
Genome Characterization and Probiotic Potential of Corynebacterium amycolatum Human Vaginal Isolates



Abstract
1. Introduction
2. Materials and Methods
2.1. DNA Preparation, Genome Sequencing and Assembly
2.2. Genome Annotation
2.3. Phylogenetic Analysis
2.4. Nucleotide Sequence Accession Numbers
3. Results
3.1. General Genome Features
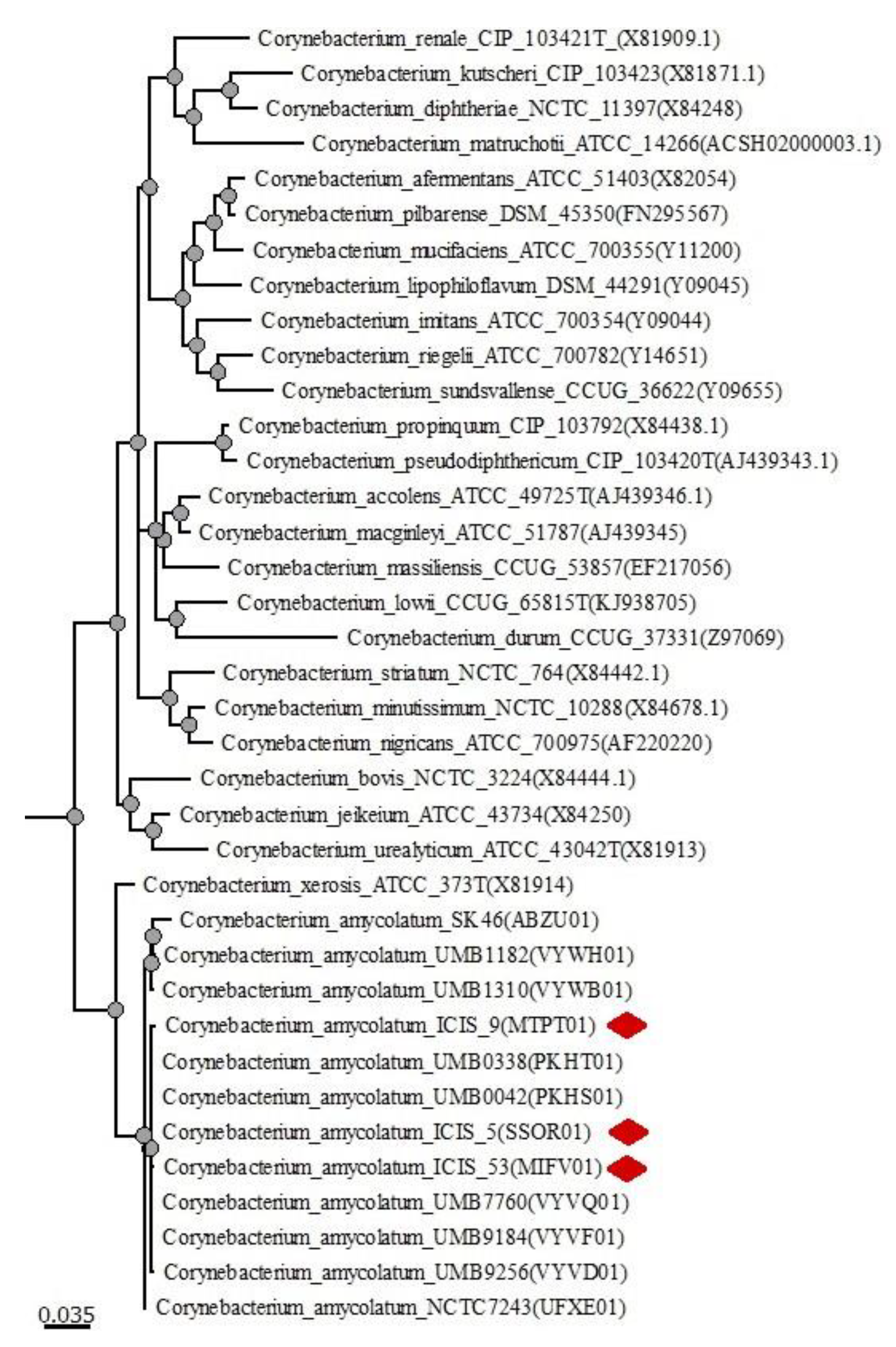
3.2. Phylogenetic Analysis
3.3. Genome Annotation of ICIS 5, ICIS 9 and ICIS 53
3.3.1. Genes and Properties Allowing, C. amycolatum to Survive in the Vaginal Ecosystem
3.3.2. Biologically Active Secondary Metabolite-Related Genes
3.3.3. Bacteriocin-Related Genes
3.3.4. H2O2-Related Genes
3.3.5. Nutrient Synthesis (Vitamins and Essential Amino Acids)-Related Genes
3.3.6. Antibiotic Resistance- and Virulence-Related Genes
3.3.7. Phage Defense Systems
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Buchta, V. Vaginal microbiome. Ceska Gynekol. 2018, 83, 371–379. [Google Scholar] [PubMed]
- Mendling, W. Vaginal microbiota. In Advances in Experimental Medicine and Biology; Springer: New York, NY, USA, 2016; Volume 902, pp. 83–93. [Google Scholar] [CrossRef]
- Smith, S.B.; Ravel, J. The vaginal microbiota, host defence and reproductive physiology. J. Physiol. 2017, 595, 451–463. [Google Scholar] [CrossRef] [PubMed]
- Tachedjian, G.; Aldunate, M.; Bradshaw, C.S.; Cone, R.A. The role of lactic acid production by probiotic Lactobacillus species in vaginal health. Res. Microbiol. 2017, 168, 782–792. [Google Scholar] [CrossRef] [PubMed]
- Hyman, R.W.; Fukushima, M.; Diamond, L.; Kumm, J.; Giudice, L.C.; Davis, R.W. Microbes on the human vaginal epithelium. Proc. Natl. Acad. Sci. USA 2005, 102, 7952–7957. [Google Scholar] [CrossRef]
- Verhelst, R.; Verstraelen, H.; Claeys, G.; Verschraegen, G.; Delanghe, J.; Van Simaey, L.; De Ganck, C.; Temmerman, M.; Vaneechoutte, M. Cloning of 16S rRNA genes amplified from normal and disturbed vaginal microflora suggests a strong association between Atopobium vaginae, Gardnerella vaginalis and bacterial vaginosis. BMC Microbiol. 2005, 4, 16. [Google Scholar] [CrossRef]
- Zhou, X.; Bent, S.J.; Schneider, M.G.; Davis, C.C.; Islam, M.R.; Forney, L.J. Characterization of vaginal microbial communities in adult healthy women using cultivation-independent methods. Microbiology 2004, 150, 2565–2573. [Google Scholar] [CrossRef]
- Zhou, X.; Brown, C.J.; Abdo, Z.; Davis, C.C.; Hansmann, M.A.; Joyce, P.; Foster, J.; Forney, L.J. Differences in the composition of vaginal microbial communities found in healthy Caucasian and black women. ISME J. 2007, 1, 121–133. [Google Scholar] [CrossRef]
- Zasada, A.A.; Mosiej, E. Contemporary microbiology and identification ofCorynebacteriaspp. causing infections in human. Lett. Appl. Microbiol. 2018, 66, 472–483. [Google Scholar] [CrossRef]
- Araújo, C.L.; Alves, J.; Lima, A.; Dias, L.; Silva, P.; Marques, J.; Azevedo, V.; Silva, A.; Folador, A.S.A.A. The Genus Corynebacterium in the Genomic Era. In Basic Biology and Applications of Actinobacteria; IntechOpen: London, UK, 2018. [Google Scholar] [CrossRef]
- Bomar, L.; Brugger, S.D.; Yost, B.H.; Davies, S.S.; Lemon, K.P. Corynebacterium accolens releases antipneumococcal free fatty acids from human nostril and skin surface triacylglycerols. mBio 2016, 7, e01725-15. [Google Scholar] [CrossRef]
- Ramsey, M.M.; Freire, M.; Gabrilska, R.A.; Rumbaugh, K.P.; Lemon, K.P. Staphylococcus aureus shifts toward commensalism in response to Corynebacterium species. Front. Microbiol. 2016, 7, 1230. [Google Scholar] [CrossRef] [PubMed]
- Hardy, B.L.; Dickey, S.W.; Plaut, R.D.; Riggins, D.P.; Stibitz, S.; Otto, M.; Merrell, D.S. Corynebacterium pseudodiphtheriticum exploits Staphylococcus aureus virulence components in a novel polymicrobial defense strategy. mBio 2019, 10, e02491-18. [Google Scholar] [CrossRef]
- Karlyshev, A.V.; Melnikov, V. Draft Genome sequence of Corynebacterium pseudodiphtheriticum strain 090104 “Sokolov”. Genome Announc. 2013, 1, e00921-13. [Google Scholar] [CrossRef]
- Gao, S.; Liu, C.; Qu, S.; Song, J.; Li, J.; Zhang, P.; Wang, Q.; Guo, C.; Gao, F.; Zhang, L. Non-cell Corynebacterium parvum generated by nanotechnology: A promising immunomodulator with less side effects. Int. Immunopharmacol. 2007, 7, 1334–1342. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ma, B.; Forney, L.J.; Ravel, J. Vaginal microbiome: Rethinking health and disease. Annu. Rev. Microbiol. 2012, 66, 371–389. [Google Scholar] [CrossRef]
- Chaban, B.; Links, M.G.; Jayaprakash, T.P.; Wagner, E.C.; Bourque, D.K.; Lohn, Z.; Albert, A.Y.; van Schalkwyk, J.; Reid, G.; Hemmingsen, S.M.; et al. Characterization of the vaginal microbiota of healthy Canadian women through the menstrual cycle. Microbiome 2014, 2, 23. [Google Scholar] [CrossRef] [PubMed]
- Pearce, M.M.; Hilt, E.E.; Rosenfeld, A.B.; Zilliox, M.J.; Thomas-White, K.; Fok, C.; Kliethermes, S.; Schreckenberger, P.C.; Brubaker, L.; Gai, X.; et al. The female urinary microbiome: A comparison of women with and without urgency urinary incontinence. mBio 2014, 5, e01283-14. [Google Scholar] [CrossRef] [PubMed]
- Hammerschlag, M.R.; Alpert, S.; Rosner, I.; Thurston, P.; Semine, D.; McComb, D.; McCormack, W.M. Microbiology of the vagina in children: Normal and potentially pathogenic organisms. Pediatrics 1978, 62, 57–62. Available online: http://pediatrics.aappublications.org/content/62/1/57.short (accessed on 19 December 2021). [CrossRef]
- Aagaard, K.; Riehle, K.; Ma, J.; Segata, N.; Mistretta, T.-A.; Coarfa, C.; Raza, S.; Rosenbaum, S.; Veyver, I.V.D.; Milosavljevic, A.; et al. A metagenomic approach to characterization of the vaginal microbiome signature in pregnancy. PLoS ONE 2012, 7, e36466. [Google Scholar] [CrossRef]
- Diop, K.; Nguyen, T.T.; Delerce, J.; Armstrong, N.; Raoult, D.; Bretelle, F.; Fenollar, F. Corynebacterium fournierii sp. nov., isolated from the female genital tract of a patient with bacterial vaginosis. Antonie Van Leeuwenhoek 2018, 111, 1165–1174. [Google Scholar] [CrossRef]
- Chen, X.; Zhao, X.; Chen, L.; Zeng, W.; Xu, H. Vaginitis Caused by Corynebacterium amycolatum in a Prepubescent Girl. J. Pediatr. Adolesc. Gynecol. 2015, 28, e165–e167. [Google Scholar] [CrossRef]
- Collins, M.D.; Burton, R.A.; Jones, D. Corynebacterium amycolatum sp. nov. a new mycolic acid-lessCorynebacteriumspecies from human skin. FEMS Microbiol. Lett. 1988, 49, 349–352. [Google Scholar] [CrossRef]
- Gladysheva, I.V.; Cherkasov, S.V.; Khlopko, Y. Antibacterial activities of metabolites from Corynebacterium spp. Strains isolated from the reproductive tract of a healthy woman against human pathogenic bacteria. Int. J. Pharma Bio Sci. 2017, 8, 549–556. [Google Scholar] [CrossRef]
- Gladysheva, I.V.; Cherkasov, S.V. Corynebacterium species in the female genital tract–pathogens or potential probiotics. Int. J. Pharma Bio Sci. 2018, 9, 265–272. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed]
- Aziz, R.K.; Bartels, D.; Best, A.A.; DeJongh, M.; Disz, T.; Edwards, R.A.; Formsma, K.; Gerdes, S.Y.; Glass, E.M.; Kubal, M.; et al. The RAST server: Rapid annotations using subsystems technology. BMC Genom. 2008, 9, 75. [Google Scholar] [CrossRef] [PubMed]
- Tatusova, T.; DiCuccio, M.; Badretdin, A.; Chetvernin, V.; Nawrocki, E.P.; Zaslavsky, L.; Lomsadze, A.; Pruitt, K.D.; Borodovsky, M.; Ostell, J. NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 2016, 44, 6614–6624. [Google Scholar] [CrossRef]
- Huerta-Cepas, J.; Szklarczyk, D.; Forslund, K.; Cook, H.; Heller, D.; Walter, M.C.; Rattei, T.; Mende, D.R.; Sunagawa, S.; Kuhn, M.; et al. eggNOG 4.5: A hierarchical orthology framework with improved functional annotations for eukaryotic, prokaryotic and viral sequences. Nucleic Acids Res. 2016, 44, D286–D293. [Google Scholar] [CrossRef]
- Van Heel, A.J.; De Jong, A.; Song, C.; Viel, J.; Kok, J.; Kuipers, O.P. BAGEL4: A user-friendly web server to thoroughly mine RiPPs and bacteriocins. Nucleic Acids Res. 2018, 46, W278–W281. [Google Scholar] [CrossRef]
- Blin, K.; Medema, M.H.; Kottmann, R.; Lee, S.Y.; Weber, T. The antiSMASH database, a comprehensive database of microbial secondary metabolite biosynthetic gene clusters. Nucleic Acids Res. 2016, 45, D555–D559. [Google Scholar] [CrossRef]
- Jia, B.; Raphenya, A.R.; Alcock, B.; Waglechner, N.; Guo, P.; Tsang, K.K.; Lago, B.A.; Dave, B.M.; Pereira, S.; Sharma, A.N.; et al. CARD 2017: Expansion and model-centric curation of the comprehensive antibiotic resistance database. Nucleic Acids Res. 2017, 45, D566–D573. [Google Scholar] [CrossRef]
- Liu, B.; Zheng, D.; Jin, Q.; Chen, L.; Yang, J. VFDB 2019: A comparative pathogenomic platform with an interactive web interface. Nucleic Acids Res. 2018, 47, D687–D692. [Google Scholar] [CrossRef] [PubMed]
- Grissa, I.; Vergnaud, G.; Pourcel, C. CRISPRFinder: A web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic Acids Res. 2007, 35, W52–W57. [Google Scholar] [CrossRef] [PubMed]
- Parte, A.C. LPSN—list of prokaryotic names with standing in nomenclature. Nucleic Acids Res. 2013, 42, D613–D616. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Okonechnikov, K.; Golosova, O.; Fursov, M.; The UGENE Team. Unipro UGENE: A unified bioinformatics toolkit. Bioinformatics 2012, 28, 1166–1167. [Google Scholar] [CrossRef]
- Lee, I.; Kim, Y.O.; Park, S.-C.; Chun, J. OrthoANI: An improved algorithm and software for calculating average nucleotide identity. Int. J. Syst. Evol. Microbiol. 2015, 66, 1100–1103. [Google Scholar] [CrossRef]
- Pascual, C.; Lawson, P.A.; Farrow, J.A.E.; Gimenez, M.N.; Collins, M.D. Phylogenetic analysis of the genus corynebacterium based on 16S rRNA gene sequences. Int. J. Syst. Bacteriol. 1995, 45, 724–728. [Google Scholar] [CrossRef]
- Ruimy, R.; Riegel, P.; Boiron, P.; Monteil, H.; Christen, R. Phylogeny of the genus corynebacterium deduced from analyses of small-subunit ribosomal DNA sequences. Int. J. Syst. Bacteriol. 1995, 45, 740–746. [Google Scholar] [CrossRef][Green Version]
- Chun, J.; Oren, A.; Ventosa, A.; Christensen, H.; Arahal, D.R.; Da Costa, M.S.; Rooney, A.P.; Yi, H.; Xu, X.-W.; De Meyer, S.; et al. Proposed minimal standards for the use of genome data for the taxonomy of prokaryotes. Int. J. Syst. Evol. Microbiol. 2018, 68, 461–466. [Google Scholar] [CrossRef]
- Deckers-Hebestreit, G.; Altendorf, K. The F0F1-type atp synthases of bacteria: Structure and function of the F0 complex. Annu. Rev. Microbiol. 1996, 50, 791–824. [Google Scholar] [CrossRef]
- Xu, N.; Wang, L.; Cheng, H.; Liu, Q.; Liu, J.; Ma, Y. In vitro functional characterization of the Na+/H+ antiporters in Corynebacterium glutamicum. FEMS Microbiol. Lett. 2015, 363, fnv237. [Google Scholar] [CrossRef]
- Toyoda, K.; Teramoto, H.; Inui, M.; Yukawa, H. The ldha gene, encoding fermentative L-lactate dehydrogenase of corynebacterium glutamicum, is under the control of positive feedback regulation mediated by LlDr. J. Bacteriol. 2009, 191, 4251–4258. [Google Scholar] [CrossRef]
- Kapse, N.G.; Engineer, A.S.; Gowdaman, V.; Wagh, S.; Dhakephalkar, P.K. Functional annotation of the genome unravels probiotic potential of Bacillus coagulans HS243. Genomics 2019, 111, 921–929. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Ji, H.; Zhang, D.; Liu, H.; Wang, S.; Wang, J.; Wang, Y. Complete genome sequencing of lactobacillus plantarum zlp001, a potential probiotic that enhances intestinal epithelial barrier function and defense against pathogens in pigs. Front. Physiol. 2018, 9, 1689. [Google Scholar] [CrossRef] [PubMed]
- Goyal, K.; Qamra, R.; Mande, S.C. Multiple gene duplication and rapid evolution in the groel gene: Functional implications. J. Mol. Evol. 2006, 63, 781–787. [Google Scholar] [CrossRef] [PubMed]
- Jaworek, M.W.; Moebitz, S.; Gao, M.; Winter, R. Stability of the chaperonin system GroEL–GroES under extreme environmental conditions. Phys. Chem. Chem. Phys. 2020, 22, 3734–3743. [Google Scholar] [CrossRef]
- Kim, S.Y.; Park, C.; Jang, H.-J.; Kim, B.-O.; Bae, H.-W.; Chung, I.-Y.; Kim, E.S.; Cho, Y.-H. Antibacterial strategies inspired by the oxidative stress and response networks. J. Microbiol. 2019, 57, 203–212. [Google Scholar] [CrossRef]
- Flohé, L.; Toppo, S.; Cozza, G.; Ursini, F. A comparison of thiol peroxidase mechanisms. Antioxid. Redox Signal. 2011, 15, 763–780. [Google Scholar] [CrossRef]
- Delaunay, A.; Pflieger, D.; Barrault, M.-B.; Vinh, J.; Toledano, M.B. A Thiol Peroxidase is an H2O2 receptor and redox-transducer in gene activation. Cell 2002, 111, 471–481. [Google Scholar] [CrossRef]
- Fridovich, I. Superoxide radical and superoxide dismutases. Annu. Rev. Biochem. 1995, 64, 97–112. [Google Scholar] [CrossRef] [PubMed]
- El Shafey, H.; Ghanem, S.; Merkamm, M.; Guyonvarch, A. Corynebacterium glutamicum superoxide dismutase is a manganese-strict non-cambialistic enzyme in vitro. Microbiol. Res. 2008, 163, 80–86. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Holmgren, A. The thioredoxin antioxidant system. Free Radic. Biol. Med. 2014, 66, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Su, T.; Si, M.; Zhao, Y.; Liu, Y.; Yao, S.; Che, C.; Chen, C. A thioredoxin-dependent peroxiredoxin Q from Corynebacterium glutamicum plays an important role in defense against oxidative stress. PLoS ONE 2018, 13, e0192674. [Google Scholar] [CrossRef]
- Newton, G.L.; Buchmeier, N.; Fahey, R.C. Biosynthesis and functions of mycothiol, the unique protective thiol of actinobacteria. Microbiol. Mol. Biol. Rev. 2008, 72, 471–494. [Google Scholar] [CrossRef]
- Koledin, T.; Newton, G.L.; Fahey, R.C. Identification of the mycothiol synthase gene (mshD) encoding the acetyltransferase producing mycothiol in actinomycetes. Arch. Microbiol. 2002, 178, 331–337. [Google Scholar] [CrossRef]
- Liu, Y.-B.; Long, M.-X.; Yin, Y.-J.; Si, M.-R.; Zhang, L.; Lu, Z.-Q.; Wang, Y.; Shen, X.-H. Physiological roles of mycothiol in detoxification and tolerance to multiple poisonous chemicals in Corynebacterium glutamicum. Arch. Microbiol. 2013, 195, 419–429. [Google Scholar] [CrossRef]
- Si, M.; Zhao, C.; Zhang, B.; Wei, D.; Chen, K.; Yang, X.; Xiao, H.; Shen, X. Overexpression of mycothiol disulfide reductase enhances Corynebacterium glutamicum robustness by modulating cellular redox homeostasis and antioxidant proteins under oxidative stress. Sci. Rep. 2016, 6, 29491. [Google Scholar] [CrossRef]
- Martin, J.F.; Demain, A.L. Control of antibiotic biosynthesis. Microbiol. Rev. 1980, 44, 230–251. [Google Scholar] [CrossRef]
- Gomes, E.S.; Schuch, V.; Lemos, E.G.D.M. Biotechnology of polyketides: New breath of life for the novel antibiotic genetic pathways discovery through metagenomics. Braz. J. Microbiol. 2013, 44, 1007–1034. [Google Scholar] [CrossRef]
- Staunton, J.; Weissman, K.J. Polyketide biosynthesis: A millennium review. Nat. Prod. Rep. 2001, 18, 380–416. [Google Scholar] [CrossRef] [PubMed]
- Kaysser, L.; Bernhardt, P.; Nam, S.-J.; Loesgen, S.; Ruby, J.G.; Skewes-Cox, P.; Jensen, P.; Fenical, W.; Moore, B.S. Merochlorins A–D, cyclic meroterpenoid antibiotics biosynthesized in divergent pathways with vanadium-dependent chloroperoxidases. J. Am. Chem. Soc. 2012, 134, 11988–11991. [Google Scholar] [CrossRef] [PubMed]
- Suroto, D.A.; Kitani, S.; Arai, M.; Ikeda, H.; Nihira, T. Characterization of the biosynthetic gene cluster for cryptic phthoxazolin A in Streptomyces avermitilis. PLoS ONE 2018, 13, e0190973. [Google Scholar] [CrossRef] [PubMed]
- Helfrich, E.J.N.; Lin, G.-M.; Voigt, C.A.; Clardy, J. Bacterial terpene biosynthesis: Challenges and opportunities for pathway engineering. Beilstein J. Org. Chem. 2019, 15, 2889–2906. [Google Scholar] [CrossRef]
- Yamada, Y.; Kuzuyama, T.; Komatsu, M.; Shin-Ya, K.; Omura, S.; Cane, D.-E.; Ikeda, H. Terpene synthases are widely distributed in bacteria. Proc. Natl. Acad. Sci. USA 2015, 112, 857–862. [Google Scholar] [CrossRef]
- Mahizan, N.A.; Yang, S.-K.; Moo, C.L.; Song, A.A.-L.; Chong, C.-M.; Chong, C.-W.; Abushelaibi, A.; Lim, S.-H.E.; Lai, K.-S. Terpene derivatives as a potential agent against antimicrobial resistance (AMR) Pathogens. Molecules 2019, 24, 2631. [Google Scholar] [CrossRef]
- Ravikumar, S.; Woo, H.M.; Choi, J.-I. Analysis of novel antioxidant sesquarterpenes (C35 terpenes) produced in recombinant Corynebacterium glutamicum. Appl. Biochem. Biotechnol. 2018, 186, 525–534. [Google Scholar] [CrossRef]
- Vasaturo, M.; Cotugno, R.; Fiengo, L.; Vinegoni, C.; Piaz, F.D.; De Tommasi, N. The anti-tumor diterpene oridonin is a direct inhibitor of Nucleolin in cancer cells. Sci. Rep. 2018, 8, 16735. [Google Scholar] [CrossRef]
- Lombard, J.; Moreira, D. Origins and early evolution of the mevalonate pathway of isoprenoid biosynthesis in the three domains of life. Mol. Biol. Evol. 2011, 28, 87–99. [Google Scholar] [CrossRef]
- Liang, P.-H.; Ko, T.-P.; Wang, A.H.-J. Structure, mechanism and function of prenyltransferases. JBIC J. Biol. Inorg. Chem. 2002, 269, 3339–3354. [Google Scholar] [CrossRef]
- Tarshis, L.C.; Proteau, P.J.; Kellogg, B.A.; Sacchettini, J.C.; Poulter, C.D. Regulation of product chain length by isoprenyl diphosphate synthases. Proc. Natl. Acad. Sci. USA 1996, 93, 15018–15023. [Google Scholar] [CrossRef]
- Riley, M.A.; Wertz, J.E. Bacteriocins: Evolution, ecology, and application. Annu. Rev. Microbiol. 2002, 56, 117–137. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Sieiro, P.; Montalbán-López, M.; Mu, D.; Kuipers, O.P. Bacteriocins of lactic acid bacteria: Extending the family. Appl. Microbiol. Biotechnol. 2016, 100, 2939–2951. [Google Scholar] [CrossRef] [PubMed]
- Balty, C.; Guillot, A.; Fradale, L.; Brewee, C.; Boulay, M.; Kubiak, X.; Benjdia, A.; Berteau, O. Ruminococcin C, an anti-clostridial sactipeptide produced by a prominent member of the human microbiota Ruminococcus gnavus. J. Biol. Chem. 2019, 294, 14512–14525. [Google Scholar] [CrossRef] [PubMed]
- Chiumento, S.; Roblin, C.; Kieffer-Jaquinod, S.; Tachon, S.; Leprètre, C.; Basset, C.; Aditiyarini, D.; Olleik, H.; Nicoletti, C.; Bornet, O.; et al. Ruminococcin C, a promising antibiotic produced by a human gut symbiont. Sci. Adv. 2019, 5, eaaw9969. [Google Scholar] [CrossRef]
- Ocaña, V.S.; de Ruiz Holgado, A.A.P.; Nader-Macías, M.E. Selection of vaginal H2O2-generating lactobacillus species for probiotic use. Curr. Microbiol. 1999, 38, 279–284. [Google Scholar] [CrossRef]
- Hertzberger, R.; Arents, J.C.; Dekker, H.L.; Pridmore, R.D.; Gysler, C.; Kleerebezem, M.; De Mattos, M.J.T. H2O2 production in species of the Lactobacillus acidophilus group: A central role for a novel NADH-dependent flavin reductase. Appl. Environ. Microbiol. 2014, 80, 2229–2239. [Google Scholar] [CrossRef]
- Leblanc, J.G.; Chain, F.; Martín, R.; Humaran, L.G.B.; Courau, S.; Langella, P. Beneficial effects on host energy metabolism of short-chain fatty acids and vitamins produced by commensal and probiotic bacteria. Microb. Cell Factories 2017, 16, 79. [Google Scholar] [CrossRef] [PubMed]
- Gu, Q.; Li, P. Biosynthesis of vitamins by probiotic bacteria. In Probiotics and Prebiotics in Human Nutrition and Health; InTechOpen: London, UK, 2016. [Google Scholar] [CrossRef]
- Ahire, J.J.; Mokashe, N.U.; Patil, H.J.; Chaudhari, B.L. Antioxidative potential of folate producing probiotic Lactobacillus helveticus CD6. J. Food Sci. Technol. 2013, 50, 26–34. [Google Scholar] [CrossRef]
- Dai, Z.; Wu, Z.; Hang, S.; Zhu, W.; Wu, G. Amino acid metabolism in intestinal bacteria and its potential implications for mammalian reproduction. Mol. Hum. Reprod. 2015, 21, 389–409. [Google Scholar] [CrossRef]
- Cherkasov, S.V.; Gladysheva, I.V. Antibiotic resistance of coryneform bacteria isolated from the reproductive tract of women. Antibiot. I Khimioterapiia 2010, 55, 45–49. Available online: http://www.ncbi.nlm.nih.gov/pubmed/21400755 (accessed on 4 April 2020).
- Tauch, A.; Burkovski, A. Molecular armory or niche factors: Virulence determinants of Corynebacterium species. FEMS Microbiol. Lett. 2015, 362, fnv185. [Google Scholar] [CrossRef] [PubMed]
- Swierczynski, A.; Ton-That, H. Type III pilus of corynebacteria: Pilus length is determined by the level of its major pilin subunit. J. Bacteriol. 2006, 188, 6318–6325. [Google Scholar] [CrossRef] [PubMed]
- Baumgart, M.; Schubert, K.; Bramkamp, M.; Frunzke, J. Impact of LytR-CpsA-Psr proteins on cell wall biosynthesis in Corynebacterium glutamicum. J. Bacteriol. 2016, 198, 3045–3059. [Google Scholar] [CrossRef]
- Wennerhold, J.; Bott, M. The DtxR regulon of Corynebacterium glutamicum. J. Bacteriol. 2006, 188, 2907–2918. [Google Scholar] [CrossRef] [PubMed]
- Bush, M.J. The actinobacterial WhiB-like (Wbl) family of transcription factors. Mol. Microbiol. 2018, 110, 663–676. [Google Scholar] [CrossRef]
- Makarova, K.S.; Wolf, Y.I.; Alkhnbashi, O.S.; Costa, F.; Shah, S.A.; Saunders, S.J.; Barrangou, R.; Brouns, S.J.; Charpentier, E.; Haft, D.H.; et al. An updated evolutionary classification of CRISPR–Cas systems. Nat. Rev. Microbiol. 2015, 13, 722–736. [Google Scholar] [CrossRef]
- Molineux, I.J. Host-parasite interactions: Recent developments in the genetics of abortive phage infections. New Boil. 1991, 3, 230–236. [Google Scholar]
- Kiliç, A.O.; Pavlova, S.I.; Alpay, S.; Kiliç, S.S.; Tao, L. Comparative study of vaginal lactobacillus phages isolated from women in the united states and turkey: Prevalence, morphology, host range, and DNA homology. Clin. Diagn. Lab. Immunol. 2001, 8, 31–39. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Statistics | ICIS 5 | ICIS 9 | ICIS 53 |
|---|---|---|---|
| Assembly | |||
| Number of contigs | 115 | 181 | 41 |
| N50 | 164,886 | 45,496 | 170,644 |
| L50 | 6 | 18 | 4 |
| Depth of coverage | 278 | 22 | 100 |
| Draft genome sequences | |||
| Genome size (b.p.) | 2,474,151 | 2,587,830 | 2,460,257 |
| GC contents (%) | 58.80 | 58.60 | 59.00 |
| Genes (total) | 2195 | 2392 | 2173 |
| CDSs (total) | 2109 | 2330 | 2110 |
| Genes (coding) | 2062 | 2277 | 2076 |
| CDSs (with protein) | 2062 | 2277 | 2076 |
| rRNAs (5S, 16S, 23S) | 30 (4, 20, 6) | 6 (4, 1, 1) | 7 (5, 1, 1) |
| complete rRNAs | 3, 1 (5S, 16S) | 1, 1, 1 (5S, 16S, 23S) | 5, 1, 1 (5S, 16S, 23S) |
| tRNAs | 53 | 53 | 53 |
| Pseudo Genes (total) | 47 | 53 | 34 |
| (OrthoANI) | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ICIS5 | ICIS9 | ICIS53 | NCTC7243 | SK46 | UMB0042 | UMB0338 | UMB1182 | UMB7760 | UMB9184 | UMB1310 | UMB9256 | |
| ICIS5 | 96.87 | 97.93 | 94.97 | 95.38 | 97.95 | 97.51 | 94.96 | 97.69 | 97.57 | 95.13 | 97.40 | |
| ICIS9 | 96.79 | 96.90 | 95.71 | 95.05 | 96.83 | 96.79 | 95.42 | 96.83 | 96.76 | 95.55 | 96.66 | |
| ICIS53 | 97.87 | 96.84 | 94.77 | 95.37 | 98.07 | 97.74 | 94.95 | 97.82 | 97.83 | 94.96 | 97.47 | |
| NCTC7243 | 94.90 | 95.66 | 94.74 | 94.30 | 94.77 | 95.13 | 96.87 | 95.03 | 95.08 | 96.77 | 95.21 | |
| SK46 | 95.20 | 94.93 | 95.29 | 94.22 | 95.35 | 95.09 | 94.42 | 95.16 | 95.17 | 94.43 | 95.12 | |
| UMB0042 | 97.85 | 96.78 | 98.05 | 94.67 | 95.29 | 97.54 | 94.73 | 97.66 | 97.55 | 94.88 | 97.26 | |
| UMB0338 | 97.48 | 96.71 | 97.67 | 95.02 | 95.05 | 97.55 | 95.04 | 97.89 | 97.85 | 95.10 | 97.66 | |
| UMB1182 | 94.83 | 95.37 | 94.75 | 96.83 | 94.26 | 94.62 | 94.90 | 95.06 | 94.96 | 98.70 | 95.20 | |
| UMB7760 | 97.69 | 96.74 | 97.82 | 95.00 | 95.12 | 97.66 | 97.81 | 94.93 | 98.10 | 95.12 | 97.98 | |
| UMB9184 | 97.58 | 96.75 | 97.79 | 94.97 | 95.11 | 97.60 | 97.80 | 94.90 | 98.03 | 95.07 | 97.68 | |
| UMB1310 | 95.02 | 95.50 | 94.85 | 96.68 | 94.28 | 94.82 | 95.07 | 98.64 | 95.06 | 95.03 | 95.23 | |
| UMB9256 | 97.33 | 96.58 | 97.47 | 95.18 | 95.06 | 97.30 | 97.58 | 95.08 | 97.95 | 97.67 | 95.18 | |
| (Original ANI) | ||||||||||||
| Stresses | Gene | Product | ICIS 5 Locus_tag | ICIS 9 Locus_tag | ICIS 53 Locus_tag |
|---|---|---|---|---|---|
| pH | atpB | F0F1 ATP synthase subunit A | E7L51_RS04555 | BXT90_RS01980 | BGC22_RS01625 |
| – | F0F1 ATP synthase subunit B | E7L51_RS04545 | BXT90_RS01970 | BGC22_RS01615 | |
| F0F1 ATP synthase subunit C | |||||
| – | F0F1 ATP synthase subunit alpha | E7L51_RS04535 | BXT90_RS01960 | BGC22_RS01605 | |
| atpD | F0F1 ATP synthase subunit beta | E7L51_RS04525 | BXT90_RS01950 | BGC22_RS01595 | |
| – | F0F1 ATP synthase subunit gamma | E7L51_RS04530 | BXT90_RS01955 | BGC22_RS01600 | |
| – | F0F1 ATP synthase subunit delta | E7L51_RS04540 | BXT90_RS01965 | BGC22_RS01610 | |
| – | F0F1 ATP synthase subunit epsilon | E7L51_RS04520 | BXT90_RS01945 | BGC22_RS01590 | |
| F0F1 ATP synthase protein I | |||||
| – | L-lactate dehydrogenase | E7L51_RS05660, E7L51_RS02260 | BXT90_RS06700, BXT90_RS07585 | BGC22_RS01270, BGC22_RS10270 | |
| – | Glucose-6-phosphate isomerase | E7L51_RS06505 | BXT90_RS00390 | BGC22_RS07690 | |
| – | GTP pyrophosphokinase | E7L51_RS00625 | BXT90_RS01675 | BGC22_RS04770 | |
| pyk | Pyruvate kinase | E7L51_RS00810, E7L51_RS02265 | BXT90_RS02290, BXT90_RS06705 | BGC22_RS04585, BGC22_RS10275 | |
| clpX | ATP-dependent Clp protease ATP-binding subunit | E7L51_RS09205 | BXT90_RS08190, BXT90_RS06490 | BGC22_RS03025, BGC22_RS06215 | |
| – | Na+/H+ antiporter subunit A | E7L51_RS03140 | BXT90_RS04255 | BGC22_RS02670 | |
| – | Na+/H+ antiporter subunit D | E7L51_RS03130 | BXT90_RS04265 | BGC22_RS02680 | |
| – | Na+/H+ antiporter subunit E | E7L51_RS03125 | BXT90_RS04270 | BGC22_RS02685 | |
| Temperature | hrcA | Heat-inducible transcriptional repressor | E7L51_RS03385 | BXT90_RS03730 | BGC22_RS05570 |
| grpE | Heat shock protein GrpE | E7L51_RS02840 | BXT90_RS09385 | BGC22_RS02385 | |
| dnaK | Heat shock protein DnaK | E7L51_RS02835 | BXT90_RS09390 | BGC22_RS02390 | |
| dnaJ | Heat shock protein DnaJ | E7L51_RS02845, E7L51_RS03380, E7L51_RS06605 | BXT90_RS03725, BXT90_RS09380 | BGC22_RS02380, BGC22_RS07585, BGC22_RS05565 | |
| – | Molecular chaperone GroES | E7L51_RS06730 | BXT90_RS00160 | BGC22_RS07460 | |
| groL | Molecular chaperone GroEL | E7L51_RS03170, E7L51_RS06725 | BXT90_RS00165, BXT90_RS06205 | BGC22_RS02645, BGC22_RS07465 | |
| Osmotic stress | betA | Choline dehydrogenase (EC 1.1.99.1) | E7L51_RS02520 | BXT90_RS09090 | BGC22_RS06865 |
| Nitrosative stress | – | Nitrate reductase | E7L51_RS05075 | BXT90_RS02975 | BGC22_RS00655 |
| Oxidative stress | – | Catalase (EC 1.11.1.6) | E7L51_RS10255 | BXT90_RS07525 | BGC22_RS03550 |
| – | Thiol peroxidase | E7L51_RS01250 | BXT90_RS05315 | BGC22_RS08510 | |
| trxA | Thioredoxin | E7L51_RS08530, E7L51_RS08670 | BXT90_RS04815, BXT90_RS10200 | BGC22_RS02030, BGC22_RS02165 | |
| trxB | Thioredoxin-disulfide reductase | E7L51_RS08665 | BXT90_RS04820 | BGC22_RS02160 | |
| – | Thioredoxin-dependent thiol peroxidase | E7L51_RS10400 | BXT90_RS08035 | BGC22_RS04010 | |
| – | Thioredoxin domain-containing protein | E7L51_RS09080, E7L51_RS10075 | BXT90_RS09275, BXT90_RS07735 | BGC22_RS03780, BGC22_RS08125 | |
| – | Glutathione peroxidase | E7L51_RS08875 | BXT90_RS09805 | BGC22_RS06430 | |
| – | Hydrogen peroxide-inducible genes activator | E7L51_RS05005 | BXT90_RS03045 | BGC22_RS00585 | |
| – | Superoxide dismutase | E7L51_RS02250 | BXT90_RS06690 | BGC22_RS10260 | |
| mshA | D-inositol-3-phosphate glycosyltransferase | E7L51_RS05705 | BXT90_RS09175 | BGC22_RS06775 | |
| mshB | N-acetyl-1-D-myo-inositol-2-amino-2-deoxy-alpha-D-glucopyranoside deacetylase | E7L51_RS04900 | BXT90_RS09615 | BGC22_RS01970 | |
| mshC | Cysteine--1-D-myo-inosityl 2-amino-2-deoxy-alpha-D-glucopyranoside ligase | E7L51_RS01445 | BXT90_RS02610 | BGC22_RS08710 | |
| mshD | Mycothiol synthase | E7L51_RS08935 | BXT90_RS09425 | BGC22_RS06490 | |
| mca | Mycothiol conjugate amidase | E7L51_RS03790 | BXT90_RS06270 | BGC22_RS05930 | |
| mtr | Mycothione reductase | E7L51_RS05350 | BXT90_RS05885 | BGC22_RS00925 | |
| nrdH | Gutaredoxin-like protein | E7L51_RS09855 | BXT90_RS10670 | BGC22_RS03865 |
| Product/EC No. | ICIS 5 Locus_tag | ICIS 9 Locus_tag | ICIS 53 Locus_tag |
|---|---|---|---|
| acetyl-CoA C-acetyltransferase (EC:2.3.1.9) | E7L51_RS02615 E7L51_RS08850 | BXT90_RS00480 | BGC22_RS02560 BGC22_RS06405 BGC22_RS07780 |
| hydroxymethylglutaryl-CoA synthase (EC:2.3.3.10) | E7L51_RS06810 | BXT90_RS00080 | BGC22_RS07380 |
| hydroxymethylglutaryl-CoA reductase (NADPH) (EC:1.1.1.34) | E7L51_RS06815 | BXT90_RS00075 | BGC22_RS07375 |
| mevalonate kinase (EC:2.7.1.36) | E7L51_RS06830 | BXT90_RS00060 | BGC22_RS07360 |
| phosphomevalonate kinase (EC:2.7.4.2) | E7L51_RS06820 | BXT90_RS00070 | BGC22_RS07370 |
| diphosphomevalonate decarboxylase (EC:4.1.1.33) | E7L51_RS06825 | BXT90_RS00065 | BGC22_RS07365 |
| isoprenyl transferase (undecaprenyl diphosphate synthase) uppS (EC:2.5.1-) | E7L51_RS03345 E7L51_RS03775 | BXT90_RS03690 BXT90_RS06255 | BGC22_RS05530 BGC22_RS05915 |
| isopentenyl-diphosphate Delta-isomerase (EC:5.3.3.2) | E7L51_RS06835 | BXT90_RS00055 | BGC22_RS07355 |
| polyprenyl synthetase family protein | E7L51_RS05960 | BXT90_RS05620 | BGC22_RS04055 |
| geranylgeranyl pyrophosphate synthase (EC:2.5.1.1 2.5.1.10 2.5.1.29) | E7L51_RS00410 | BXT90_RS01380 | BGC22_RS04980 |
| farnesyl-diphosphate farnesyltransferase (EC:2.5.1.21) | E7L51_RS00395 | BXT90_RS01365 | BGC22_RS04995 |
| Vitamin | Biosynthesis Protein/Gene/EC No. |
|---|---|
| Biotin | Transmembrane component BioN of energizing module of biotin ECF transporter Predicted biotin repressor from TetR family Substrate-specific component BioY of biotin ECF transporter Adenosylmethionine-8-amino-7-oxononanoate aminotransferase (EC 2.6.1.62) 8-amino-7-oxononanoate synthase (EC 2.3.1.47) Dethiobiotin synthetase (EC 6.3.3.3) Biotin synthase (EC 2.8.1.6) Long-chain-fatty-acid--CoA ligase (EC 6.2.1.3) Biotin synthesis protein BioC 3-ketoacyl-CoA thiolase (EC 2.3.1.16) ATPase component BioM of energizing module of biotin ECF transporter |
| Cobalamin | Cobalt-precorrin-6x reductase (EC 1.3.1.54) Cobalamin biosynthesis protein BluB L-threonine 3-O-phosphate decarboxylase (EC 4.1.1.81) Adenosylcobinamide-phosphate guanylyltransferase (EC 2.7.7.62) Cobalt-precorrin-8x methylmutase (EC 5.4.1.2) Cobalt-precorrin-2 C20-methyltransferase (EC 2.1.1.130) Cobyric acid synthase (EC 6.3.5.10) Cobalt-precorrin-4 C11-methyltransferase (EC 2.1.1.133) Cobalt-precorrin-3b C17-methyltransferase Nicotinate-nucleotide--dimethylbenzimidazole phosphoribosyltransferase (EC 2.4.2.21) Adenosylcobinamide-phosphate synthase (EC 6.3.1.10) Cob(I)alamin adenosyltransferase (EC 2.5.1.17) Cobyrinic acid A,C-diamide synthase |
| Riboflavin | FMN adenylyltransferase (EC 2.7.7.2) hypothetical protein YebC C-terminal domain of CinA type S 6,7-dimethyl-8-ribityllumazine synthase (EC 2.5.1.78) Riboflavin transporter PnuX 5-amino-6-(5-phosphoribosylamino)uracil reductase (EC 1.1.1.193) tRNA pseudouridine synthase B (EC 4.2.1.70) Riboflavin kinase (EC 2.7.1.26) GTP cyclohydrolase II (EC 3.5.4.25) Diaminohydroxyphosphoribosylaminopyrimidine deaminase (EC 3.5.4.26) 3,4-dihydroxy-2-butanone 4-phosphate synthase (EC 4.1.99.12) FMN adenylyltransferase (EC 2.7.7.2) Riboflavin synthase eubacterial/eukaryotic (EC 2.5.1.9) |
| Pyridoxine | Pyridoxine biosynthesis glutamine amidotransferase, synthase subunit (EC 2.4.2.-) D-3-phosphoglycerate dehydrogenase (EC 1.1.1.95) Pyridoxal kinase (EC 2.7.1.35) Phosphoserine aminotransferase (EC 2.6.1.52) NAD-dependent glyceraldehyde-3-phosphate dehydrogenase (EC 1.2.1.12) |
| Folate | Dihydropteroate synthase (EC 2.5.1.15) tRNA(Ile)-lysidine synthetase (EC 6.3.4.19) Aspartate 1-decarboxylase (EC 4.1.1.11) Cell division protein FtsH (EC 3.4.24.-) GTP cyclohydrolase I (EC 3.5.4.16) type 1 Pantoate--beta-alanine ligase (EC 6.3.2.1) 2-amino-4-hydroxy-6-hydroxymethyldihydropteridine pyrophosphokinase (EC 2.7.6.3) Dihydroneopterin aldolase (EC 4.1.2.25) 5-formyltetrahydrofolate cyclo-ligase (EC 6.3.3.2) Dihydrofolate reductase (EC 1.5.1.3) Thymidylate synthase thyX (EC 2.1.1.-) Dihydrofolate synthase (EC 6.3.2.12) Para-aminobenzoate synthase, aminase component (EC 2.6.1.85) |
| Amino Acid | Biosynthesis Protein/Gene/EC No. |
|---|---|
| Histidine | Phosphoribosylformimino-5-aminoimidazole carboxamide ribotide isomerase (EC 5.3.1.16) Phosphoribosyl-ATP pyrophosphatase (EC 3.6.1.31) Imidazole glycerol phosphate synthase amidotransferase subunit (EC 2.4.2.-) Histidinol-phosphatase [alternative form] (EC 3.1.3.15) Histidinol-phosphate aminotransferase (EC 2.6.1.9) Imidazoleglycerol-phosphate dehydratase (EC 4.2.1.19) Imidazole glycerol phosphate synthase cyclase subunit (EC 4.1.3.-) Phosphoribosyl-AMP cyclohydrolase (EC 3.5.4.19) ATP phosphoribosyltransferase (EC 2.4.2.17) Histidinol dehydrogenase (EC 1.1.1.23) |
| Arginine | N-succinyl-L,L-diaminopimelate desuccinylase (EC 3.5.1.18) Glutamate N-acetyltransferase (EC 2.3.1.35) Acetylglutamate kinase (EC 2.7.2.8) Arginine pathway regulatory protein ArgR Argininosuccinate lyase (EC 4.3.2.1) N-acetylglutamate synthase (EC 2.3.1.1) Argininosuccinate synthase (EC 6.3.4.5) N-acetyl-gamma-glutamyl-phosphate reductase (EC 1.2.1.38) Ornithine carbamoyltransferase (EC 2.1.3.3) Acetylornithine aminotransferase (EC 2.6.1.11) |
| Methionine | Methionine ABC transporter ATP-binding protein S-adenosylmethionine synthetase (EC 2.5.1.6) O-succinylhomoserine sulfhydrylase (EC 2.5.1.48) Serine acetyltransferase (EC 2.3.1.30) Homoserine kinase (EC 2.7.1.39) 5-methyltetrahydropteroyltriglutamate--homocysteine methyltransferase (EC 2.1.1.14) 5-methyltetrahydrofolate--homocysteine methyltransferase (EC 2.1.1.13) Cystathionine beta-lyase, type II (EC 4.4.1.8) Cysteine synthase (EC 2.5.1.47) O-acetylhomoserine sulfhydrylase (EC 2.5.1.49) 5,10-methylenetetrahydrofolate reductase (EC 1.5.1.20) Homoserine dehydrogenase (EC 1.1.1.3) Methionine ABC transporter substrate-binding protein Homoserine O-acetyltransferase (EC 2.3.1.31) Methionine ABC transporter permease protein Adenosylhomocysteinase (EC 3.3.1.1) |
| Threonine | Homoserine dehydrogenase (EC 1.1.1.3) Aspartate-semialdehyde dehydrogenase (EC 1.2.1.11) Aspartate aminotransferase (EC 2.6.1.1) Threonine synthase (EC 4.2.3.1) Homoserine kinase (EC 2.7.1.39) Aspartokinase (EC 2.7.2.4) |
| Lysine | N-succinyl-L,L-diaminopimelate desuccinylase (EC 3.5.1.18) N-acetyl-L,L-diaminopimelate deacetylase (EC 3.5.1.47) 2,3,4,5-tetrahydropyridine-2,6-dicarboxylate N-succinyltransferase (EC 2.3.1.117) Diaminopimelate epimerase (EC 5.1.1.7) Aspartate-semialdehyde dehydrogenase (EC 1.2.1.11) 4-hydroxy-tetrahydrodipicolinate synthase (EC 4.3.3.7) Meso-diaminopimelate D-dehydrogenase (EC 1.4.1.16) Diaminopimelate decarboxylase (EC 4.1.1.20) N-succinyl-L,L-diaminopimelate aminotransferase alternative (EC 2.6.1.17) 2,3,4,5-tetrahydropyridine-2,6-dicarboxylate N-acetyltransferase (EC 2.3.1.89) 4-hydroxy-tetrahydrodipicolinate reductase (EC 1.17.1.8) Aspartokinase (EC 2.7.2.4) |
| Leucine | 3-isopropylmalate dehydratase small subunit (EC 4.2.1.33) 3-isopropylmalate dehydratase large subunit (EC 4.2.1.33) 2-isopropylmalate synthase (EC 2.3.3.13) Branched-chain amino acid aminotransferase (EC 2.6.1.42) 3-isopropylmalate dehydrogenase (EC 1.1.1.85) |
| Tryptophan | Anthranilate synthase, amidotransferase component (EC 4.1.3.27) Aminodeoxychorismate lyase (EC 4.1.3.38) Tryptophan-associated membrane protein Tryptophan synthase alpha chain (EC 4.2.1.20) Anthranilate phosphoribosyltransferase (EC 2.4.2.18) Tryptophan synthase beta chain (EC 4.2.1.20) Acting phosphoribosylanthranilate isomerase (EC 5.3.1.24) Indole-3-glycerol phosphate synthase (EC 4.1.1.48) Anthranilate synthase, aminase component (EC 4.1.3.27) Para-aminobenzoate synthase, aminase component (EC 2.6.1.85) Para-aminobenzoate synthase, amidotransferase component (EC 2.6.1.85) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gladysheva, I.V.; Cherkasov, S.V.; Khlopko, Y.A.; Plotnikov, A.O. Genome Characterization and Probiotic Potential of Corynebacterium amycolatum Human Vaginal Isolates. Microorganisms 2022, 10, 249. https://doi.org/10.3390/microorganisms10020249
Gladysheva IV, Cherkasov SV, Khlopko YA, Plotnikov AO. Genome Characterization and Probiotic Potential of Corynebacterium amycolatum Human Vaginal Isolates. Microorganisms. 2022; 10(2):249. https://doi.org/10.3390/microorganisms10020249
Chicago/Turabian StyleGladysheva, Irina V., Sergey V. Cherkasov, Yuriy A. Khlopko, and Andrey O. Plotnikov. 2022. "Genome Characterization and Probiotic Potential of Corynebacterium amycolatum Human Vaginal Isolates" Microorganisms 10, no. 2: 249. https://doi.org/10.3390/microorganisms10020249
APA StyleGladysheva, I. V., Cherkasov, S. V., Khlopko, Y. A., & Plotnikov, A. O. (2022). Genome Characterization and Probiotic Potential of Corynebacterium amycolatum Human Vaginal Isolates. Microorganisms, 10(2), 249. https://doi.org/10.3390/microorganisms10020249