
Improved Cladocopium goreaui Genome Assembly Reveals Features of a Facultative Coral Symbiont and the Complex Evolutionary History of Dinoflagellate Genes

 , , ,
, , ,
Abstract
:1. Introduction
2. Results and Discussion
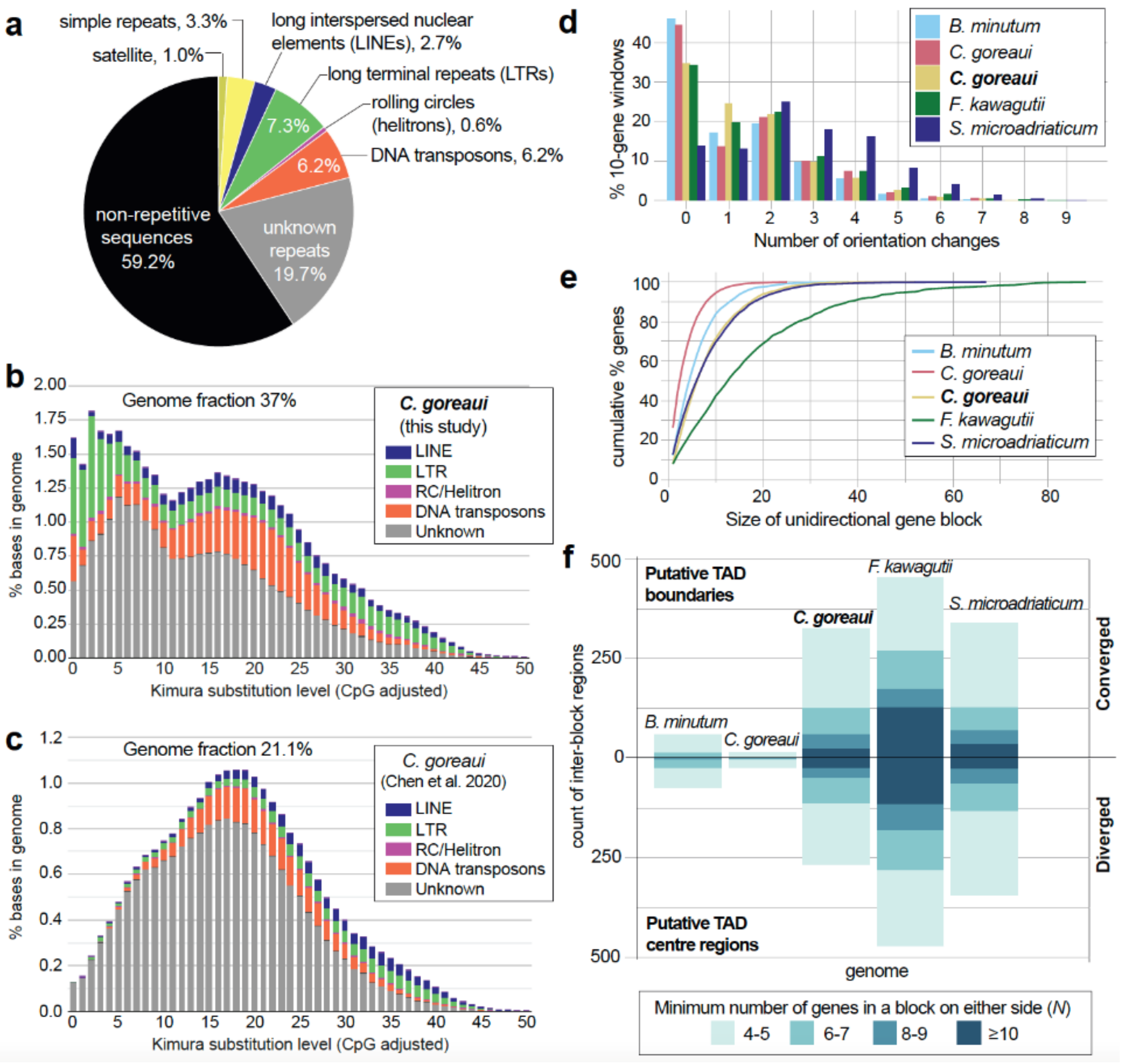
2.1. Improved C. goreaui Genome Assembly Reveals More Repeats and More Duplicated Genes
2.2. Topologically Associated Domains (TADs) and Unidirectional Gene Blocks
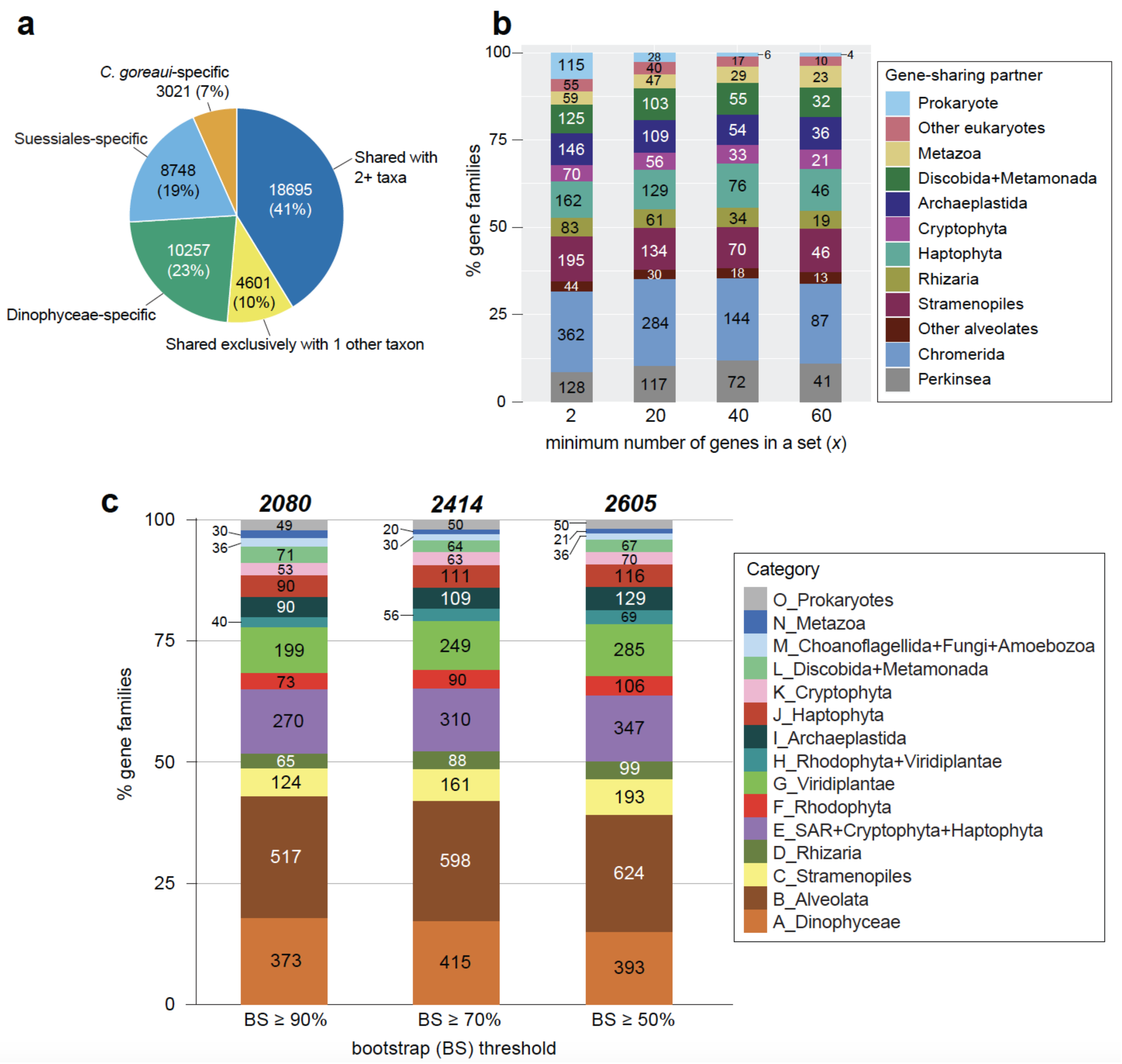
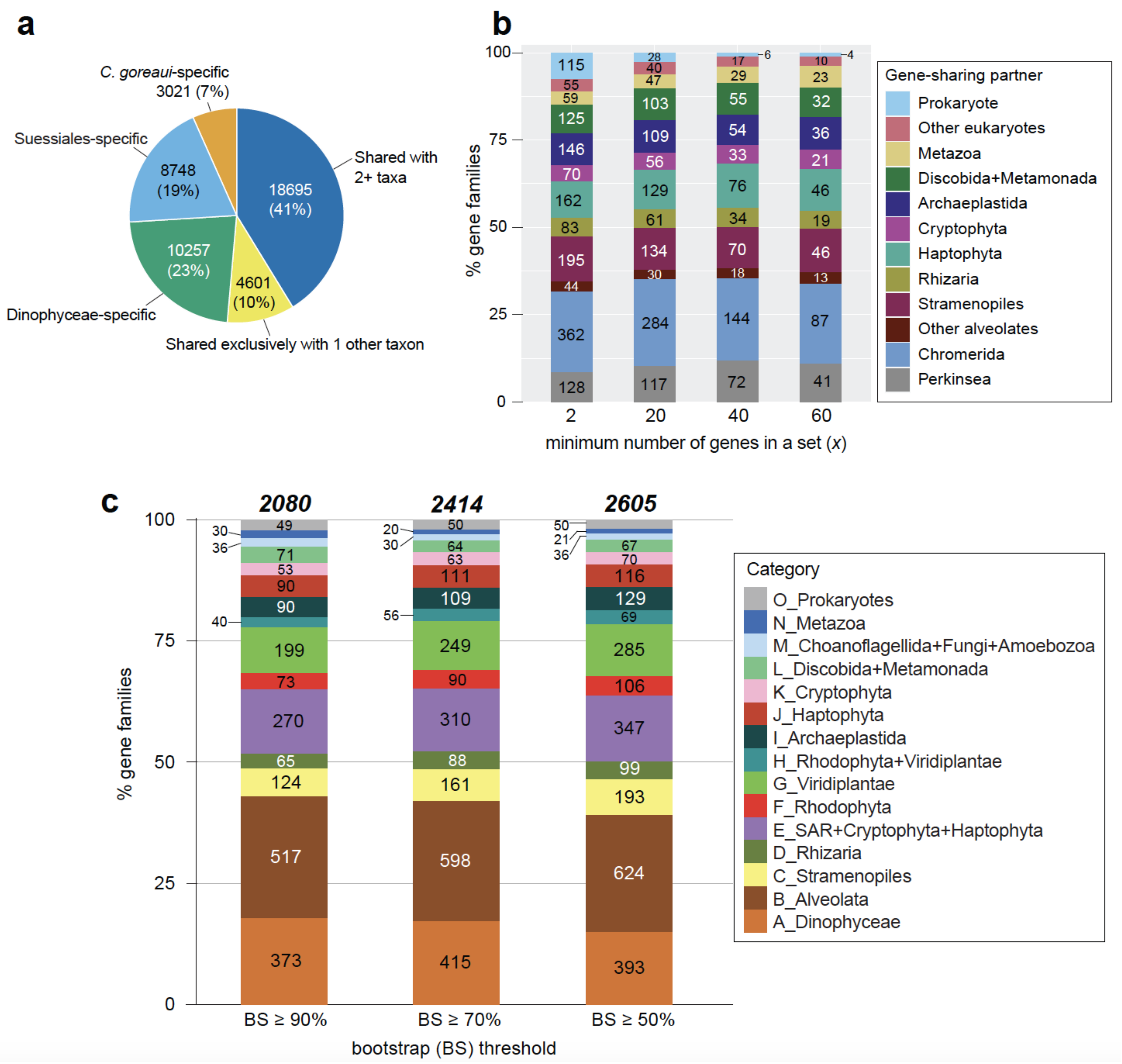
2.3. Evolutionary Origin of C. goreaui Genes
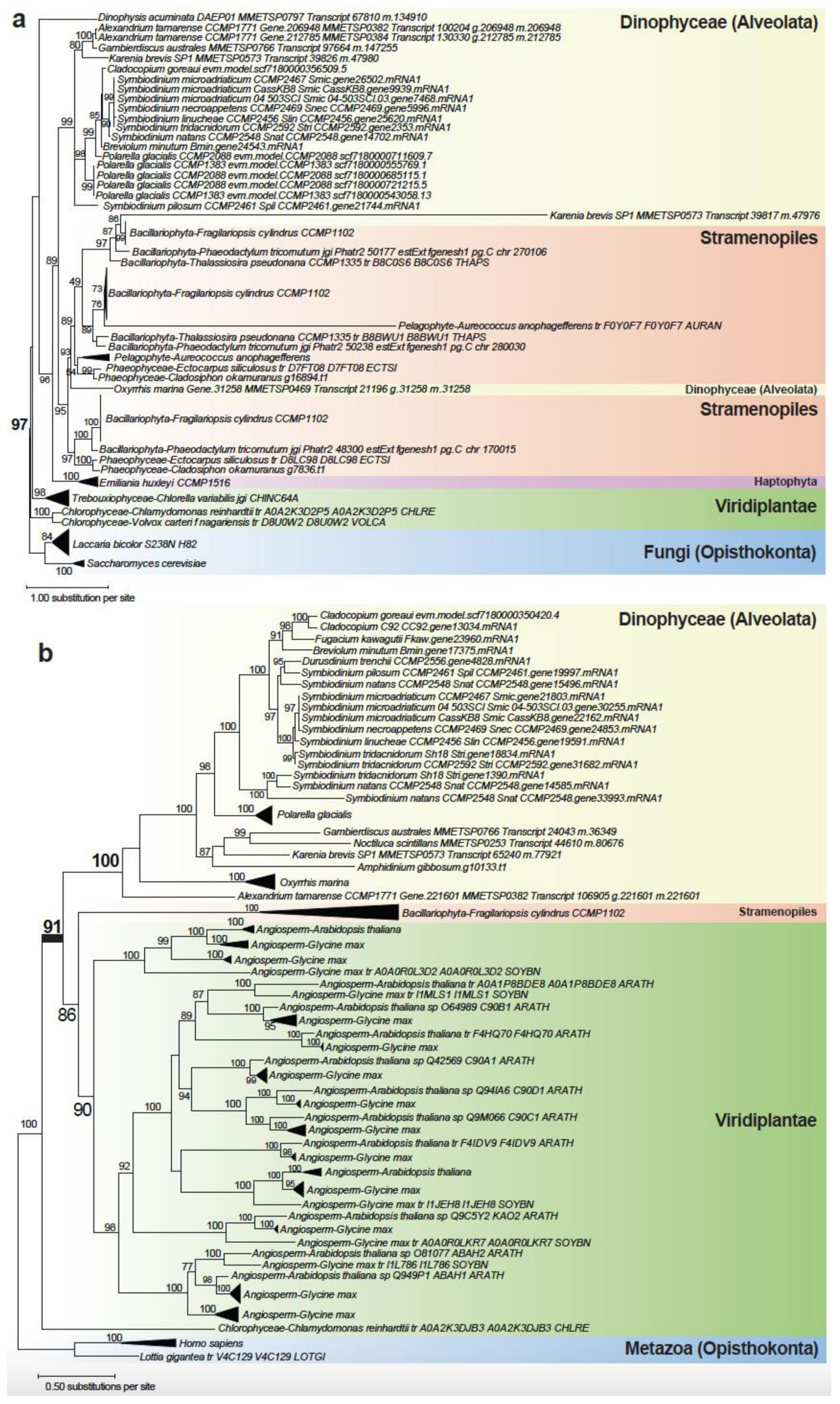
2.4. Genes Implicating a History of Horizontal Transfer
2.5. Genes Implicating Vertical Inheritance
3. Conclusions
4. Materials and Methods
4.1. Generation of Long-Read Genome and Transcriptome Data
4.2. De Novo Genome Assembly Combining Short- and Long-Read Sequence Data
4.3. Estimation of Genome Size Based on Sequencing Data
4.4. Annotation of Repeat Content
4.5. Ab Initio Prediction of Protein-Coding Genes
4.6. Functional Annotation of C. goreaui Genes
4.7. Analysis of Duplicated Genes
4.8. GO Enrichment Analysis
4.9. Analysis of Unidirectional Gene Blocks and TADs
4.10. Phylogenomic Analysis of C. goreaui Genes
4.11. Inference of the Dinoflagellate Species Tree
4.12. Inference of C. goreaui Gene Origins
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- LaJeunesse, T.C.; Parkinson, J.E.; Gabrielson, P.W.; Jeong, H.J.; Reimer, J.D.; Voolstra, C.R.; Santos, S.R. Systematic revision of Symbiodiniaceae highlights the antiquity and diversity of coral endosymbionts. Curr. Biol. 2018, 28, 2570–2580. [Google Scholar] [CrossRef] [PubMed]
- Muscatine, L.; Falkowski, P.G.; Porter, J.W.; Dubinsky, Z.; Smith, D.C. Fate of photosynthetic fixed carbon in light-and shade-adapted colonies of the symbiotic coral Stylophora pistillata. Proc. R. Soc. B Biol. Sci. 1984, 222, 181–202. [Google Scholar]
- Kopp, C.; Pernice, M.; Domart-Coulon, I.; Djediat, C.; Spangenberg, J.E.; Alexander, D.T.L.; Hignette, M.; Meziane, T.; Meibom, A.; Orphan, V.; et al. Highly dynamic cellular-level response of symbiotic coral to a sudden increase in environmental nitrogen. mBio 2013, 4, e00052-13. [Google Scholar] [CrossRef] [PubMed]
- Hoegh-Guldberg, O. Climate change, coral bleaching and the future of the world’s coral reefs. Mar. Freshw. Res. 1999, 50, 839–866. [Google Scholar] [CrossRef]
- Lin, S.; Cheng, S.; Song, B.; Zhong, X.; Lin, X.; Li, W.; Li, L.; Zhang, Y.; Zhang, H.; Ji, Z. The Symbiodinium kawagutii genome illuminates dinoflagellate gene expression and coral symbiosis. Science 2015, 350, 691–694. [Google Scholar] [CrossRef] [PubMed]
- Aranda, M.; Li, Y.; Liew, Y.J.; Baumgarten, S.; Simakov, O.; Wilson, M.C.; Piel, J.; Ashoor, H.; Bougouffa, S.; Bajic, V.B. Genomes of coral dinoflagellate symbionts highlight evolutionary adaptations conducive to a symbiotic lifestyle. Sci. Rep. 2016, 6, 39734. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Stephens, T.G.; González-Pech, R.A.; Beltran, V.H.; Lapeyre, B.; Bongaerts, P.; Cooke, I.; Aranda, M.; Bourne, D.G.; Forêt, S.; et al. Symbiodinium genomes reveal adaptive evolution of functions related to coral-dinoflagellate symbiosis. Commun. Biol. 2018, 1, 95. [Google Scholar]
- Shoguchi, E.; Beedessee, G.; Tada, I.; Hisata, K.; Kawashima, T.; Takeuchi, T.; Arakaki, N.; Fujie, M.; Koyanagi, R.; Roy, M.C. Two divergent Symbiodinium genomes reveal conservation of a gene cluster for sunscreen biosynthesis and recently lost genes. BMC Genomics 2018, 19, 458. [Google Scholar] [CrossRef]
- Dougan, K.E.; González-Pech, R.A.; Stephens, T.G.; Shah, S.; Chen, Y.; Ragan, M.A.; Bhattacharya, D.; Chan, C.X. Genome-powered classification of microbial eukaryotes: Focus on coral algal symbionts. Trends Microbiol. 2022, 30, 831–840. [Google Scholar] [CrossRef]
- González-Pech, R.A.; Stephens, T.G.; Chen, Y.; Mohamed, A.R.; Cheng, Y.; Shah, S.; Dougan, K.E.; Fortuin, M.D.A.; Lagorce, R.; Burt, D.W.; et al. Comparison of 15 dinoflagellate genomes reveals extensive sequence and structural divergence in family Symbiodiniaceae and genus Symbiodinium. BMC Biol. 2021, 19, 73. [Google Scholar] [CrossRef]
- Lo, R.; Dougan, K.E.; Chen, Y.; Shah, S.; Bhattacharya, D.; Chan, C.X. Alignment-free analysis of whole-genome sequences from Symbiodiniaceae reveals different phylogenetic signals in distinct regions. Front. Plant Sci. 2022, 13, 815714. [Google Scholar] [CrossRef] [PubMed]
- González-Pech, R.A.; Bhattacharya, D.; Ragan, M.A.; Chan, C.X. Genome evolution of coral reef symbionts as intracellular residents. Trends Ecol. Evol. 2019, 34, 799–806. [Google Scholar] [CrossRef] [PubMed]
- Bongaerts, P.; Carmichael, M.; Hay, K.B.; Tonk, L.; Frade, P.R.; Hoegh-Guldberg, O. Prevalent endosymbiont zonation shapes the depth distributions of scleractinian coral species. R. Soc. Open Sci. 2015, 2, 140297. [Google Scholar] [CrossRef]
- LaJeunesse, T.C. “Species” radiations of symbiotic dinoflagellates in the Atlantic and Indo-Pacific since the Miocene-Pliocene transition. Mol. Biol. Evol. 2005, 22, 570–581. [Google Scholar] [CrossRef]
- Chen, Y.; González-Pech, R.A.; Stephens, T.G.; Bhattacharya, D.; Chan, C.X. Evidence that inconsistent gene prediction can mislead analysis of dinoflagellate genomes. J. Phycol. 2020, 56, 6–10. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.X.; Soares, M.B.; Bonaldo, M.F.; Wisecaver, J.H.; Hackett, J.D.; Anderson, D.M.; Erdner, D.L.; Bhattacharya, D. Analysis of Alexandrium tamarense (Dinophyceae) genes reveals the complex evolutionary history of a microbial eukaryote J. Phycol. 2012, 48, 1130–1142. [Google Scholar] [CrossRef] [PubMed]
- Manni, M.; Berkeley, M.R.; Seppey, M.; Simão, F.A.; Zdobnov, E.M. BUSCO update: Novel and streamlined workflows along with broader and deeper phylogenetic coverage for scoring of eukaryotic, prokaryotic, and viral genomes. Mol. Biol. Evol. 2021, 38, 4647–4654. [Google Scholar] [CrossRef]
- Stephens, T.G.; González-Pech, R.A.; Cheng, Y.; Mohamed, A.R.; Burt, D.W.; Bhattacharya, D.; Ragan, M.A.; Chan, C.X. Genomes of the dinoflagellate Polarella glacialis encode tandemly repeated single-exon genes with adaptive functions. BMC Biol. 2020, 18, 56. [Google Scholar] [CrossRef]
- Li, T.; Yu, L.; Song, B.; Song, Y.; Li, L.; Lin, X.; Lin, S. Genome improvement and core gene set refinement of Fugacium kawagutii. Microorganisms 2020, 8, 102. [Google Scholar] [CrossRef]
- Nand, A.; Zhan, Y.; Salazar, O.R.; Aranda, M.; Voolstra, C.R.; Dekker, J. Genetic and spatial organization of the unusual chromosomes of the dinoflagellate Symbiodinium microadriaticum. Nat. Genet. 2021, 53, 618–629. [Google Scholar] [CrossRef]
- Marinov, G.K.; Trevino, A.E.; Xiang, T.; Kundaje, A.; Grossman, A.R.; Greenleaf, W.J. Transcription-dependent domain-scale three-dimensional genome organization in the dinoflagellate Breviolum minutum. Nat. Genet. 2021, 53, 613–617. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Song, B.; Morse, D. Spatial organization of dinoflagellate genomes: Novel insights and remaining critical questions. J. Phycol. 2021, 57, 1674–1678. [Google Scholar] [CrossRef]
- Price, D.C.; Bhattacharya, D. Robust Dinoflagellata phylogeny inferred from public transcriptome databases. J. Phycol. 2017, 53, 725–729. [Google Scholar] [CrossRef] [PubMed]
- Stephens, T.G.; Ragan, M.A.; Bhattacharya, D.; Chan, C.X. Core genes in diverse dinoflagellate lineages include a wealth of conserved dark genes with unknown functions. Sci. Rep. 2018, 8, 17175. [Google Scholar] [CrossRef] [PubMed]
- Burki, F.; Roger, A.J.; Brown, M.W.; Simpson, A.G.B. The new tree of eukaryotes. Trends Ecol. Evol. 2020, 35, 43–55. [Google Scholar] [CrossRef]
- Hackett, J.D.; Yoon, H.S.; Li, S.; Reyes-Prieto, A.; Rümmele, S.E.; Bhattacharya, D. Phylogenomic analysis supports the monophyly of cryptophytes and haptophytes and the association of rhizaria with chromalveolates. Mol. Biol. Evol. 2007, 24, 1702–1713. [Google Scholar] [CrossRef]
- Adl, S.M.; Bass, D.; Lane, C.E.; Lukeš, J.; Schoch, C.L.; Smirnov, A.; Agatha, S.; Berney, C.; Brown, M.W.; Burki, F.; et al. Revisions to the classification, nomenclature, and diversity of eukaryotes. J. Eukaryot. Microbiol. 2019, 66, 4–119. [Google Scholar] [CrossRef]
- Ishida, K.-I.; Green, B.R. Second- and third-hand chloroplasts in dinoflagellates: Phylogeny of oxygen-evolving enhancer 1 (PsbO) protein reveals replacement of a nuclear-encoded plastid gene by that of a haptophyte tertiary endosymbiont. Proc. Natl. Acad. Sci. USA 2002, 99, 9294–9299. [Google Scholar] [CrossRef]
- Chan, C.X.; Gross, J.; Yoon, H.S.; Bhattacharya, D. Plastid origin and evolution: New models provide insights into old problems. Plant Physiol. 2011, 155, 1552–1560. [Google Scholar] [CrossRef]
- Yoon, H.S.; Hackett, J.D.; Van Dolah, F.M.; Nosenko, T.; Lidie, K.L.; Bhattacharya, D. Tertiary endosymbiosis driven genome evolution in dinoflagellate algae. Mol. Biol. Evol. 2005, 22, 1299–1308. [Google Scholar] [CrossRef]
- Stephens, T.G.; Bhattacharya, D.; Ragan, M.A.; Chan, C.X. PhySortR: A fast, flexible tool for sorting phylogenetic trees in R. PeerJ 2016, 4, e2038. [Google Scholar] [CrossRef] [PubMed]
- Gabrielsen, T.M.; Minge, M.A.; Espelund, M.; Tooming-Klunderud, A.; Patil, V.; Nederbragt, A.J.; Otis, C.; Turmel, M.; Shalchian-Tabrizi, K.; Lemieux, C. Genome evolution of a tertiary dinoflagellate plastid. PLoS ONE 2011, 6, e19132. [Google Scholar] [CrossRef] [PubMed]
- Janouškovec, J.; Horák, A.; Oborník, M.; Lukeš, J.; Keeling, P.J. A common red algal origin of the apicomplexan, dinoflagellate, and heterokont plastids. Proc. Natl. Acad. Sci. USA 2010, 107, 10949–10954. [Google Scholar] [CrossRef] [PubMed]
- Moustafa, A.; Beszteri, B.; Maier, U.G.; Bowler, C.; Valentin, K.; Bhattacharya, D. Genomic footprints of a cryptic plastid endosymbiosis in diatoms. Science 2009, 324, 1724–1726. [Google Scholar] [CrossRef]
- Chan, C.X.; Reyes-Prieto, A.; Bhattacharya, D. Red and green algal origin of diatom membrane transporters: Insights into environmental adaptation and cell evolution. PLoS ONE 2011, 6, e29138. [Google Scholar] [CrossRef]
- Nevo, Z.; Sharon, N. The cell wall of Peridinium westii, a non cellulosic glucan. Biochim. Biophys. Acta 1969, 173, 161–175. [Google Scholar] [CrossRef]
- Salmeán, A.A.; Duffieux, D.; Harholt, J.; Qin, F.; Michel, G.; Czjzek, M.; Willats, W.G.T.; Hervé, C. Insoluble (1→3), (1→4)-β-D-glucan is a component of cell walls in brown algae (Phaeophyceae) and is masked by alginates in tissues. Sci. Rep. 2017, 7, 2880. [Google Scholar] [CrossRef]
- Footitt, S.; Douterelo-Soler, I.; Clay, H.; Finch-Savage, W.E. Dormancy cycling in Arabidopsis seeds is controlled by seasonally distinct hormone-signaling pathways. Proc. Natl. Acad. Sci. USA 2011, 108, 20236–20241. [Google Scholar] [CrossRef]
- Krochko, J.E.; Abrams, G.D.; Loewen, M.K.; Abrams, S.R.; Cutler, A.J. (+)-Abscisic acid 8’-hydroxylase is a cytochrome P450 monooxygenase. Plant Physiol. 1998, 118, 849–860. [Google Scholar] [CrossRef]
- Deng, Y.; Hu, Z.; Shang, L.; Peng, Q.; Tang, Y.Z. Transcriptomic analyses of Scrippsiella trochoidea reveals processes regulating encystment and dormancy in the life cycle of a dinoflagellate, with a particular attention to the role of abscisic acid. Front. Microbiol. 2017, 8, 2450. [Google Scholar] [CrossRef]
- Rosic, N.N.; Pernice, M.; Dunn, S.; Dove, S.; Hoegh-Guldberg, O. Differential regulation by heat stress of novel cytochrome P450 genes from the dinoflagellate symbionts of reef-building corals. Appl. Environ. Microbiol. 2010, 76, 2823–2829. [Google Scholar] [CrossRef] [PubMed]
- Melis, A.; Chen, H.-C. Chloroplast sulfate transport in green algae–genes, proteins and effects. Photosynth. Res. 2005, 86, 299–307. [Google Scholar] [CrossRef] [PubMed]
- Depuydt, T.; Vandepoele, K. Multi-omics network-based functional annotation of unknown Arabidopsis genes. Plant J. 2021, 108, 1193–1212. [Google Scholar] [CrossRef] [PubMed]
- Kaundal, R.; Saini, R.; Zhao, P.X. Combining machine learning and homology-based approaches to accurately predict subcellular localization in Arabidopsis. Plant Physiol. 2010, 154, 36–54. [Google Scholar] [CrossRef]
- Sarai, C.; Tanifuji, G.; Nakayama, T.; Kamikawa, R.; Takahashi, K.; Yazaki, E.; Matsuo, E.; Miyashita, H.; Ishida, K.-I.; Iwataki, M.; et al. Dinoflagellates with relic endosymbiont nuclei as models for elucidating organellogenesis. Proc. Natl. Acad. Sci. USA 2020, 117, 5364–5375. [Google Scholar] [CrossRef]
- Yoon, H.S.; Hackett, J.D.; Bhattacharya, D. A single origin of the peridinin- and fucoxanthin-containing plastids in dinoflagellates through tertiary endosymbiosis. Proc. Natl. Acad. Sci. USA 2002, 99, 11724–11729. [Google Scholar] [CrossRef]
- Morozov, A.A.; Galachyants, Y.P. Diatom genes originating from red and green algae: Implications for the secondary endosymbiosis models. Mar Genomics 2019, 45, 72–78. [Google Scholar] [CrossRef]
- Archibald, J.M.; Keeling, P.J. Recycled plastids: A ‘green movement’ in eukaryotic evolution. Trends Genet. 2002, 18, 577–584. [Google Scholar] [CrossRef]
- Kamikawa, R.; Tanifuji, G.; Kawachi, M.; Miyashita, H.; Hashimoto, T.; Inagaki, Y. Plastid genome-based phylogeny pinpointed the origin of the green-colored plastid in the dinoflagellate Lepidodinium chlorophorum. Genome Biol. Evol. 2015, 7, 1133–1140. [Google Scholar] [CrossRef]
- Rowan, R.; Whitney, S.M.; Fowler, A.; Yellowlees, D. Rubisco in marine symbiotic dinoflagellates: Form II enzymes in eukaryotic oxygenic phototrophs encoded by a nuclear multigene family. Plant Cell 1996, 8, 539–553. [Google Scholar]
- Howells, E.; Beltran, V.; Larsen, N.; Bay, L.; Willis, B.; Van Oppen, M. Coral thermal tolerance shaped by local adaptation of photosymbionts. Nat. Clim. Chang. 2012, 2, 116–120. [Google Scholar] [CrossRef]
- Wilson, K.; Li, Y.; Whan, V.; Lehnert, S.; Byrne, K.; Moore, S.; Pongsomboon, S.; Tassanakajon, A.; Rosenberg, G.; Ballment, E.; et al. Genetic mapping of the black tiger shrimp Penaeus monodon with amplified fragment length polymorphism. Aquaculture 2002, 204, 297–309. [Google Scholar] [CrossRef]
- Rosic, N.N.; Hoegh-Guldberg, O. A method for extracting a high-quality RNA from Symbiodinium sp. J. Appl. Phycol. 2010, 22, 139–146. [Google Scholar] [CrossRef]
- Zimin, A.V.; Puiu, D.; Luo, M.-C.; Zhu, T.; Koren, S.; Marçais, G.; Yorke, J.A.; Dvořák, J.; Salzberg, S.L. Hybrid assembly of the large and highly repetitive genome of Aegilops tauschii, a progenitor of bread wheat, with the MaSuRCA mega-reads algorithm. Genome Res. 2017, 27, 787–792. [Google Scholar] [CrossRef] [PubMed]
- Iha, C.; Dougan, K.E.; Varela, J.A.; Avila, V.; Jackson, C.J.; Bogaert, K.A.; Chen, Y.; Judd, L.M.; Wick, R.; Holt, K.E. Genomic adaptations to an endolithic lifestyle in the coral-associated alga Ostreobium. Curr. Biol. 2021, 31, 1393–1402. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Laetsch, D.; Blaxter, M. BlobTools: Interrogation of genome assemblies. F1000Research 2017, 6, 1287. [Google Scholar] [CrossRef]
- Levin, R.A.; Beltran, V.H.; Hill, R.; Kjelleberg, S.; McDougald, D.; Steinberg, P.D.; van Oppen, M.J.H. Sex, scavengers, and chaperones: Transcriptome secrets of divergent Symbiodinium thermal tolerances. Mol. Biol. Evol. 2016, 33, 2201–2215. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q. Trinity: Reconstructing a full-length transcriptome without a genome from RNA-Seq data. Nat. Biotechnol. 2011, 29, 644. [Google Scholar] [CrossRef]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef]
- Marçais, G.; Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 2011, 27, 764–770. [Google Scholar] [CrossRef] [PubMed]
- Ranallo-Benavidez, T.R.; Jaron, K.S.; Schatz, M.C. GenomeScope 2.0 and Smudgeplot for reference-free profiling of polyploid genomes. Nat. Commun. 2020, 11, 1432. [Google Scholar] [CrossRef] [PubMed]
- Haas, B.J.; Delcher, A.L.; Mount, S.M.; Wortman, J.R.; Smith, R.K., Jr.; Hannick, L.I.; Maiti, R.; Ronning, C.M.; Rusch, D.B.; Town, C.D. Improving the Arabidopsis genome annotation using maximal transcript alignment assemblies. Nucleic Acids Res. 2003, 31, 5654–5666. [Google Scholar] [CrossRef] [PubMed]
- Remmert, M.; Biegert, A.; Hauser, A.; Söding, J. HHblits: Lightning-fast iterative protein sequence searching by HMM-HMM alignment. Nat. Methods 2012, 9, 173–175. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 1658–1659. [Google Scholar] [CrossRef] [PubMed]
- Korf, I. Gene finding in novel genomes. BMC Bioinform. 2004, 5, 59. [Google Scholar] [CrossRef]
- Stanke, M.; Keller, O.; Gunduz, I.; Hayes, A.; Waack, S.; Morgenstern, B. AUGUSTUS: Ab initio prediction of alternative transcripts. Nucleic Acids Res. 2006, 34, W435–W439. [Google Scholar] [CrossRef]
- Lomsadze, A.; Ter-Hovhannisyan, V.; Chernoff, Y.O.; Borodovsky, M. Gene identification in novel eukaryotic genomes by self-training algorithm. Nucleic Acids Res. 2005, 33, 6494–6506. [Google Scholar] [CrossRef]
- Holt, C.; Yandell, M. MAKER2: An annotation pipeline and genome-database management tool for second-generation genome projects. BMC Bioinform. 2011, 12, 491. [Google Scholar] [CrossRef]
- Haas, B.J.; Salzberg, S.L.; Zhu, W.; Pertea, M.; Allen, J.E.; Orvis, J.; White, O.; Buell, C.R.; Wortman, J.R. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 2008, 9, R7. [Google Scholar] [CrossRef]
- Wang, Y.; Li, J.; Paterson, A.H. MCScanX-transposed: Detecting transposed gene duplications based on multiple colinearity scans. Bioinformatics 2013, 29, 1458–1460. [Google Scholar] [CrossRef] [PubMed]
- Alexa, A.; Rahnenführer, J. topGO: Enrichment analysis for Gene Ontology. R package version 2.48.0. 2022. Available online: https://bioconductor.org/packages/release/bioc/html/topGO.html (accessed on 30 June 2022).
- Emms, D.M.; Kelly, S. OrthoFinder: Phylogenetic orthology inference for comparative genomics. Genome Biol. 2019, 20, 238. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Capella-Gutiérrez, S.; Silla-Martínez, J.M.; Gabaldón, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef] [PubMed]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Metric | Earlier Assembly [15] | Revised Assembly (This Study) | |
|---|---|---|---|
| Assembly size (Gbp) | 1.0 | 1.2 | |
| Number of scaffolds | 41,235 | 6843 | |
| Genome scaffolds N50 (Kbp) | 91 | 354 | |
| Genome GC-content (%) | 44.76 | 44.38 | |
| Number of predicted genes | 39,006 | 45,322 | |
| Percentage of BUSCO proteins recovered (alveolata_odb10) | 67.3 | 82.4 | |
| Genes with transcript support (%) | 76.5 | 82.5 | |
| Average gene length (bp) | 8428 | 15,745 | |
| Average CDS length (bp) | 1625 | 2018 | |
| Total gene length (Mbp) | 328.7 | 713.6 | |
| Total CDS length (Mbp) | 63.4 | 91.5 | |
| Average number of exons per gene | 12.4 | 17.2 | |
| Average exon length (bp) | 130.4 | 120.4 | |
| Genes with introns (%) | 95.9 | 95.9 | |
| Number of introns per gene | 11.4 | 16.2 | |
| Average intron length (bp) | 593.5 | 838.8 | |
| Splice donor motif (%) | GT | 35.7 | 36.6 |
| GC | 43.3 | 43.6 | |
| GA | 20.8 | 19.8 | |
| Splice acceptor with AGG motif (%) | 96.3 | 96.1 | |
| Number of intergenic regions | 24,243 | 39,720 | |
| Average length of intergenic regions (bp) | 9539 | 7388 | |
| Duplication Type | Earlier Assembly [15] | Revised Assembly (This Study) |
|---|---|---|
| Singleton | 13,456 (34.5%) | 10,203 (22.5%) |
| Dispersed | 24,441 (62.6%) | 31,827 (70.2%) |
| Proximal | 273 (0.7%) | 907 (2.0%) |
| Tandem | 836 (2.1%) | 1998 (4.4%) |
| Segmental | 0 (0.0%) | 387 (0.9%) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Y.; Shah, S.; Dougan, K.E.; van Oppen, M.J.H.; Bhattacharya, D.; Chan, C.X. Improved Cladocopium goreaui Genome Assembly Reveals Features of a Facultative Coral Symbiont and the Complex Evolutionary History of Dinoflagellate Genes. Microorganisms 2022, 10, 1662. https://doi.org/10.3390/microorganisms10081662
Chen Y, Shah S, Dougan KE, van Oppen MJH, Bhattacharya D, Chan CX. Improved Cladocopium goreaui Genome Assembly Reveals Features of a Facultative Coral Symbiont and the Complex Evolutionary History of Dinoflagellate Genes. Microorganisms. 2022; 10(8):1662. https://doi.org/10.3390/microorganisms10081662
Chicago/Turabian StyleChen, Yibi, Sarah Shah, Katherine E. Dougan, Madeleine J. H. van Oppen, Debashish Bhattacharya, and Cheong Xin Chan. 2022. "Improved Cladocopium goreaui Genome Assembly Reveals Features of a Facultative Coral Symbiont and the Complex Evolutionary History of Dinoflagellate Genes" Microorganisms 10, no. 8: 1662. https://doi.org/10.3390/microorganisms10081662
APA StyleChen, Y., Shah, S., Dougan, K. E., van Oppen, M. J. H., Bhattacharya, D., & Chan, C. X. (2022). Improved Cladocopium goreaui Genome Assembly Reveals Features of a Facultative Coral Symbiont and the Complex Evolutionary History of Dinoflagellate Genes. Microorganisms, 10(8), 1662. https://doi.org/10.3390/microorganisms10081662