
Molecular Evolution of the Pseudomonas aeruginosa DNA Gyrase gyrA Gene


 ,
,  , , , , and
, , , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Strain selection
2.2. Phylogenetic-Tree Construction and Estimation of Evolutionary Rate Using the Bayesian Markov Chain Monte Carlo Method
2.3. Analysis of Selective Pressure
2.4. Bayesian Skyline Plot Analysis
2.5. Structural Modeling
2.6. Docking Simulation Analysis
2.7. Statistical Analyses
3. Results
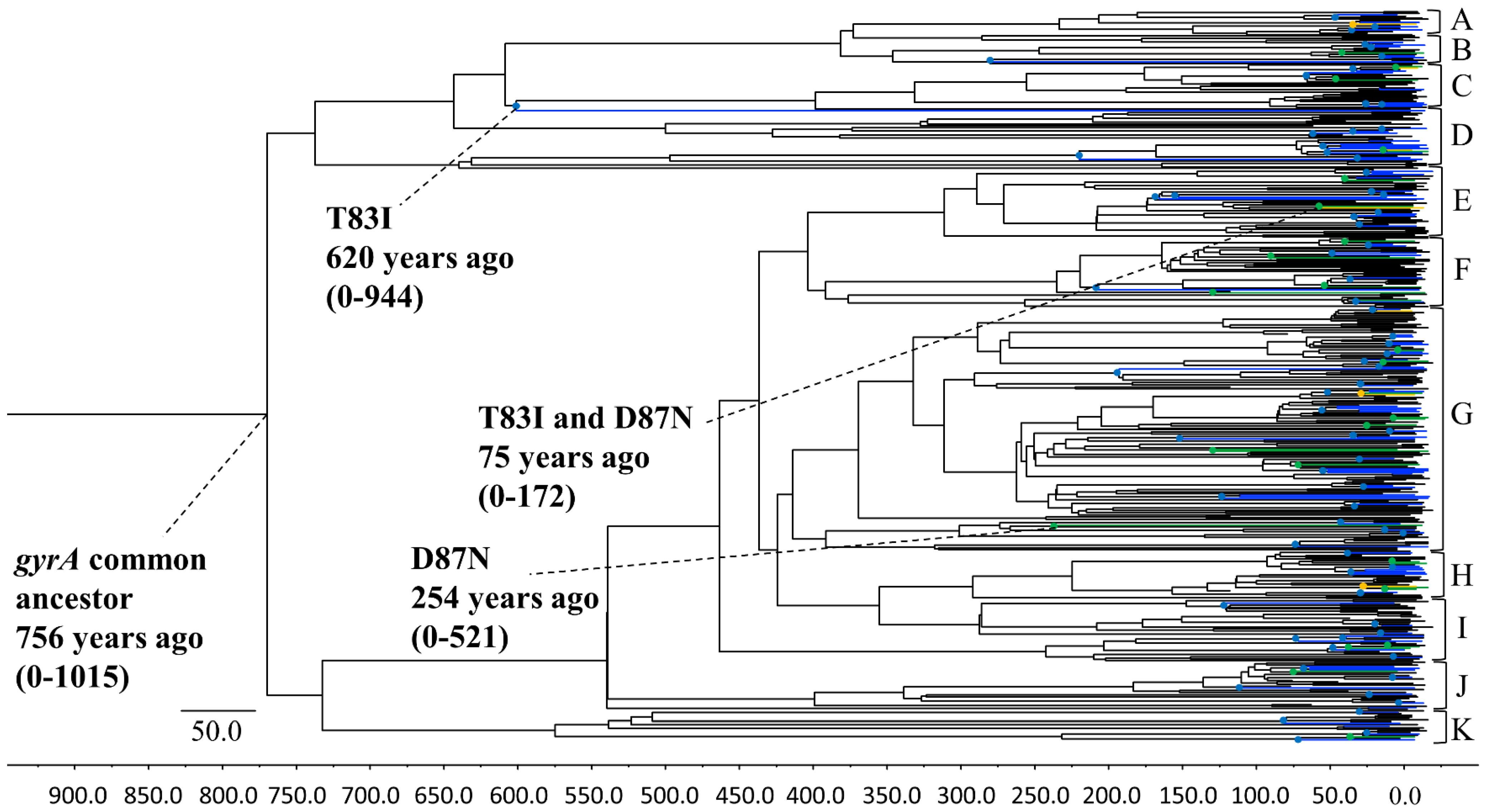
3.1. Phylogenetic Tree Construction Using the Bayesian MCMC Method
3.2. Selective Pressure Analysis Using the SLAC, FEL, IFEL, FUBAR, and MEME Methods
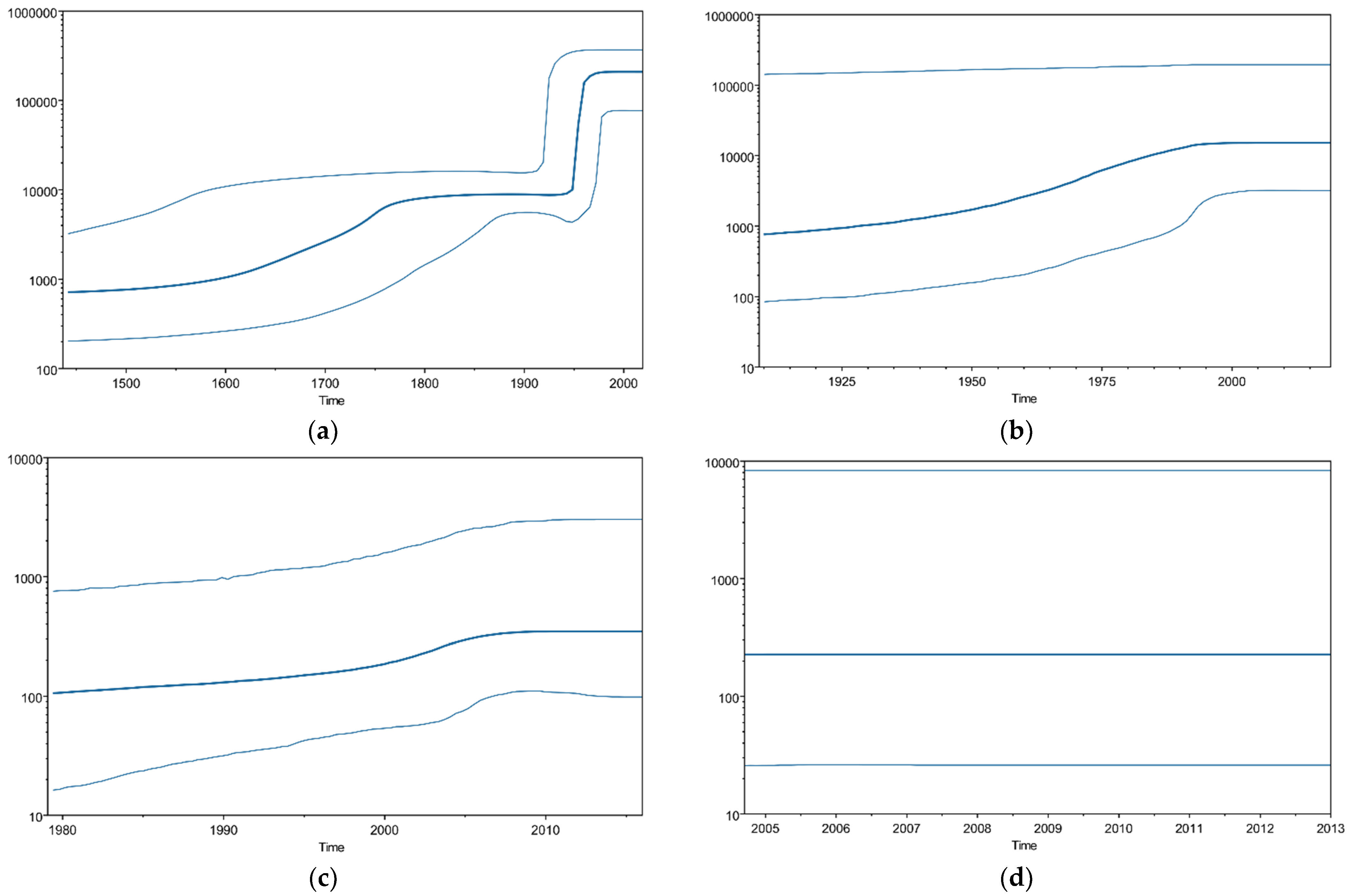
3.3. P. aeruginosa gyrA Gene Phylodynamics Using the Bayesian Skyline Plot Method
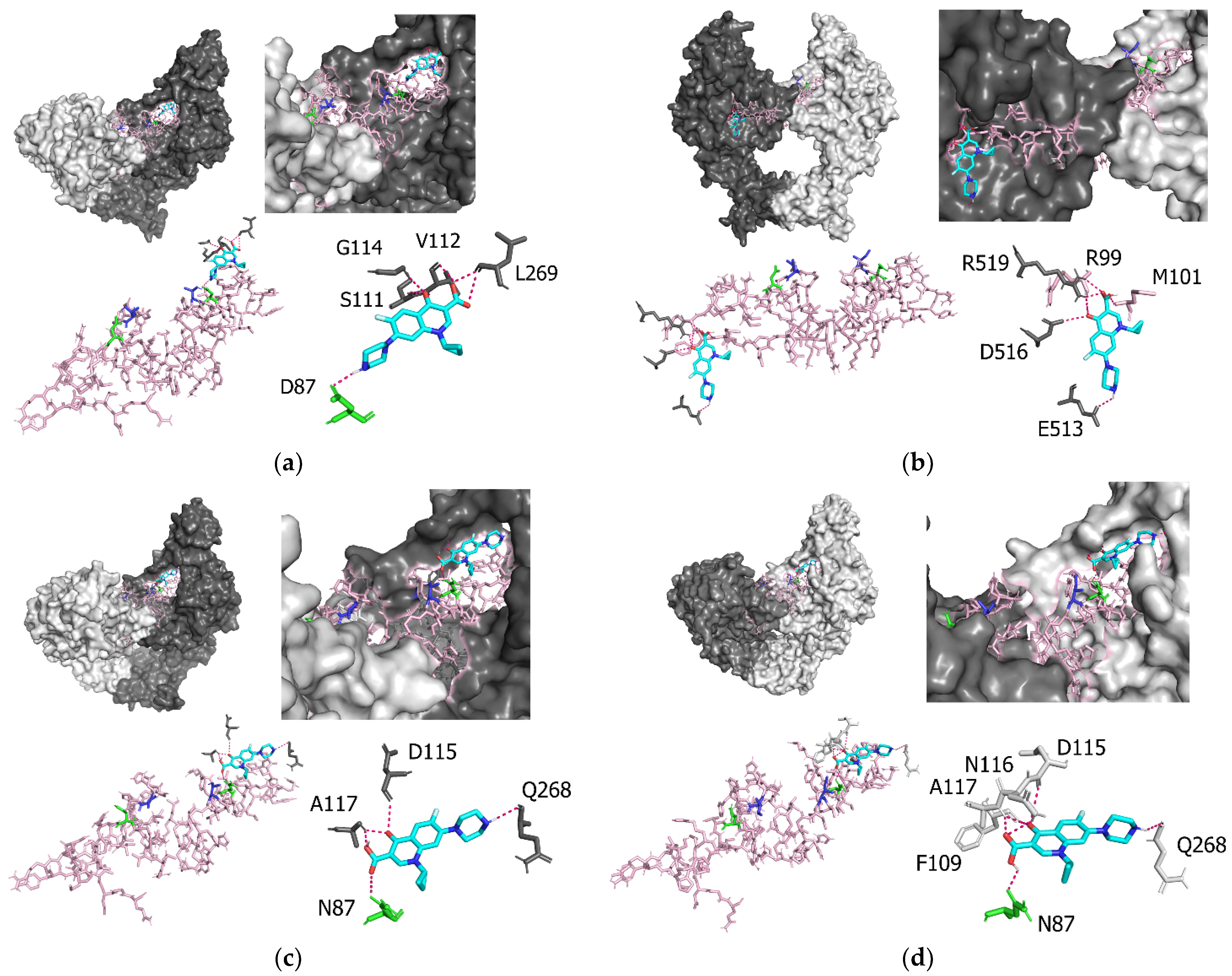
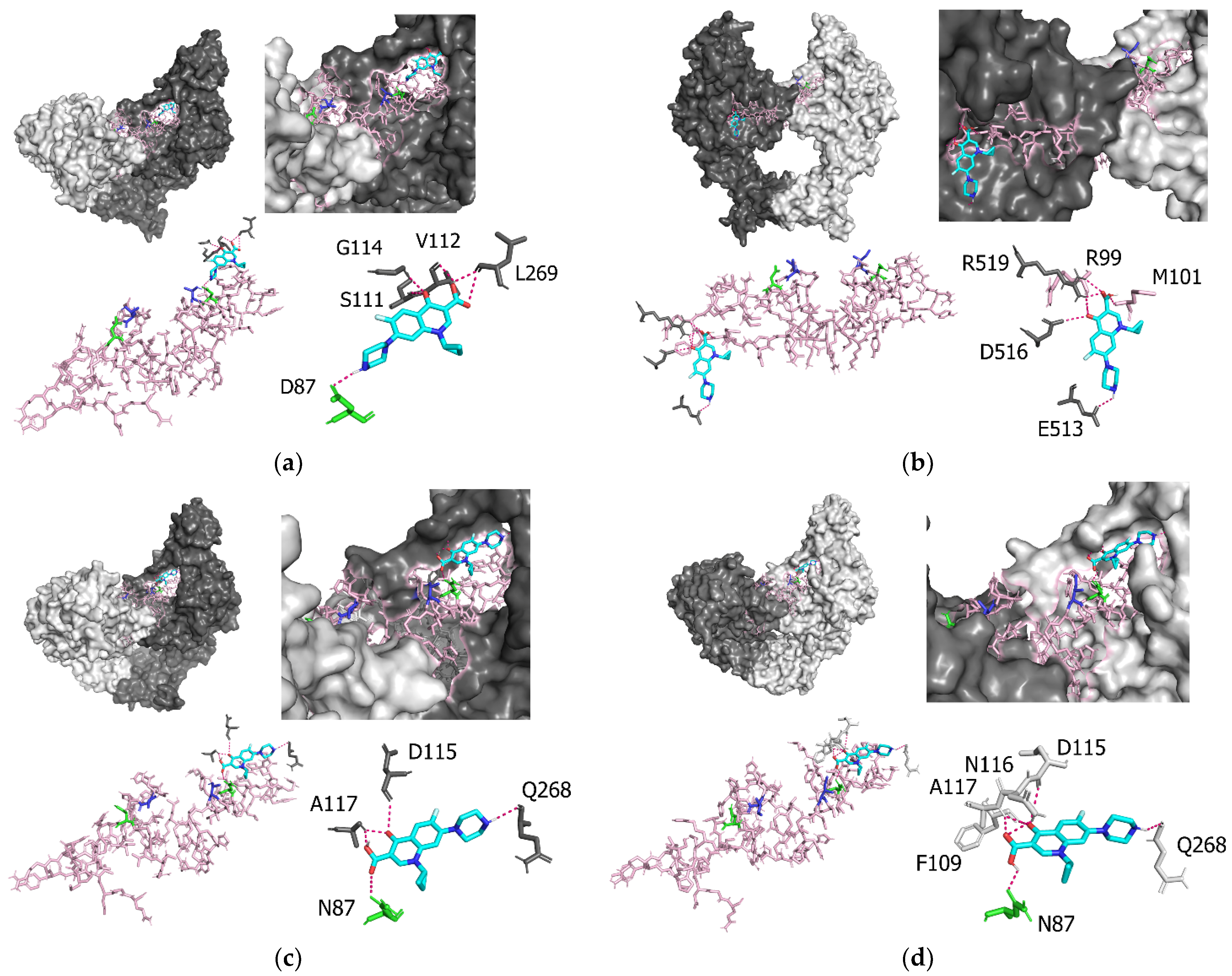
3.4. Molecular Interactions between P. aeruginosa gyrA and Ciprofloxacin
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Park, S.Y.; Park, H.J.; Moon, S.M.; Park, K.H.; Chong, Y.P.; Kim, M.N.; Kim, S.H.; Lee, S.O.; Kim, Y.S.; Woo, J.H.; et al. Impact of adequate empirical combination therapy on mortality from bacteremic Pseudomonas aeruginosa pneumonia. BMC Infect. Dis. 2012, 12, 308. [Google Scholar] [CrossRef] [PubMed]
- Michelena, J.; Altamirano, J.; Abraldes, J.G.; Affò, S.; Morales-Ibanez, O.; Sancho-Bru, P.; Dominguez, M.; Garcia-Pagan, J.C.; Fernández, J.; Arroyo, V.; et al. Systemic inflammatory response and serum lipopolysaccharide levels predict multiple organ failure and death in alcoholic hepatitis. Hepatology 2015, 62, 762–772. [Google Scholar] [CrossRef] [PubMed]
- Pachori, P.; Gothalwal, R.; Gandhi, P. Emergence of antibiotic resistance Pseudomonas aeruginosa in intensive care unit; a critical review. Gene Funct. Dis. 2019, 6, 109–119. [Google Scholar] [CrossRef] [PubMed]
- Viedma, E.; Juan, C.; Acosta, J.; Zamorano, L.; Otero, J.R.; Sanz, F.; Chaves, F.; Oliver, A. Nosocomial spread of colistin-only-sensitive sequence type 235 Pseudomonas aeruginosa isolates producing the extended-spectrum beta-lactamases GES-1 and GES-5 in Spain. Antimicrob. Agents Chemother. 2009, 53, 4930–4933. [Google Scholar] [CrossRef]
- Raman, G.; Avendano, E.E.; Chan, J.; Merchant, S.; Puzniak, L. Risk factors for hospitalized patients with resistant or multidrug-resistant Pseudomonas aeruginosa infections: A systematic review and meta-analysis. Antimicrob. Resist. Infect. Control 2018, 7, 1–14. [Google Scholar] [CrossRef]
- Hsu, D.I.; Okamoto, M.P.; Murthy, R.; Wong-Beringer, A. Fluoroquinolone-resistant Pseudomonas aeruginosa: Risk factors for acquisition and impact on outcomes. J. Antimicrob. Chemother. 2005, 55, 535–541. [Google Scholar] [CrossRef]
- Hooper, D.C. Expanding uses of fluoroquinolones: Opportunities and challenges. Ann. Intern. Med. 1998, 129, 908–910. [Google Scholar] [CrossRef]
- Correia, S.; Poeta, P.; Hébraud, M.; Capelo, J.L.; Igrejas, G. Mechanisms of quinolone action and resistance: Where do we stand? J. Med. Microbiol. 2017, 66, 551–559. [Google Scholar] [CrossRef]
- Jogula, S.; Krishna, V.S.; Meda, N.; Balraju, V.; Sriram, D. Design, synthesis and biological evaluation of novel Pseudomonas aeruginosa DNA gyrase B inhibitors. Bioorganic Chem. 2020, 100, 103905. [Google Scholar] [CrossRef]
- Bush, N.G.; Evans Roberts, K.; Maxwell, A. DNA Topoisomerases. EcoSal Plus 2015, 6. [Google Scholar] [CrossRef]
- Rehman, A.; Patrick, W.; Lamont, I.L. Mechanisms of ciprofloxacin resistance in Pseudomonas aeruginosa: New approaches to an old problem. J. Med. Microbiol. 2019, 68, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, H.; Bogaki, M.; Nakamura, M. Quinolone resistance-determining region in the DNA gyrase gyrA gene of Escherichia coli. Antimicrob. Agents Chemother. 1990, 34, 1271–1272. [Google Scholar] [CrossRef] [PubMed]
- Madurga, S.; Cespedes, J.S.; Belda, I.; Vila, J.; Giralt, E. Mechanism of Binding of Fluoroquinolones to the Quinolone Resistance-Determining Region of DNA Gyrase: Towards an Understanding of the Molecular Basis of Quinolone Resistance. ChemBioChem 2008, 9, 2081–2086. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.K.; Lee, Y.S.; Park, Y.K.; Kim, B.S. Alterations in the GyrA and GyrB subunits of topoisomerase II and the ParC and ParE subunits of topoisomerase IV in ciprofloxacin-resistant clinical isolates of Pseudomonas aeruginosa. Int. J. Antimicrob. Agents 2005, 25, 290–295. [Google Scholar] [CrossRef] [PubMed]
- Ostrer, L.; Khodursky, R.F.; Johnson, J.R.; Hiasa, H.; Khodursky, A. Analysis of mutational patterns in quinolone resistance-determining regions of GyrA and ParC of clinical isolates. Int. J. Antimicrob. Agents 2019, 53, 318–324. [Google Scholar] [CrossRef] [PubMed]
- Akasaka, T.; Tanaka, M.; Yamaguchi, A.; Sato, K. Type II Topoisomerase Mutations in Fluoroquinolone-Resistant Clinical Strains of Pseudomonas aeruginosa Isolated in 1998 and 1999: Role of Target Enzyme in Mechanism of Fluoroquinolone Resistance. Antimicrob. Agents Chemother. 2001, 45, 2263–2268. [Google Scholar] [CrossRef] [PubMed]
- Bruchmann, S.; Dötsch, A.; Nouri, B.; Chaberny, I.F.; Häussler, S. Quantitative Contributions of Target Alteration and Decreased Drug Accumulation to Pseudomonas aeruginosa Fluoroquinolone Resistance. Antimicrob. Agents Chemother. 2013, 57, 1361–1368. [Google Scholar] [CrossRef]
- Hughes, D.; Andersson, D.I. Evolutionary Trajectories to Antibiotic Resistance. Annu. Rev. Microbiol. 2017, 71, 579–596. [Google Scholar] [CrossRef]
- Thafar, M.; Raies, A.; Albaradei, S.; Essack, M.; Bajic, V.B. Comparison Study of Computational Prediction Tools for Drug-Target Binding Affinities. Front. Chem. 2019, 7, 782. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Suyama, M.; Torrents, D.; Bork, P. PAL2NAL: Robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res. 2006, 34, W609–W612. [Google Scholar] [CrossRef] [PubMed]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef] [PubMed]
- Bouckaert, R.; Heled, J.; Kühnert, D.; Vaughan, T.; Wu, C.-H.; Xie, D.; Suchard, M.A.; Rambaut, A.; Drummond, A.J. BEAST 2: A Software Platform for Bayesian Evolutionary Analysis. PLoS Comput. Biol. 2014, 10, e1003537. [Google Scholar] [CrossRef] [PubMed]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. jModelTest 2: More models, new heuristics and parallel computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef] [PubMed]
- Baele, G.; Lemey, P.; Bedford, T.; Rambaut, A.; Suchard, M.A.; Alekseyenko, A.V. Improving the Accuracy of Demographic and Molecular Clock Model Comparison While Accommodating Phylogenetic Uncertainty. Mol. Biol. Evol. 2012, 29, 2157–2167. [Google Scholar] [CrossRef]
- Baele, G.; Li, W.L.S.; Drummond, A.J.; Suchard, M.A.; Lemey, P. Accurate Model Selection of Relaxed Molecular Clocks in Bayesian Phylogenetics. Mol. Biol. Evol. 2012, 30, 239–243. [Google Scholar] [CrossRef]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior Summarization in Bayesian Phylogenetics Using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef]
- Weaver, S.; Shank, S.D.; Spielman, S.J.; Li, M.; Muse, S.V.; Kosakovsky Pond, S.L. Datamonkey 2.0: A Modern Web Application for Characterizing Selective and Other Evolutionary Processes. Mol. Biol. Evol. 2018, 35, 773–777. [Google Scholar] [CrossRef]
- Webb, B.; Sali, A. Comparative Protein Structure Modeling Using MODELLER. Curr. Protoc. Bioinform. 2016, 54, 5.6.1–5.6.37. [Google Scholar] [CrossRef]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef] [PubMed]
- Soares, T.A.; Hünenberger, P.H.; Kastenholz, M.A.; Kräutler, V.; Lenz, T.; Lins, R.D.; Oostenbrink, C.; van Gunsteren, W.F. An improved nucleic acid parameter set for the GROMOS force field. J. Comput. Chem. 2005, 26, 725–737. [Google Scholar] [CrossRef] [PubMed]
- Guex, N.; Peitsch, M.C. SWISS-MODEL and the Swiss-PdbViewer: An environment for comparative protein modeling. Electrophoresis 1997, 18, 2714–2723. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- DeLano, W.L. The PyMOL Molecular Graphics System; DeLano Scientific: San Carlos, CA, USA, 2002. [Google Scholar]
- Kanda, Y. Investigation of the freely available easy-to-use software ‘EZR’ for medical statistics. Bone Marrow Transplant. 2013, 48, 452–458. [Google Scholar] [CrossRef]
- Lesher, G.Y.; Froelich, E.J.; Gruett, M.D.; Bailey, J.H.; Brundage, R.P. 1,8-Naphthyridine derivatives. A new class of chemotherapeutic agents. J. Med. Pharm. Chem. 1962, 91, 1063–1065. [Google Scholar] [CrossRef]
- Stein, G.E. The 4-quinolone antibiotics: Past, present, and future. Pharmacotherapy 1988, 8, 301–314. [Google Scholar] [CrossRef]
- Emmerson, A.M.; Jones, A.M. The quinolones: Decades of development and use. J. Antimicrob. Chemother. 2003, 51 (Suppl. 1), 13–20. [Google Scholar] [CrossRef]
- Karam, G.; Chastre, J.; Wilcox, M.H.; Vincent, J.-L. Antibiotic strategies in the era of multidrug resistance. Crit. Care 2016, 20, 1–9. [Google Scholar] [CrossRef]
- Vernon, J.J.; Wilcox, M.H.; Freeman, J. Effect of fluoroquinolone resistance mutation Thr-82→Ile on Clostridioides difficile fitness. J. Antimicrob. Chemother. 2018, 74, 877–884. [Google Scholar] [CrossRef]
- Kureishi, A.; Diver, J.M.; Beckthold, B.; Schollaardt, T.; Bryan, L.E. Cloning and nucleotide sequence of Pseudomonas aeruginosa DNA gyrase gyrA gene from strain PAO1 and quinolone-resistant clinical isolates. Antimicrob. Agents Chemother. 1994, 38, 1944–1952. [Google Scholar] [CrossRef] [PubMed]
- Mehla, K.; Ramana, J. Structural signature of Ser83Leu and Asp87Asn mutations in DNA gyrase from enterotoxigenic Escherichia coli and impact on quinolone resistance. Gene 2016, 576, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Shorr, A.F. Epidemiology of Staphylococcal Resistance. Clin. Infect. Dis. 2007, 45, S171–S176. [Google Scholar] [CrossRef] [PubMed]
- Aguileta, G.; Refrégier, G.; Yockteng, R.; Fournier, E.; Giraud, T. Rapidly evolving genes in pathogens: Methods for detecting positive selection and examples among fungi, bacteria, viruses and protists. Infect. Genet. Evol. 2009, 9, 656–670. [Google Scholar] [CrossRef] [PubMed]
- Deguchi, T.; Fukuoka, A.; Yasuda, M.; Nakano, M.; Ozeki, S.; Kanematsu, E.; Nishino, Y.; Ishihara, S.; Ban, Y.; Kawada, Y. Alterations in the GyrA subunit of DNA gyrase and the ParC subunit of topoisomerase IV in quinolone-resistant clinical isolates of Klebsiella pneumoniae. Antimicrob. Agents Chemother. 1997, 41, 699–701. [Google Scholar] [CrossRef]
- Weigel, L.M.; Anderson, G.J.; Tenover, F.C. DNA Gyrase and Topoisomerase IV Mutations Associated with Fluoroquinolone Resistance in Proteus mirabilis. Antimicrob. Agents Chemother. 2002, 46, 2582–2587. [Google Scholar] [CrossRef]
- Muggeo, A.; Cambau, E.; Amara, M.; Micaëlo, M.; Pangon, B.; Bajolet, O.; Benmansour, H.; de Champs, C.; Guillard, T. Phenotypic and genotypic quinolone resistance in Escherichia coli underlining GyrA83/87 mutations as a target to detect ciprofloxacin resistance. J. Antimicrob. Chemother. 2020, 75, 2466–2470. [Google Scholar] [CrossRef]
- Neuhauser, M.M.; Weinstein, R.A.; Rydman, R.; Danziger, L.H.; Karam, G.; Quinn, J.P. Antibiotic resistance among gram-negative bacilli in US intensive care units: Implications for fluoroquinolone use. JAMA J. Am. Med. Assoc. 2003, 289, 885–888. [Google Scholar] [CrossRef]
- Pakyz, A.L.; Lee, J.A.; Ababneh, M.A.; Harpe, S.E.; Oinonen, M.J.; Polk, R.E. Fluoroquinolone use and fluoroquinolone-resistant Pseudomonas aeruginosa is declining in US academic medical centre hospitals. J. Antimicrob. Chemother. 2012, 67, 1562–1564. [Google Scholar] [CrossRef]
- Maeda, Y.; Murayama, M.; Goldsmith, C.E.; Coulter, W.A.; Mason, C.; Millar, B.C.; Dooley, J.S.G.; Lowery, C.J.; Matsuda, M.; Rendall, J.C.; et al. Molecular characterization and phylogenetic analysis of quinolone resistance-determining regions (QRDRs) of gyrA, gyrB, parC and parE gene loci in viridans group streptococci isolated from adult patients with cystic fibrosis. J. Antimicrob. Chemother. 2010, 66, 476–486. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Amino acid Residues of GyrA | Atoms Involved in GyrA–Ciprofloxacin Interactions | Intermolecular Distance (Å) |
|---|---|---|
| Prototype | ||
| D87 | O-H | 2.1 |
| S111 | O-O | 2.9 |
| V112 | O-H | 2.3 |
| G114 | H-O | 2.8 |
| L269 | O-H | 2.4 |
| L269 | O-O | 3.3 |
| T83I | ||
| R99 | O-O | 3.3 |
| M101 | H-O | 2.2 |
| E513 | O-H | 2.6 |
| D516 | O-O | 3.2 |
| R519 | H-O | 2.1 |
| R519 | H-O | 2.5 |
| H-O | 2.6 | |
| D87N | ||
| N87 | O-O | 3.3 |
| D115 | O-O | 3.3 |
| A117 | H-O | 2.2 |
| A117 | H-O | 2.2 |
| Q268 | O-H | 2.4 |
| D83I and D87N | ||
| N87 | O-H | 2.2 |
| F109 | O-O | 3.3 |
| D115 | O-O | 3.5 |
| N116 | H-O | 2.6 |
| A117 | H-O | 2.3 |
| A117 | H-O | 2.4 |
| Q268 | O-H | 2.5 |
| P. aeruginosa GyrA | ||||
|---|---|---|---|---|
| Mutation patterns at amino acid 83 and amino acid 87 | Prototype | T83I | D87N | T83I and D87N |
| Molecular affinity (kcal/mol) | −6.98 ± 0.42 | −6.2 ± 0.19 | −6.9 ± 0.46 | −6.5 ± 0.52 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sada, M.; Kimura, H.; Nagasawa, N.; Akagawa, M.; Okayama, K.; Shirai, T.; Sunagawa, S.; Kimura, R.; Saraya, T.; Ishii, H.; et al. Molecular Evolution of the Pseudomonas aeruginosa DNA Gyrase gyrA Gene. Microorganisms 2022, 10, 1660. https://doi.org/10.3390/microorganisms10081660
Sada M, Kimura H, Nagasawa N, Akagawa M, Okayama K, Shirai T, Sunagawa S, Kimura R, Saraya T, Ishii H, et al. Molecular Evolution of the Pseudomonas aeruginosa DNA Gyrase gyrA Gene. Microorganisms. 2022; 10(8):1660. https://doi.org/10.3390/microorganisms10081660
Chicago/Turabian StyleSada, Mitsuru, Hirokazu Kimura, Norika Nagasawa, Mao Akagawa, Kaori Okayama, Tatsuya Shirai, Soyoka Sunagawa, Ryusuke Kimura, Takeshi Saraya, Haruyuki Ishii, and et al. 2022. "Molecular Evolution of the Pseudomonas aeruginosa DNA Gyrase gyrA Gene" Microorganisms 10, no. 8: 1660. https://doi.org/10.3390/microorganisms10081660
APA StyleSada, M., Kimura, H., Nagasawa, N., Akagawa, M., Okayama, K., Shirai, T., Sunagawa, S., Kimura, R., Saraya, T., Ishii, H., Kurai, D., Tsugawa, T., Nishina, A., Tomita, H., Okodo, M., Hirai, S., Ryo, A., Ishioka, T., & Murakami, K. (2022). Molecular Evolution of the Pseudomonas aeruginosa DNA Gyrase gyrA Gene. Microorganisms, 10(8), 1660. https://doi.org/10.3390/microorganisms10081660