X. Sequences of flavivirus-related RNA viruses persist in DNA form integrated in the genome of Aedes spp. Mosquitoes. J. Gen. Virol. 2004, 85, 1971–1980. [Google Scholar] [CrossRef]
- Holmes, E.C. The Evolution of Endogenous Viral Elements. Cell Host Microbe 2011, 10, 368–377. [Google Scholar] [CrossRef]
- Horie, M.; Kobayashi, Y.; Honda, T.; Fujino, K.; Akasaka, T.; Kohl, C.; Wibbelt, G.; Mühldorfer, K.; Kurth, A.; Müller, M.A.; et al. An RNA-dependent RNA polymerase gene in bat genomes derived from an ancient negative-strand RNA virus. Sci. Rep. 2016, 6, 25873. [Google Scholar] [CrossRef]
- Wilson, D.E.; Reeder, D.M. (Eds.) Mammal Species of the World: A Taxonomic and Geographic Reference, 3rd ed.; Johns Hopkins University Press: Baltimore, MD, USA, 2005; ISBN 978-0-8018-8221-0. OCLC 62265494. [Google Scholar]
- Brook, C.E.; Dobson, A.P. Bats as ‘special’ reservoirs for emerging zoonotic pathogens. Trends Microbiol. 2015, 23, 172–180. [Google Scholar] [CrossRef]
- Skirmuntt, E.C.; Escalera-Zamudio, M.; Teeling, E.C.; Smith, A.; Katzourakis, A. The Potential Role of Endogenous Viral Elements in the Evolution of Bats as Reservoirs for Zoonotic Viruses. Annu. Rev. Virol. 2020, 7, 103–119. [Google Scholar] [CrossRef] [PubMed]
- Coker, R.J.; Hunter, B.M.; Rudge, J.W.; Liverani, M.; Hanvoravongchai, P. Emerging infectious diseases in southeast Asia: Regional challenges to control. Lancet 2011, 377, 599–609. [Google Scholar] [CrossRef]
- Wood, J.L.; Leach, M.; Waldman, L.; Macgregor, H.; Fooks, A.R.; Jones, K.E.; Restif, O.; Dechmann, D.; Hayman, D.T.; Baker, K.S.; et al. A framework for the study of zoonotic disease emergence and its drivers: Spillover of bat pathogens as a case study. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2012, 367, 2881–2892. [Google Scholar] [CrossRef] [PubMed]
- Brierley, L.; Vonhof, M.J.; Olival, K.J.; Daszak, P.; Jones, K.E. Quantifying Global Drivers of Zoonotic Bat Viruses: A Process-Based Perspective. Am. Nat. 2016, 187, E53–E64. [Google Scholar] [CrossRef] [PubMed]
- Haelewaters, D.; Hiller, T.; Dick, C.W. Bats, Bat Flies, and Fungi: A Case of Hyperparasitism. Trends Parasitol. 2018, 34, 784–799. [Google Scholar] [CrossRef]
- Vicente-Santos, A.; Moreira-Soto, A.; Soto-Garita, C.; Chaverri, L.G.; Chaves, A.; Drexler, J.F.; Morales, J.A.; Alfaro-Alarcón, A.; Rodríguez-Herrera, B.; Corrales-Aguilar, E. Neotropical bats that co-habit with humans function as dead-end hosts for dengue virus. PLoS Negl. Trop. Dis. 2017, 11, e0005537. [Google Scholar] [CrossRef]
- Bennett, A.J.; Bushmaker, T.; Cameron, K.; Ondzie, A.; Niama, F.R.; Parra, H.J.; Mombouli, J.V.; Olson, S.H.; Munster, V.J.; Goldberg, T.L. Diverse RNA viruses of arthropod origin in the blood of fruit bats suggest a link between bat and arthropod viromes. Virology 2019, 528, 64–72. [Google Scholar] [CrossRef]
- Hayman, D.T.S. Bats as Viral Reservoirs. Ann. Rev. Vir. 2016, 3, 77–99. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).