
Metagenomic Assessment of DNA Viral Diversity in Freshwater Sponges, Baikalospongia bacillifera





Abstract
1. Introduction
2. Materials and Methods
2.1. Sampling and Sample Processing
2.2. Library Preparation and Sequencing
2.3. Initial Shotgun Metagenomic Data on DNA Viruses in Marine Sponges and Water Samples
2.4. Primary Processing of Virome Reads
2.5. Assembly of Virome Reads, Identification, and Taxonomic Assignment of Viral Scaffolds
2.6. Statistical Analysis of Taxonomic Diversity
2.7. Functional Assignment of Viral Communities
2.8. Viral Hosts Prediction
2.9. Bacterial Defense Mechanisms against Viruses
3. Results
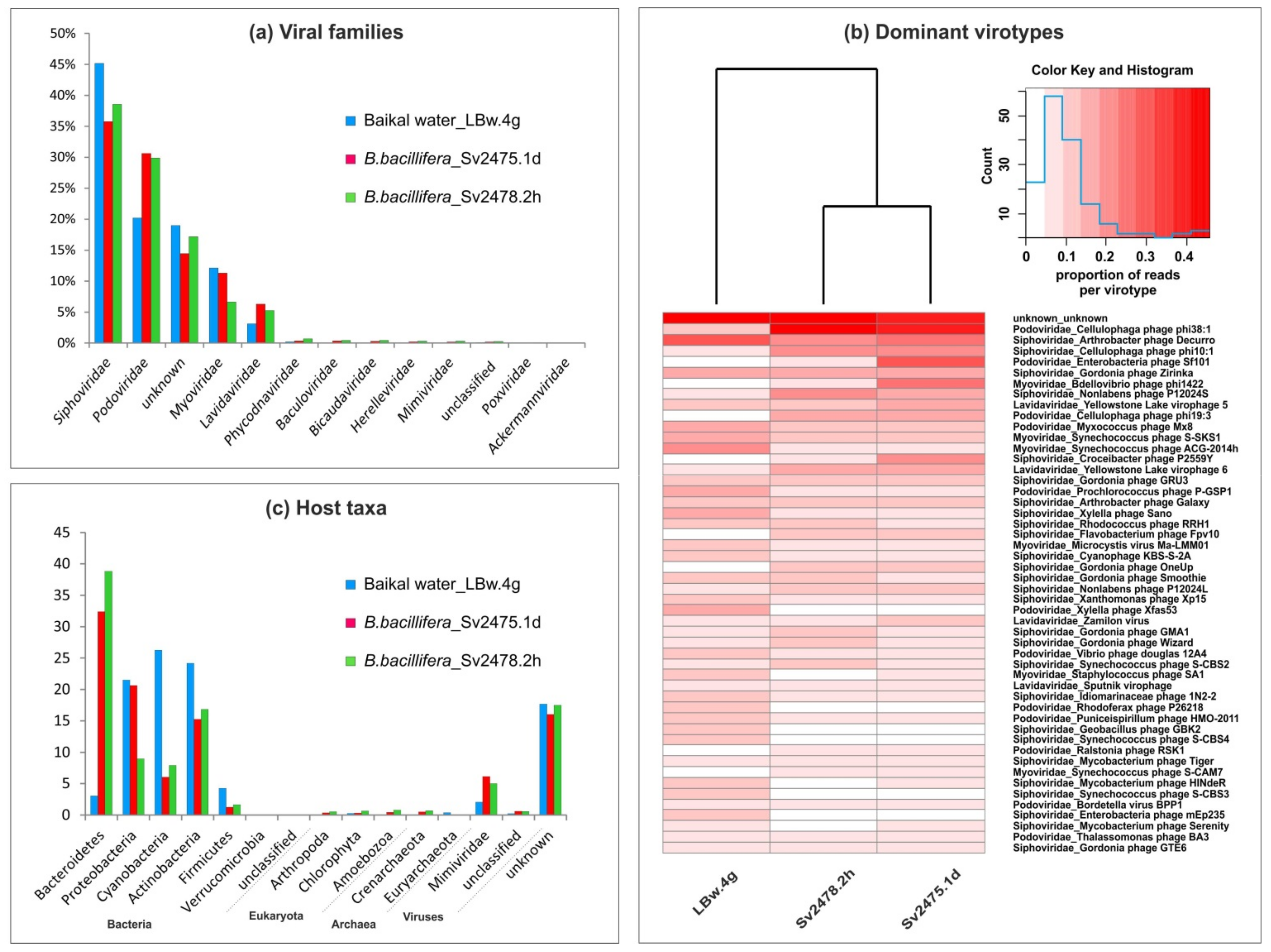
3.1. Taxonomic Affiliation of Viral Sequences in Baikal Samples
3.2. Virotypes Diversity
3.3. Putative Range of Viral Hosts
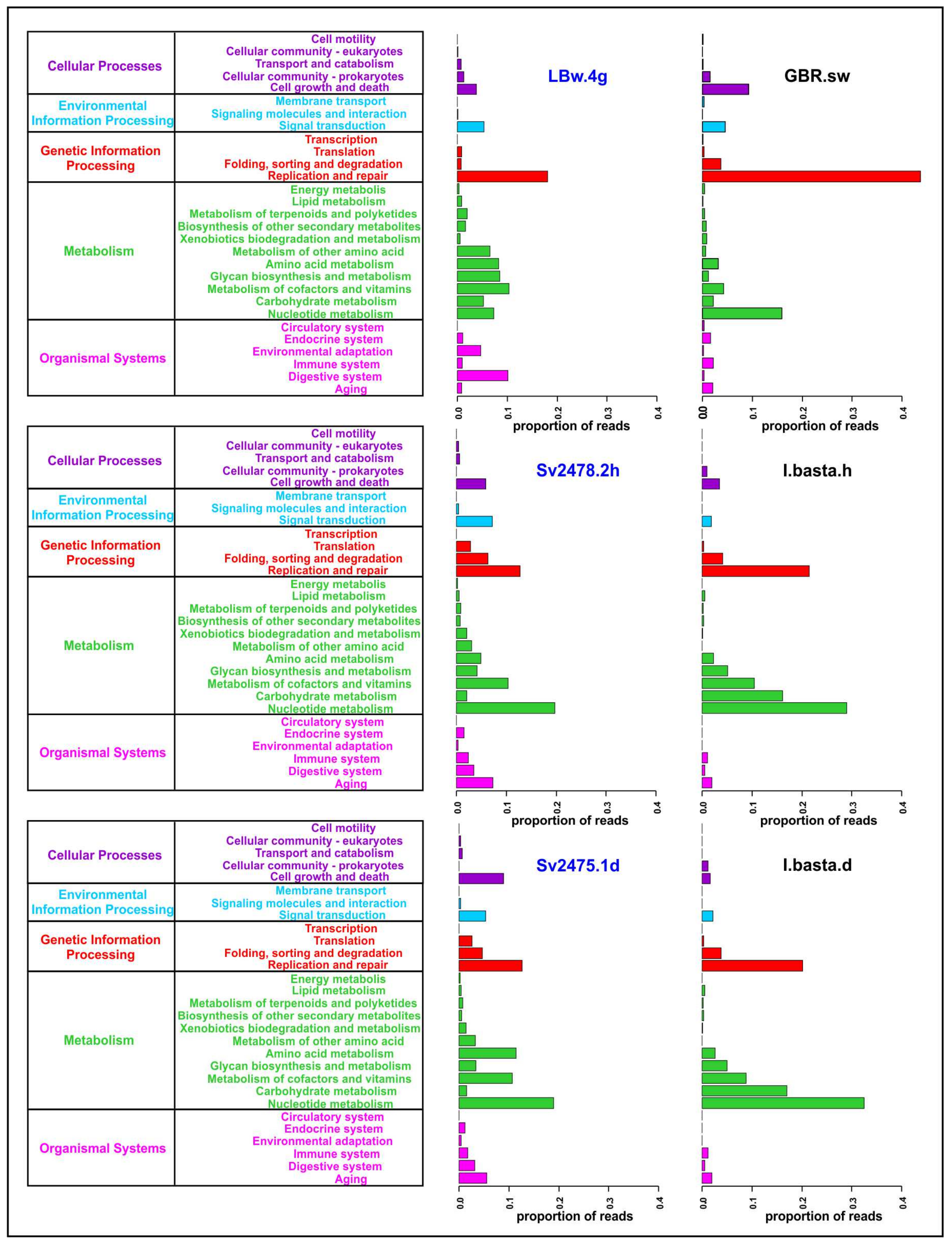
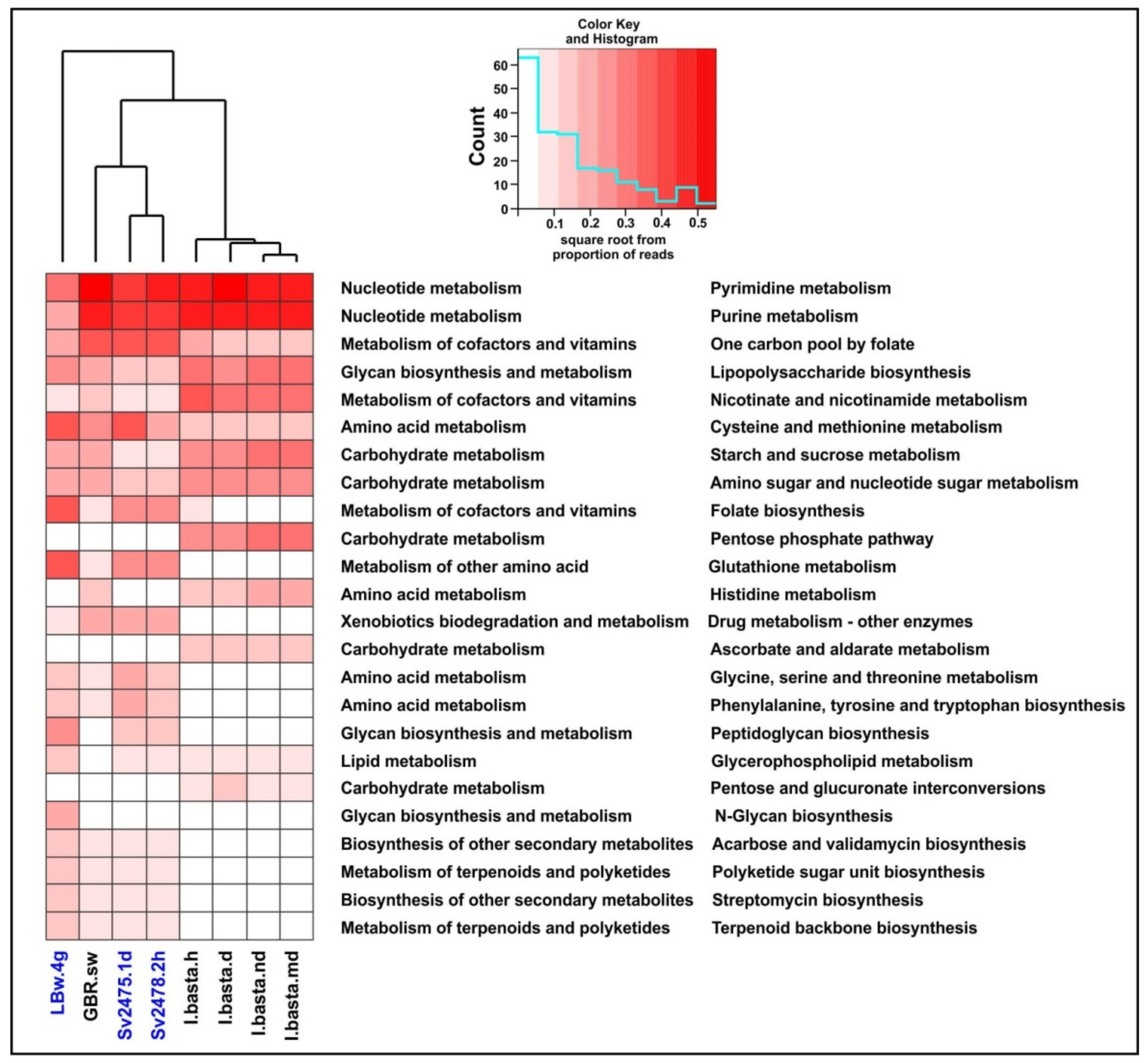
3.4. Functional Analysis of Baikal Viromes
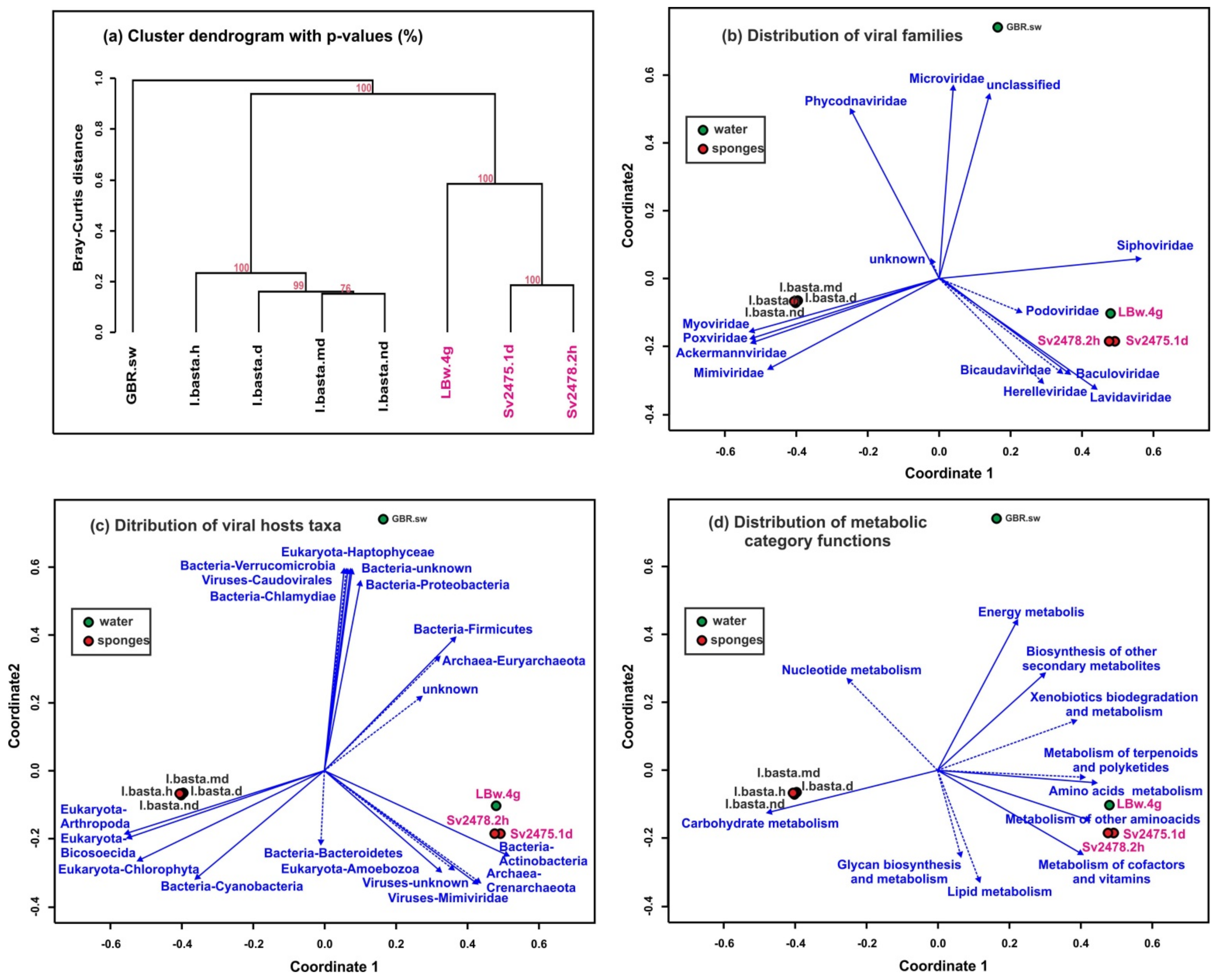
3.5. Comparative Analysis of Freshwater and Marine Viromes
4. Discussion
4.1. Analysis of the Reads Assembly of Marine and Freshwater Virome Samples
4.2. The Diversity of Viral Communities in the B. bacillifera
4.3. Comparative Analysis of Viromes of Diseased and Healthy B. bacillifera and the Surrounding Baikal Water
4.4. Putative Viral Hosts for Baikal Viruses
4.5. Comparative Analysis of Marine and Freshwater Viromes
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hooper, J.N.A.; Van Soest, R.W.M. Systema Porifera. A Guide to the Classification of Sponges. In Systema Porifera; Springer: Boston, MA, USA, 2002; pp. 1–7. [Google Scholar]
- Wulff, J. Ecological Interactions and the Distribution, Abundance, and Diversity of Sponges. Adv. Mar. Biol. 2012, 61, 273–344. [Google Scholar] [PubMed]
- Webster, N.S.; Taylor, M.W. Marine sponges and their microbial symbionts: Love and other relationships. Environ. Microbiol. 2012, 14, 335–346. [Google Scholar] [CrossRef]
- Loxdale, H.D.; Wilmer, P. Invertebrate Relationships: Patterns in Animal Evolution. J. Anim. Ecol. 1991, 60, 375–376. [Google Scholar] [CrossRef]
- Diaz, M.C.; Rützler, K. Sponges: An essential component of Caribbean coral reefs. Bull. Mar. Sci. 2001, 69, 535–546. [Google Scholar]
- Bell, J.J. The functional roles of marine sponges. Estuar. Coast. Shelf Sci. 2008, 79, 341–353. [Google Scholar] [CrossRef]
- Kumar, S.M.; Pal, K.A. A review of bioactive compounds from marine organisms with special mention on the potential of marine sponges in pharmacological applications. J. Mar. Biol. Assoc. India 2016, 58, 84. [Google Scholar] [CrossRef][Green Version]
- Steinert, G.; Stauffer, C.H.; Aas-Valleriani, N.; Borchert, E.; Bhushan, A.; Campbell, A.; De Mares, M.C.; Costa, M.; Gutleben, J.; Knobloch, S.; et al. BluePharmTrain: Biology and Biotechnology of Marine Sponges. In Grand Challenges in Biology and Biotechnology; Springer: Cham, Switzerland, 2018; pp. 505–553. [Google Scholar]
- Manconi, R.; Pronzato, R. Phylum Porifera. In Thorp and Covich’s Freshwater Invertebrates; Elsevier: Amsterdam, The Netherlands, 2019; pp. 43–92. [Google Scholar]
- Itskovich, V.B.; Kaluzhnaya, O.V.; Veynberg, E.; Erpenbeck, D. Endemic Lake Baikal sponges from deep water. 2: Taxonomy and Bathymetric Distribution. Zootaxa 2017, 4236, 335–342. [Google Scholar] [CrossRef]
- Bukshuk, N.A.; Maikova, O.O. A new species of baikal endemic sponges (Porifera, demospongiae, spongillida, lubomirskiidae). Zookeys 2020, 2020, 113–130. [Google Scholar] [CrossRef]
- Khanaev, I.V.; Kravtsova, L.S.; Maikova, O.O.; Bukshuk, N.A.; Sakirko, M.V.; Kulakova, N.V.; Butina, T.V.; Nebesnykh, I.A.; Belikov, S.I. Current state of the sponge fauna (Porifera: Lubomirskiidae) of Lake Baikal: Sponge disease and the problem of conservation of diversity. J. Great Lakes Res. 2018, 44, 77–85. [Google Scholar] [CrossRef]
- Dutilh, B.E.; Varsani, A.; Tong, Y.; Simmonds, P.; Sabanadzovic, S.; Rubino, L.; Roux, S.; Muñoz, A.R.; Lood, C.; Lefkowitz, E.J.; et al. Perspective on taxonomic classification of uncultivated viruses. Curr. Opin. Virol. 2021, 51, 207–215. [Google Scholar] [CrossRef]
- Pascelli, C.; Laffy, P.W.; Botté, E.; Kupresanin, M.; Rattei, T.; Lurgi, M.; Ravasi, T.; Webster, N.S. Viral ecogenomics across the Porifera. Microbiome 2020, 8, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, M.; Wemheuer, B.; Laffy, P.W.; Webster, N.S.; Thomas, T. Taxonomic, functional and expression analysis of viral communities associated with marine sponges. PeerJ 2021, 9, e10715. [Google Scholar] [CrossRef] [PubMed]
- Ambalavanan, L.; Iehata, S.; Fletcher, R.; Stevens, E.H.; Zainathan, S.C. A review of marine viruses in coral ecosystem. J. Mar. Sci. Eng. 2021, 9, 711. [Google Scholar] [CrossRef]
- Vacelet, J.; Gallissian, M.F. Virus-like particles in cells of the sponge Verongia cavernicola (demospongiae, dictyoceratida) and accompanying tissues changes. J. Invertebr. Pathol. 1978, 31, 246–254. [Google Scholar] [CrossRef]
- Johnson, P.T. Viral diseases of marine invertebrates. Helgoländer Meeresunters. 1984, 37, 65–98. [Google Scholar] [CrossRef]
- Claverie, J.M.; Grzela, R.; Lartigue, A.; Bernadac, A.; Nitsche, S.; Vacelet, J.; Ogata, H.; Abergel, C. Mimivirus and Mimiviridae: Giant viruses with an increasing number of potential hosts, including corals and sponges. J. Invertebr. Pathol. 2009, 101, 172–180. [Google Scholar] [CrossRef]
- Pascelli, C.; Laffy, P.W.; Kupresanin, M.; Ravasi, T.; Webster, N.S. Morphological characterization of virus-like particles in coral reef sponges. PeerJ 2018, 6, e5625. [Google Scholar] [CrossRef]
- Laffy, P.W.; Wood-Charlson, E.M.; Turaev, D.; Weynberg, K.D.; Botté, E.S.; Van Oppen, M.J.H.; Webster, N.S.; Rattei, T. HoloVir: A workflow for investigating the diversity and function of viruses in invertebrate holobionts. Front. Microbiol. 2016, 7, 822. [Google Scholar] [CrossRef]
- Laffy, P.W.; Wood-Charlson, E.M.; Turaev, D.; Jutz, S.; Pascelli, C.; Botté, E.S.; Bell, S.C.; Peirce, T.E.; Weynberg, K.D.; van Oppen, M.J.H.; et al. Reef invertebrate viromics: Diversity, host specificity and functional capacity. Environ. Microbiol. 2018, 20, 2125–2141. [Google Scholar] [CrossRef]
- Batista, D.; Costa, R.; Carvalho, A.P.; Batista, W.R.; Rua, C.P.J.; de Oliveira, L.; Leomil, L.; Fróes, A.M.; Thompson, F.L.; Coutinho, R.; et al. Environmental conditions affect activity and associated microorganisms of marine sponges. Mar. Environ. Res. 2018, 142, 59–68. [Google Scholar] [CrossRef]
- Butina, T.V.; Bukin, Y.S.; Khanaev, I.V.; Kravtsova, L.S.; Maikova, O.O.; Tupikin, A.E.; Kabilov, M.R.; Belikov, S.I. Metagenomic analysis of viral communities in diseased Baikal sponge Lubomirskia baikalensis. Limnol. Freshw. Biol. 2019, 155–162. [Google Scholar] [CrossRef]
- Butina, T.V.; Khanaev, I.V.; Kravtsova, L.S.; Maikova, O.O.; Bukin, Y.S. Metavirome datasets from two endemic Baikal sponges Baikalospongia bacillifera. Data Br. 2020, 29, 105260. [Google Scholar] [CrossRef] [PubMed]
- Waldron, F.M.; Stone, G.N.; Obbard, D.J. Metagenomic sequencing suggests a diversity of RNA interference-like responses to viruses across multicellular eukaryotes. PLoS Genet. 2018, 14, e1007533. [Google Scholar] [CrossRef] [PubMed]
- Urayama, S.I.; Takaki, Y.; Hagiwara, D.; Nunoura, T. DsRNA-seq reveals novel RNA virus and virus-like putative complete genome sequences from hymeniacidon sp. Sponge. Microbes Environ. 2020, 35, ME19132. [Google Scholar] [CrossRef] [PubMed]
- Harrington, C.; Del Casale, A.; Kennedy, J.; Neve, H.; Picton, B.E.; Mooij, M.J.; O’Gara, F.; Kulakov, L.A.; Larkin, M.J.; Dobson, A.D.W. Evidence of bacteriophage-mediated horizontal transfer of bacterial 16S rRNA genes in the viral metagenome of the marine sponge Hymeniacidon perlevis. Microbiology 2012, 158, 2789–2795. [Google Scholar] [CrossRef]
- Kravtsova, L.S.; Izhboldina, L.A.; Khanaev, I.V.; Pomazkina, G.V.; Domysheva, V.M.; Kravchenko, O.S.; Grachev, M.A. Disturbances of the vertical zoning of green algae in the coastal part of the Listvennichnyi gulf of Lake Baikal. Dokl. Biol. Sci. 2012, 447, 350–352. [Google Scholar] [CrossRef]
- Kravtsova, L.S.; Izhboldina, L.A.; Khanaev, I.V.; Pomazkina, G.V.; Rodionova, E.V.; Domysheva, V.M.; Sakirko, M.V.; Tomberg, I.V.; Kostornova, T.Y.; Kravchenko, O.S.; et al. Nearshore benthic blooms of filamentous green algae in Lake Baikal. J. Great Lakes Res. 2014, 40, 441–448. [Google Scholar] [CrossRef]
- Timoshkin, O.A.; Samsonov, D.P.; Yamamuro, M.; Moore, M.V.; Belykh, O.I.; Malnik, V.V.; Sakirko, M.V.; Shirokaya, A.A.; Bondarenko, N.A.; Domysheva, V.M.; et al. Rapid ecological change in the coastal zone of Lake Baikal (East Siberia): Is the site of the world’s greatest freshwater biodiversity in danger? J. Great Lakes Res. 2016, 42, 487–497. [Google Scholar] [CrossRef]
- Bondarenko, N.A.; Logacheva, N.F. Structural changes in phytoplankton of the littoral zone of Lake Baikal. Hydrobiol. J. 2017, 53, 16–24. [Google Scholar] [CrossRef]
- Webster, N.S. Sponge disease: A global threat? Environ. Microbiol. 2007, 9, 1363–1375. [Google Scholar] [CrossRef]
- Luter, H.M.; Webster, N.S. Sponge Disease and Climate Change. In Climate Change, Ocean Acidification and Sponges; Springer: Cham, Switzerland, 2017; pp. 411–428. [Google Scholar]
- Pita, L.; Rix, L.; Slaby, B.M.; Franke, A.; Hentschel, U. The sponge holobiont in a changing ocean: From microbes to ecosystems. Microbiome 2018, 6, 46. [Google Scholar] [CrossRef] [PubMed]
- Luter, H.M.; Bannister, R.J.; Whalan, S.; Kutti, T.; Pineda, M.-C.; Webster, N.S. Microbiome analysis of a disease affecting the deep-sea sponge Geodia barretti. FEMS Microbiol. Ecol. 2017, 93, 74. [Google Scholar] [CrossRef] [PubMed]
- Denikina, N.N.; Dzyuba, E.V.; Bel’kova, N.L.; Khanaev, I.V.; Feranchuk, S.I.; Makarov, M.M.; Granin, N.G.; Belikov, S.I. The first case of disease of the sponge Lubomirskia baicalensis: Investigation of its microbiome. Biol. Bull. 2016, 43, 263–270. [Google Scholar] [CrossRef]
- Kulakova, N.V.; Sakirko, M.V.; Adelshin, R.V.; Khanaev, I.V.; Nebesnykh, I.A.; Pérez, T. Brown Rot Syndrome and Changes in the Bacterial Community of the Baikal Sponge Lubomirskia baicalensis. Microb. Ecol. 2018, 75, 1024–1034. [Google Scholar] [CrossRef] [PubMed]
- Belikov, S.; Belkova, N.; Butina, T.; Chernogor, L.; Van Kley, A.M.; Nalian, A.; Rorex, C.; Khanaev, I.; Maikova, O.; Feranchuk, S. Diversity and shifts of the bacterial community associated with Baikal sponge mass mortalities. PLoS ONE 2019, 14, e0213926. [Google Scholar] [CrossRef]
- Itskovich, V.; Kaluzhnaya, O.; Glyzina, O.; Prathiviraj, R.; Kiran, G.S.; Selvin, J. Microbiome Changes of Endemic Lake Baikal Sponges during Bleaching Syndrome Development. Diversity 2021, 13, 653. [Google Scholar] [CrossRef]
- Choudhury, J.D.; Pramanik, A.; Webster, N.S.; Llewellyn, L.E.; Gachhui, R.; Mukherjee, J. The Pathogen of the Great Barrier Reef Sponge Rhopaloeides odorabile Is a New Strain of Pseudoalteromonas agarivorans Containing Abundant and Diverse Virulence-Related Genes. Mar. Biotechnol. 2015, 17, 463–478. [Google Scholar] [CrossRef]
- Fan, L.; Reynolds, D.; Liu, M.; Stark, M.; Kjelleberg, S.; Webster, N.S.; Thomas, T. Functional equivalence and evolutionary convergence in complex communities of microbial sponge symbionts. Proc. Natl. Acad. Sci. USA 2012, 109, E1878–E1887. [Google Scholar] [CrossRef]
- Butina, T.V.; Petrushin, I.S.; Khanaev, I.V.; Bukin, Y.S. Virome Analysis of Near-Bottom Coastal Water of Lake Baikal. Microbiol. Resour. Announc. 2020, 9, 1–3. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Butina, T.V.; Bukin, Y.S.; Petrushin, I.S.; Tupikin, A.E.; Kabilov, M.R.; Belikov, S.I. Extended evaluation of viral diversity in lake baikal through metagenomics. Microorganisms 2021, 9, 760. [Google Scholar] [CrossRef] [PubMed]
- Nurk, S.; Meleshko, D.; Korobeynikov, A.; Pevzner, P.A. MetaSPAdes: A new versatile metagenomic assembler. Genome Res. 2017, 27, 824–834. [Google Scholar] [CrossRef] [PubMed]
- Roux, S.; Enault, F.; Hurwitz, B.L.; Sullivan, M.B. VirSorter: Mining viral signal from microbial genomic data. PeerJ 2015, 3, e985. [Google Scholar] [CrossRef] [PubMed]
- Pruitt, K.D.; Tatusova, T.; Maglott, D.R. NCBI Reference Sequence (RefSeq): A curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 2005, 33, D501–D504. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- O’Hara, R.B. Species richness estimators: How many species can dance on the head of a pin? J. Anim. Ecol. 2005, 74, 375–386. [Google Scholar] [CrossRef]
- Colwell, R.K.; Coddington, J.A. Estimating terrestrial biodiversity through extrapolation. Biodivers. Meas. Estim. 1995, 345, 101–118. [Google Scholar]
- Hill, M.O. Diversity and Evenness: A Unifying Notation and Its Consequences. Ecology 1973, 54, 427–432. [Google Scholar] [CrossRef]
- Suzuki, R.; Shimodaira, H. Pvclust: An R package for assessing the uncertainty in hierarchical clustering. Bioinformatics 2006, 22, 1540–1542. [Google Scholar] [CrossRef]
- Oksanen, J. Vegan: Ecological Diversity. 2018. Available online: https://cran.r-project.org/web/packages/vegan/vignettes/diversity-vegan.pdf (accessed on 20 December 2021).
- Warnes, G.R.; Bolker, B.; Bonebakker, L.; Gentleman, R.; Liaw, W.H.A.; Lumley, T.; Maechler, M.; Magnusson, A.; Moeller, S.; Schwartz, M.; et al. Package “gplots”: Various R Programming Tools for Plotting Data; R Package Version 2.17.0; ScienceOpen: Berlin, Germany, 2015; pp. 1–68. [Google Scholar]
- Bairoch, A.; Apweiler, R.; Wu, C.H.; Barker, W.C.; Boeckmann, B.; Ferro, S.; Gasteiger, E.; Huang, H.; Lopez, R.; Magrane, M.; et al. The Universal Protein Resource (UniProt). Nucleic Acids Res. 2005, 33, D154–D159. [Google Scholar] [CrossRef] [PubMed]
- Mistry, J.; Chuguransky, S.; Williams, L.; Qureshi, M.; Salazar, G.A.; Sonnhammer, E.L.L.; Tosatto, S.C.E.; Paladin, L.; Raj, S.; Richardson, L.J.; et al. Pfam: The protein families database in 2021. Nucleic Acids Res. 2021, 49, D412–D419. [Google Scholar] [CrossRef] [PubMed]
- Madeira, F.; Park, Y.M.; Lee, J.; Buso, N.; Gur, T.; Madhusoodanan, N.; Basutkar, P.; Tivey, A.R.N.; Potter, S.C.; Finn, R.D.; et al. The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res. 2019, 47, W636–W641. [Google Scholar] [CrossRef]
- Aramaki, T.; Blanc-Mathieu, R.; Endo, H.; Ohkubo, K.; Kanehisa, M.; Goto, S.; Ogata, H. KofamKOALA: KEGG Ortholog assignment based on profile HMM and adaptive score threshold. Bioinformatics 2020, 36, 2251–2252. [Google Scholar] [CrossRef] [PubMed]
- Mao, X.; Cai, T.; Olyarchuk, J.G.; Wei, L. Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics 2005, 21, 3787–3793. [Google Scholar] [CrossRef] [PubMed]
- Tenenbaum, D. KEGGREST: Client-Side REST Access to KEGG. R Package Version 1.28.0. 2020. Available online: http://bioconductor.org/packages/release/bioc/html/KEGGREST.html (accessed on 20 December 2021).
- Mihara, T.; Nishimura, Y.; Shimizu, Y.; Nishiyama, H.; Yoshikawa, G.; Uehara, H.; Hingamp, P.; Goto, S.; Ogata, H. Linking virus genomes with host taxonomy. Viruses 2016, 8, 66. [Google Scholar] [CrossRef]
- Petrushin, I.; Belikov, S.; Chernogor, L. Cooperative interaction of janthinobacterium sp. Slb01 and flavobacterium sp. slb02 in the diseased sponge lubomirskia baicalensis. Int. J. Mol. Sci. 2020, 21, 8128. [Google Scholar] [CrossRef]
- Payne, L.J.; Todeschini, T.C.; Wu, Y.; Perry, B.J.; Ronson, C.W.; Fineran, P.C.; Nobrega, F.L.; Jackson, S.A. Identification and classification of antiviral defence systems in bacteria and archaea with PADLOC reveals new system types. Nucleic Acids Res. 2021, 49, 10868–10878. [Google Scholar] [CrossRef]
- Couvin, D.; Bernheim, A.; Toffano-Nioche, C.; Touchon, M.; Michalik, J.; Néron, B.; Rocha, E.P.C.; Vergnaud, G.; Gautheret, D.; Pourcel, C. CRISPRCasFinder, an update of CRISRFinder, includes a portable version, enhanced performance and integrates search for Cas proteins. Nucleic Acids Res. 2018, 46, W246–W251. [Google Scholar] [CrossRef]
- Edwards, R.A.; McNair, K.; Faust, K.; Raes, J.; Dutilh, B.E. Computational approaches to predict bacteriophage-host relationships. FEMS Microbiol. Rev. 2016, 40, 258–272. [Google Scholar] [CrossRef]
- La Scola, B.; Desnues, C.; Pagnier, I.; Robert, C.; Barrassi, L.; Fournous, G.; Merchat, M.; Suzan-Monti, M.; Forterre, P.; Koonin, E.; et al. The virophage as a unique parasite of the giant mimivirus. Nature 2008, 455, 100–104. [Google Scholar] [CrossRef] [PubMed]
- Doron, S.; Melamed, S.; Ofir, G.; Leavitt, A.; Lopatina, A.; Keren, M.; Amitai, G.; Sorek, R. Systematic discovery of antiphage defense systems in the microbial pangenome. Science 2018, 359, eaar4120. [Google Scholar] [CrossRef] [PubMed]
- Compeau, P.E.C.; Pevzner, P.A.; Tesler, G. How to apply de Bruijn graphs to genome assembly. Nat. Biotechnol. 2011, 29, 987–991. [Google Scholar] [CrossRef]
- Breitbart, M.; Salamon, P.; Andresen, B.; Mahaffy, J.M.; Segall, A.M.; Mead, D.; Azam, F.; Rohwer, F. Genomic analysis of uncultured marine viral communities. Proc. Natl. Acad. Sci. USA 2002, 99, 14250–14255. [Google Scholar] [CrossRef] [PubMed]
- De Cárcer, D.A.; López-Bueno, A.; Alonso-Lobo, J.M.; Quesada, A.; Alcamí, A. Metagenomic analysis of lacustrine viral diversity along a latitudinal transect of the Antarctic Peninsula. FEMS Microbiol. Ecol. 2016, 92, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Butina, T.V.; Bukin, Y.S.; Krasnopeev, A.S.; Belykh, O.I.; Tupikin, A.E.; Kabilov, M.R.; Sakirko, V.; Belikov, S.I. Estimate of the diversity of viral and bacterial assemblage in the coastal water of Lake Baikal. FEMS Microbiol. Lett. 2019, 366, fnz094. [Google Scholar] [CrossRef] [PubMed]
- Potapov, S.A.; Tikhonova, I.V.; Krasnopeev, A.Y.; Kabilov, M.R.; Tupikin, A.E.; Chebunina, N.S.; Zhuchenko, N.A.; Belykh, O.I. Metagenomic analysis of virioplankton from the pelagic zone of Lake Baikal. Viruses 2019, 11, 991. [Google Scholar] [CrossRef]
- Park, E.-J.; Kim, K.-H.; Abell, G.C.J.; Kim, M.-S.; Roh, S.W.; Bae, J.-W. Metagenomic Analysis of the Viral Communities in Fermented Foods. Appl. Environ. Microbiol. 2011, 77, 1284–1291. [Google Scholar] [CrossRef]
- Santos-Medellin, C.; Zinke, L.A.; ter Horst, A.M.; Gelardi, D.L.; Parikh, S.J.; Emerson, J.B. Viromes outperform total metagenomes in revealing the spatiotemporal patterns of agricultural soil viral communities. ISME J. 2021, 15, 1956–1970. [Google Scholar] [CrossRef]
- Hentschel, U.; Piel, J.; Degnan, S.M.; Taylor, M.W. Genomic insights into the marine sponge microbiome. Nat. Rev. Microbiol. 2012, 10, 641–654. [Google Scholar] [CrossRef]
- Albuquerque, L.; da Costa, M.S. The Family Idiomarinaceae. In The Prokaryotes; Rosenberg, E., DeLong, E.F., Lory, S., Stackebrandt, E., Thompson, F., Eds.; Springer: Berlin/Heidelberg, Germany, 2014; pp. 361–385. [Google Scholar]
- Crone, S.; Vives-Flórez, M.; Kvich, L.; Saunders, A.M.; Malone, M.; Nicolaisen, M.H.; Martínez-García, E.; Rojas-Acosta, C.; Catalina Gomez-Puerto, M.; Calum, H.; et al. The environmental occurrence of Pseudomonas aeruginosa. Apmis 2020, 128, 220–231. [Google Scholar] [CrossRef] [PubMed]
- Evseev, P.; Sykilinda, N.; Gorshkova, A.; Kurochkina, L.; Ziganshin, R.; Drucker, V.; Miroshnikov, K. Pseudomonas Phage PaBG-A Jumbo Member of an Old Parasite Family. Viruses 2020, 12, 721. [Google Scholar] [CrossRef]
- Evseev, P.V.; Gorshkova, A.S.; Sykilinda, N.N.; Drucker, V.V.; Miroshnikov, K.A. Pseudomonas bacteriophage AN14 a Baikal-borne representative of Yuavirus. Limnol. Freshw. Biol. 2020, 5, 1055–1066. [Google Scholar] [CrossRef]
- Zhou, J.; Sun, D.; Childers, A.; McDermott, T.R.; Wang, Y.; Liles, M.R. Three Novel Virophage Genomes Discovered from Yellowstone Lake Metagenomes. J. Virol. 2015, 89, 1278–1285. [Google Scholar] [CrossRef]
- Afonso, C.L.; Tulman, E.R.; Lu, Z.; Oma, E.; Kutish, G.F.; Rock, D.L. The Genome of Melanoplus sanguinipes Entomopoxvirus. J. Virol. 1999, 73, 533–552. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.M.; Wang, Y.; Tian, R.M.; Lee, O.O.; Wong, Y.H.; Batang, Z.B.; Al-Suwailem, A.; Lafi, F.F.; Bajic, V.B.; Qian, P.Y. Pyrosequencing revealed shifts of prokaryotic communities between healthy and disease-like tissues of the Red Sea sponge Crella cyathophora. PeerJ 2015, 3, e890. [Google Scholar] [CrossRef]
- Blanquer, A.; Uriz, M.J.; Cebrian, E.; Galand, P.E. Snapshot of a bacterial microbiome shift during the early symptoms of a massive sponge die-offin the western Mediterranean. Front. Microbiol. 2016, 7, 1–10. [Google Scholar] [CrossRef]
- Laffy, P.W.; Botté, E.S.; Wood-Charlson, E.M.; Weynberg, K.D.; Rattei, T.; Webster, N.S. Thermal stress modifies the marine sponge virome. Environ. Microbiol. Rep. 2019, 11, 690–698. [Google Scholar] [CrossRef] [PubMed]
- Butina, T.V.; Potapov, S.A.; Belykh, O.I.; Belikov, S.I. Genetic diversity of cyanophages of the myoviridae family as a constituent of the associated community of the Baikal sponge Lubomirskia baicalensis. Russ. J. Genet. 2015, 51, 313–317. [Google Scholar] [CrossRef]
- Gladkikh, A.S.; Kalyuzhnaya, O.V.; Belykh, O.I.; Ahn, T.S.; Parfenova, V.V. Analysis of bacterial communities of two Lake Baikal endemic sponge species. Microbiology 2014, 83, 787–797. [Google Scholar] [CrossRef]
- Seo, E.Y.; Jung, D.; Belykh, O.I.; Bukshuk, N.A.; Parfenova, V.V.; Joung, Y.; Kim, I.C.; Yim, J.H.; Ahn, T.S. Comparison of bacterial diversity and species composition in three endemic Baikalian sponges. Ann. Limnol. 2016, 52, 27–32. [Google Scholar] [CrossRef]
- Lipko, I.; Krasnopeev, A.; Tikhonova, I.; Timoshkin, O.; Kabilov, M.; Belykh, O. Genetic diversity of Actinobacteria inhabiting water and sponges of Lake Baikal. Limnol. Freshw. Biol. 2020, 4, 998–999. [Google Scholar] [CrossRef]
- Bukin, Y.S.; Galachyants, Y.P.; Morozov, I.V.; Bukin, S.V.; Zakharenko, A.S.; Zemskaya, T.I. The effect of 16s rRNA region choice on bacterial community metabarcoding results. Sci. Data 2019, 6, 190007. [Google Scholar] [CrossRef] [PubMed]
- Coutinho, F.H.; Cabello-Yeves, P.J.; Gonzalez-Serrano, R.; Rosselli, R.; López-Pérez, M.; Zemskaya, T.I.; Zakharenko, A.S.; Ivanov, V.G.; Rodriguez-Valera, F. New viral biogeochemical roles revealed through metagenomic analysis of Lake Baikal. Microbiome 2020, 8, 163. [Google Scholar] [CrossRef]
- Newton, R.J.; Jones, S.E.; Eiler, A.; McMahon, K.D.; Bertilsson, S. A Guide to the Natural History of Freshwater Lake Bacteria. Microbiol. Mol. Biol. Rev. 2011, 75, 14–49. [Google Scholar] [CrossRef] [PubMed]
- Kaluzhnaya, O.V.; Lipko, I.A.; Itskovich, V.B. PCR-screening of bacterial strains isolated from the microbiome of the Lubomirskia baicalensis sponge for the presence of secondary metabolite synthesis genes. Limnol. Freshw. Biol. 2021, 2021, 1137–1142. [Google Scholar] [CrossRef]
- Zhou, K.; Qian, P.Y.; Zhang, T.; Xu, Y.; Zhang, R. Unique phage–bacterium interplay in sponge holobionts from the southern Okinawa Trough hydrothermal vent. Environ. Microbiol. Rep. 2021, 13, 675–683. [Google Scholar] [CrossRef]
- Jahn, M.T.; Lachnit, T.; Markert, S.M.; Stigloher, C.; Pita, L.; Ribes, M.; Dutilh, B.E.; Hentschel, U. Lifestyle of sponge symbiont phages by host prediction and correlative microscopy. ISME J. 2021, 15, 2001–2011. [Google Scholar] [CrossRef]
- Rezvoi, P.D. The Fauna of the USSR. Freshwater Sponges; AS USSR: Moscow, Russia, 1936. [Google Scholar]
- Kamaltynov, R.M.; Chernykh, V.I.; Slugina, Z.V.; Karabanov, E.B. The consortium of the sponge Lubomirskia baicalensis in Lake Baikal, East Siberia. Hydrobiologia 1993, 271, 179–189. [Google Scholar] [CrossRef]
- Gaikwad, S.; Shouche, Y.S.; Gade, W.N. Microbial community structure of two freshwater sponges using Illumina MiSeq sequencing revealed high microbial diversity. Amb Express 2016, 6, 40. [Google Scholar] [CrossRef]
- Laport, M.S.; Pinheiro, U.; Rachid, C.T.C.D.C. Freshwater sponge Tubella variabilis presents richer microbiota than marine sponge species. Front. Microbiol. 2019, 10, 2799. [Google Scholar] [CrossRef] [PubMed]
- Kozhova, O.M.; Izmesteva, L.R. Lake Baikal: Evolution and Biodiversity; Backhuys Publisher: Leiden, The Netherlands, 1998. [Google Scholar]
- Williams, T.; Bergoin, M.; van Oers, M.M. Diversity of large DNA viruses of invertebrates. J. Invertebr. Pathol. 2017, 147, 4–22. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dataset Name | Sample Description | Geographic Location | Latitude and Longitude | Data | BioProject | Experiments | Reference 1 |
|---|---|---|---|---|---|---|---|
| GBR.sw | Seawater control | Australia: GBR, Davies Reef | 18.83 S 147.63 E | 2014-10 | PRJNA388297 | SRX2883300, SRX2883301, SRX2883298 | - - - |
| I.basta.h | Ianthella basta, disease-free sponge | Australia: GBR, Davies Reef | 18.83 S 147.63 E | 2014-10 | PRJNA388007 | SRX2864027, SRX2864026, SRX2864019 | - [22] [22] |
| I.basta.nd | Ianthella basta, non-diseased region of diseased sponge | Australia: GBR, Davies Reef | 18.83 S 147.63 E | 2014-10 | PRJNA388007 | SRX2864023, SRX2864022, SRX2864016 | [22] [22] [22] |
| I.basta.d | Ianthella basta, disease lesion of diseased sponge | Australia: GBR, Davies Reef | 18.83 S 147.63 E | 2014-10 | PRJNA388007 | SRX2864021, SRX2864020, SRX2864018 | - - - |
| I.basta.md | Ianthella basta, lesion interface of diseased sponge | Australia: GBR, Davies Reef | 18.83 S 147.63 E | 2014-10 | PRJNA388007 | SRX2864025, SRX2864024, SRX2864017 | - - - |
| Sv2475.1d | Baikalospongia bacillifera, diseased sponge | Russia: Lake Baikal | 51.90 N 105.10 E | 2018-06 | PRJNA577390 | SRX6994059 | This study |
| Sv2478.2h | Baikalospongia bacillifera, disease-free sponge | Russia: Lake Baikal | 51.90 N 105.10 E | 2018-06 | PRJNA577390 | SRX6994055 | This study |
| LBw.4g | Lake Baikal water control | Russia: Lake Baikal | 51.90 N 105.10 E | 2018-06 | PRJNA577390 | SRX9228319 | This study |
| Samples | Reads_total | Viral Reads in Viral Scaffolds | Viral Scaffolds | Viral Scaffolds with Taxonomic Assignment | Virotypes | Chao1 (Scaffolds/Virotypes) | ACE (Scaffolds/Virotypes) | Shannon (Scaffolds/Virotypes) | Simpson (Scaffolds/Virotypes) | Reference 1 |
|---|---|---|---|---|---|---|---|---|---|---|
| Sv2475.1d | 4,348,746 | 637,148 (14.7%) | 404 | 318 (78.7%) | 168 | 408/173 | 407/173 | 4.5/3.2 | 0.97/0.91 | This study |
| Sv2478.2h | 3,574,388 | 681,061 (19.1%) | 417 | 325 (77.9%) | 171 | 417/172 | 418/173 | 4.8/3.2 | 0.98/0.87 | This study |
| LBw.4g | 9,477,618 | 805,244 (8.5%) | 428 | 338 (79.0%) | 183 | 429/186 | 433/187 | 5.1/4.1 | 0.99/0.97 | This study |
| I.basta.d | 15,774,944 | 788,317 (5.0%) | 208 | 161 (77.4%) | 98 | 211/99 | 213/99 | 4.3/3.2 | 0.98/0.93 | - |
| I.basta.h | 17,123,842 | 1,307,528 (7.6%) | 225 | 175 (77.8%) | 109 | 228/110 | 231/111 | 4.1/3.1 | 0.97/0.92 | [22] 2 |
| I.basta.md | 15,050,992 | 711,400 (4.7%) | 190 | 147 (77.4%) | 89 | 190/89 | 192/91 | 4.3/3.2 | 0.98/0.93 | - |
| I.basta.nd | 15,377,078 | 770,232 (5.0%) | 197 | 156 (79.2%) | 99 | 197/100 | 198/101 | 4.3/3.3 | 0.98/0.94 | [22] |
| GBR.sw | 9,359,144 | 1,107,475 (11.8%) | 384 | 297 (77.3%) | 178 | 405/182 | 407/185 | 4.7/3.9 | 0.98/0.96 | - |
| Over-Represented Viral Taxa or Functions | Baikal Water vs. Sponges | Healthy vs. Diseased | ||
|---|---|---|---|---|
| LBw.4 g | B. bacillifera | 2478.2 h | 2475.1 d | |
| Families | Siphoviridae | Podoviridae, Lavidaviridae, Mimiviridae, Baculoviridae, Bicaudaviridae, Herelleviridae | Siphoviridae, Phycodnaviridae, Mimiviridae, Baculoviridae, Bicaudaviridae, Herelleviridae | Myoviridae |
| Virotypes | Arthrobacter phage Decurro, Synechococcus phages S-SKS1 and ACG-2014h, Prochlorococcus phage P-GSP1, Xylella phages Sano and Xfas53 | Cellulophaga phages, (phi38:1, phi10:1, and phi19:3), Yellowstone lake virophage 6 and others | Nonlabens phage P12024S, Gordonia phages (GMA1, Wizard, etc.), Synechococcus phage S-CBS2 | Enterobacteria phage Sf101, Bdellovibrio phage phi1422, Croceibacter phage P2559Y |
| Putative hosts | Cyanobacteria, Actinobacteria, Firmicutes, Euryarchaeota | Bacteroidetes, Crenarchaeota, Amoebozoa, Arthropoda, Mimiviridae | Chlorophyta, Amoebozoa, | Proteobacteria |
| Functional categories (except “Metabolism”) | ‘Replication and repair’, ‘Environmental adaptation’, ‘Digestive system’ | ‘Folding, sorting and degradation’, ‘Translation’, ‘Cell growth and death’, ‘Aging’ | ‘Signal transduction’ | ‘Cell growth and death’ |
| Metabolic functions | ‘Metabolism of terpenoids and polyketides’, ‘Glycan biosynthesis and metabolism’, ‘Carbohydrate metabolism’ | ‘Nucleotide metabolism’, ‘Xenobiotics biodegradation and metabolism’ | Almost all (except ‘Amino acid metabolism’), ‘Folate biosynthesis’ | ‘Amino acid metabolism’, ‘Riboflavin metabolism’ |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Butina, T.V.; Petrushin, I.S.; Khanaev, I.V.; Bukin, Y.S. Metagenomic Assessment of DNA Viral Diversity in Freshwater Sponges, Baikalospongia bacillifera. Microorganisms 2022, 10, 480. https://doi.org/10.3390/microorganisms10020480
Butina TV, Petrushin IS, Khanaev IV, Bukin YS. Metagenomic Assessment of DNA Viral Diversity in Freshwater Sponges, Baikalospongia bacillifera. Microorganisms. 2022; 10(2):480. https://doi.org/10.3390/microorganisms10020480
Chicago/Turabian StyleButina, Tatyana V., Ivan S. Petrushin, Igor V. Khanaev, and Yurij S. Bukin. 2022. "Metagenomic Assessment of DNA Viral Diversity in Freshwater Sponges, Baikalospongia bacillifera" Microorganisms 10, no. 2: 480. https://doi.org/10.3390/microorganisms10020480
APA StyleButina, T. V., Petrushin, I. S., Khanaev, I. V., & Bukin, Y. S. (2022). Metagenomic Assessment of DNA Viral Diversity in Freshwater Sponges, Baikalospongia bacillifera. Microorganisms, 10(2), 480. https://doi.org/10.3390/microorganisms10020480