
A Potential Predictive Role of the Scalp Microbiome Profiling in Patients with Alopecia Areata: Staphylococcus caprae, Corynebacterium, and Cutibacterium Species



Abstract
:1. Introduction
2. Materials and Methods
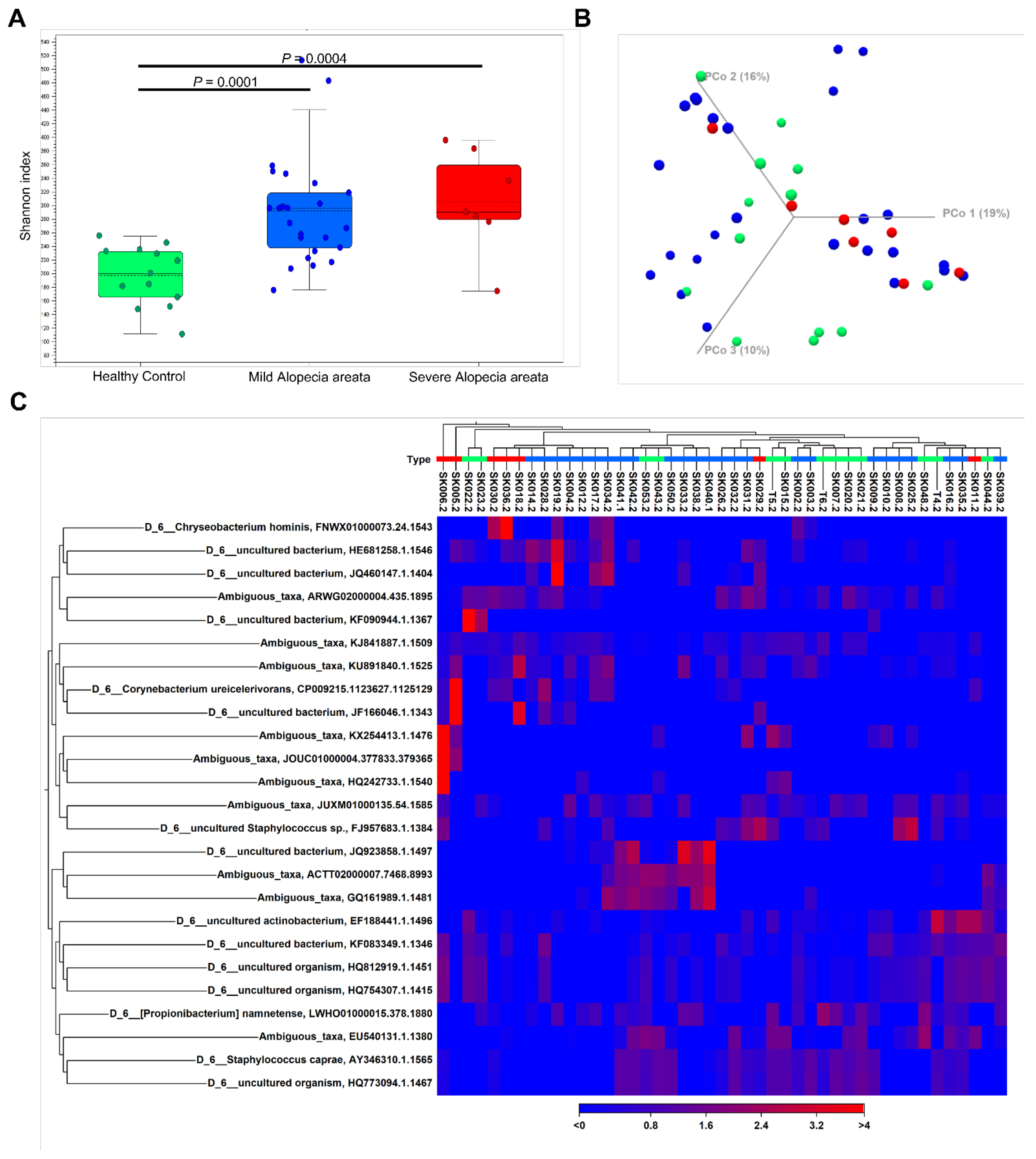
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Odom, R.B.; Davidsohn, I.J.; William, D.; Henry, J.B.; Berger, T.G. Clinical diagnosis by laboratory methods. In Andrews’ Diseases of the Skin: Clinical Dermatology; Elston, D.M., Ed.; Saunders Elsevier: Amsterdam, The Netherlands, 2006. [Google Scholar]
- Camacho, F. Alopecia areata. Clinical characteristics and dermatopathology. In Trichology: Diseases of the Pilosebaceous Follicle; Aula Medical Group SA: Madrid, Spain, 1997; pp. 440–471. [Google Scholar]
- Wang, E.C.E.; Christiano, A.M. The changing landscape of alopecia areata: The translational landscape. Adv. Ther. 2017, 34, 1586–1593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Migacz-Gruszka, K.; Branicki, W.; Obtulowicz, A.; Pirowska, M.; Gruszka, K.; Wojas-Pelc, A. What’s new in the pathophysiology of alopecia areata? the possible contribution of skin and gut microbiome in the pathogenesis of alopecia—Big opportunities, big challenges, and novel perspectives. Int. J. Trichol. 2019, 11, 185–188. [Google Scholar] [CrossRef] [PubMed]
- Borde, A.; Åstrand, A. Alopecia areata and the gut-the link opens up for novel therapeutic interventions. Expert Opin. Ther. Targets 2018, 22, 503–511. [Google Scholar] [CrossRef] [PubMed]
- Human Microbiome Project Consortium. Structure, function and diversity of the healthy human microbiome. Nature 2012, 486, 207–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rinaldi, F.; Pinto, D.; Marzani, B.; Rucco, M.; Giuliani, G.; Sorbellini, E. Human microbiome: What’s new in scalp diseases. J. Transl. Sci. 2018, 4, 1–4. [Google Scholar]
- Pinto, D.; Sorbellini, E.; Marzani, B.; Rucco, M.; Giuliani, G.; Rinaldi, F. Scalp bacterial shift in Alopecia areata. PLoS ONE 2019, 14, e0215206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ho, B.S.-Y.; Ho, E.X.P.; Chu, C.W.; Ramasamy, S.; Bigliardi-Qi, M.; de Sessions, P.F.; Bigliardi, P.L. Microbiome in the hair follicle of androgenetic alopecia patients. PLoS ONE 2019, 14, e0216330. [Google Scholar]
- Suzuki, K.; Inoue, M.; Cho, O.; Mizutani, R.; Shimizu, Y.; Nagahama, T.; Sugita, T. Scalp Microbiome and Sebum Composition in Japanese Male Individuals with and without Androgenetic Alopecia. Microorganisms 2021, 9, 2132. [Google Scholar] [CrossRef] [PubMed]
- Grice, E.A.; Kong, H.H.; Conlan, S.; Deming, C.B.; Davis, J.; Young, A.C.; NISC Comparative Sequencing Program; Bouffard, G.G.; Blakesley, R.W.; Murraye, P.R.; et al. Topographical and Temporal Diversity of the Human Skin Microbiome. Science 2009, 324, 1190–1192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, Z.; Perez-Perez, G.I.; Chen, Y.; Blaser, M.J. Quantitation of Major Human Cutaneous Bacterial and Fungal Populations. J. Clin. Microbiol. 2010, 48, 3575–3581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, M.J.; Lee, Y.J.; Kim, T.J.; Won, E.J. Gut Microbiome Profiles in Colonizations with the Enteric Protozoa Blastocystis in Korean Populations. Microorganisms 2021, 10, 34. [Google Scholar] [CrossRef] [PubMed]
- Clavaud, C.; Jourdain, R.; Bar-Hen, A.; Tichit, M.; Bouchier, C.; Pouradier, F.; el Rawadi, C.; Guillot, J.; Ménard-Szczebara, F.; Breton, L.; et al. Dandruff is associated with disequilibrium in the proportion of the major bacterial and fungal populations colonizing the scalp. PLoS ONE 2013, 8, e58203. [Google Scholar] [CrossRef]
- Shuttleworth, R.; Behme, R.J.; McNabb, A.; Colby, W.D. Human isolates of Staphylococcus caprae: Association with bone and joint infections. J. Clin. Microbiol. 1997, 35, 2537–2541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dimitriu, P.A.; Iker, B.; Malik, K.; Leung, H.; Mohn, W.W.; Hillebrand, G.G. New Insights into the Intrinsic and Extrinsic Factors That Shape the Human Skin Microbiome. mBio 2019, 10, e00839-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mignard, S.; Flandrois, J.P. 16S rRNA sequencing in routine bacterial identification: A 30-month experiment. J. Microbiol. Methods 2006, 67, 574–581. [Google Scholar] [CrossRef] [PubMed]
- Winand, R.; Bogaerts, B.; Hoffman, S.; Lefevre, L.; Delvoye, M.; Braekel, J.V.; Fu, Q.; Roosens, N.H.; Keersmaecker, S.C.; Vanneste, K. Targeting the 16S rRNA Gene for Bacterial Identification in Complex Mixed Samples: Comparative Evaluation of Second (Illumina) and Third (Oxford Nanopore Technologies) Generation Sequencing Technologies. Int. J. Mol. Sci. 2019, 21, 298. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
| Mild Alopecia Areata (N = 26) | Severe Alopecia Areata (N = 7) * | |
|---|---|---|
| Male (%) | 5 (71.4) | 17 (65.4) |
| Mean age | 19.5 (10–43) | 33.9 (9–60) |
| Ranges of hair loss involvement (%) | 5–85% | 95–100% |
| Average of hair loss involvement (%) | 40.0 | 98.3 |
| Progressive hair loss or non-response to therapy, No. (%) | 7 (26.9) | 7 (100.0) |
| Size increment, No. (%) | 17 (65.4) | 4 (57.1) |
| Spreading tendency, No. (%) | 19 (73.1) | 3 (42.9) |
| Hair growth, No. (%) | 18 (69.2) | 2 (28.6) |
| Median duration (Day) | 151.5 | 746 |
| Presence of autoimmune disorders, No. (%) | 3 (11.5) | 3 (42.9) |
| HC | Mild AA | Severe AA | |
|---|---|---|---|
| Staphylococcus caprae/Staphylococcus species | 49.5% (115,707/233,865) a | 42.1% (176,043/418,857) | 10.5% (3739/35,653) a |
| Corynebacterium species/Total | 0.3% (2307/698,287) b | 0.6% (8860/443,367) a | 6.3% (23,134/116,344) a,b |
| Cutibacterium species/S. caprae | 0.97 (113,005/115,707) | 2.13 (375,500/176,043) | 16.01 (59,877/3739) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Won, E.J.; Jang, H.H.; Park, H.; Kim, S.J. A Potential Predictive Role of the Scalp Microbiome Profiling in Patients with Alopecia Areata: Staphylococcus caprae, Corynebacterium, and Cutibacterium Species. Microorganisms 2022, 10, 864. https://doi.org/10.3390/microorganisms10050864
Won EJ, Jang HH, Park H, Kim SJ. A Potential Predictive Role of the Scalp Microbiome Profiling in Patients with Alopecia Areata: Staphylococcus caprae, Corynebacterium, and Cutibacterium Species. Microorganisms. 2022; 10(5):864. https://doi.org/10.3390/microorganisms10050864
Chicago/Turabian StyleWon, Eun Jeong, Hyun Hee Jang, Hansoo Park, and Seong Jin Kim. 2022. "A Potential Predictive Role of the Scalp Microbiome Profiling in Patients with Alopecia Areata: Staphylococcus caprae, Corynebacterium, and Cutibacterium Species" Microorganisms 10, no. 5: 864. https://doi.org/10.3390/microorganisms10050864
APA StyleWon, E. J., Jang, H. H., Park, H., & Kim, S. J. (2022). A Potential Predictive Role of the Scalp Microbiome Profiling in Patients with Alopecia Areata: Staphylococcus caprae, Corynebacterium, and Cutibacterium Species. Microorganisms, 10(5), 864. https://doi.org/10.3390/microorganisms10050864