
Comparative Genomic Analyses of the Genus Nesterenkonia Unravels the Genomic Adaptation to Polar Extreme Environments


Abstract
:1. Introduction
2. Materials and Methods
2.1. Sampling, Isolation, and Physiological Measurement
2.2. Whole Genome Sequencing, Assembly, and Reference Genomes Collection
2.3. Genome Quality Estimation, Gene Annotation, and Phylogenetic Analysis
2.4. Comparative Genomic Analysis
3. Results and Discussion
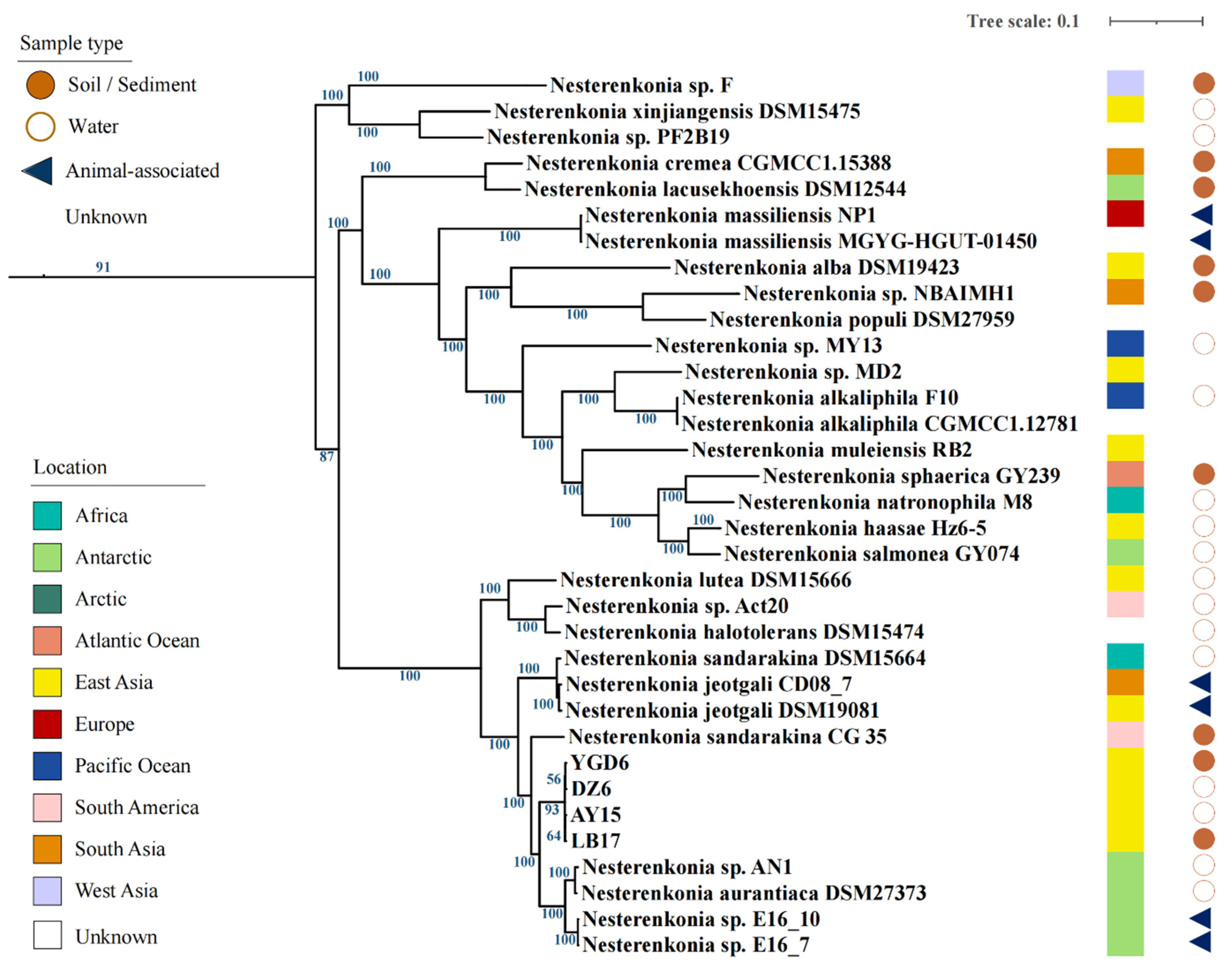
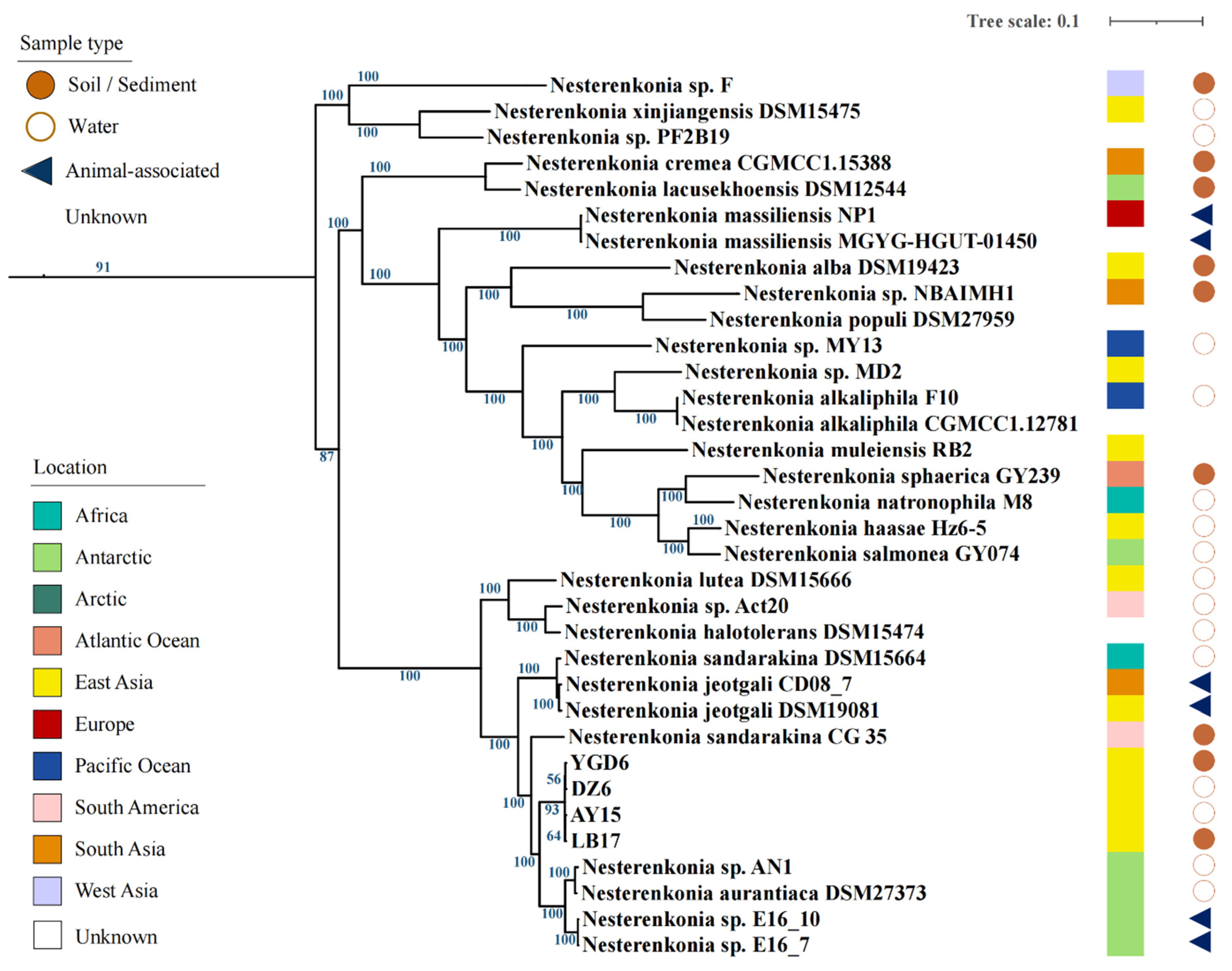
3.1. Tibetan Isolates Show High Similarities with Antarctic Isolates
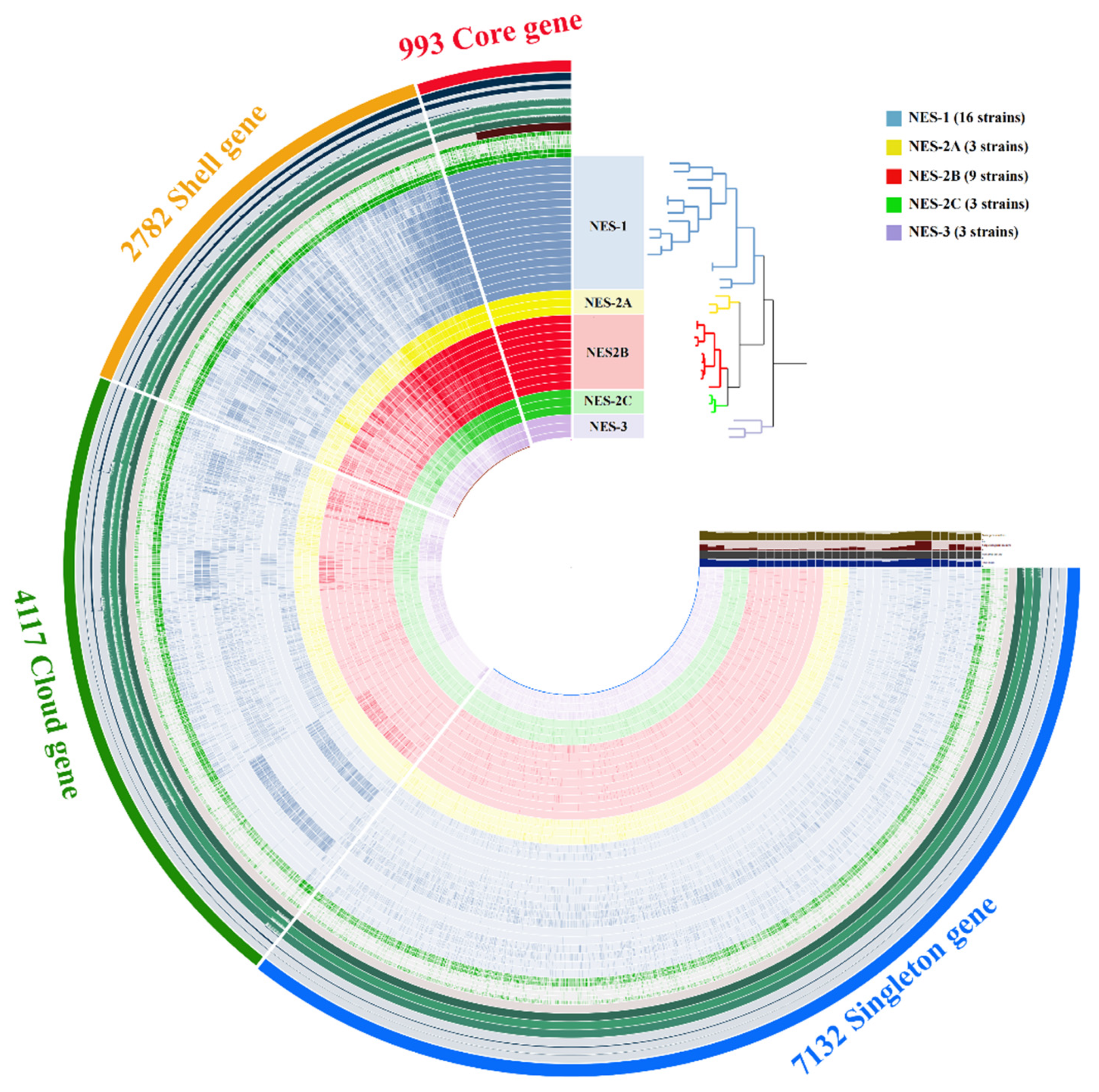
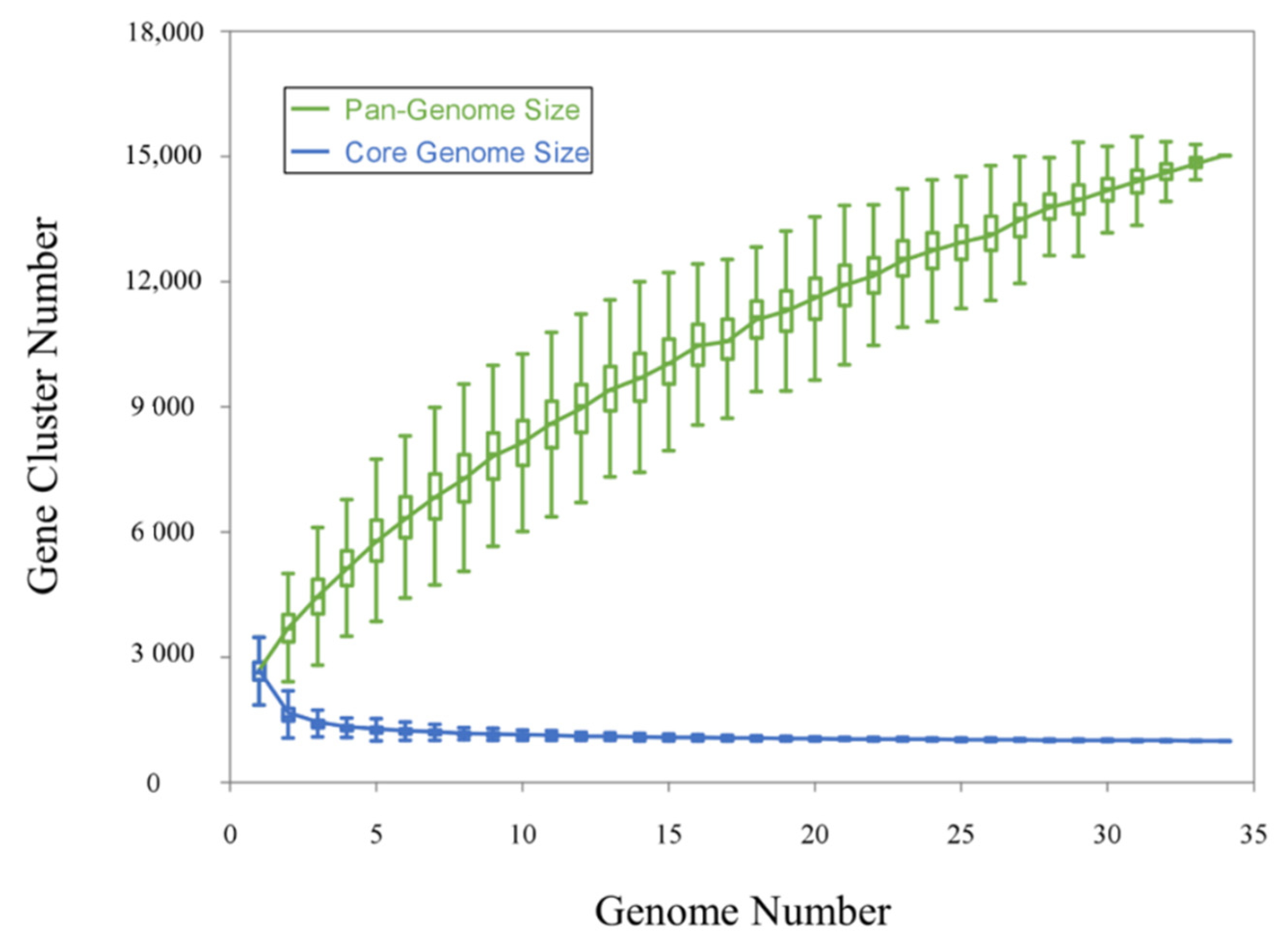
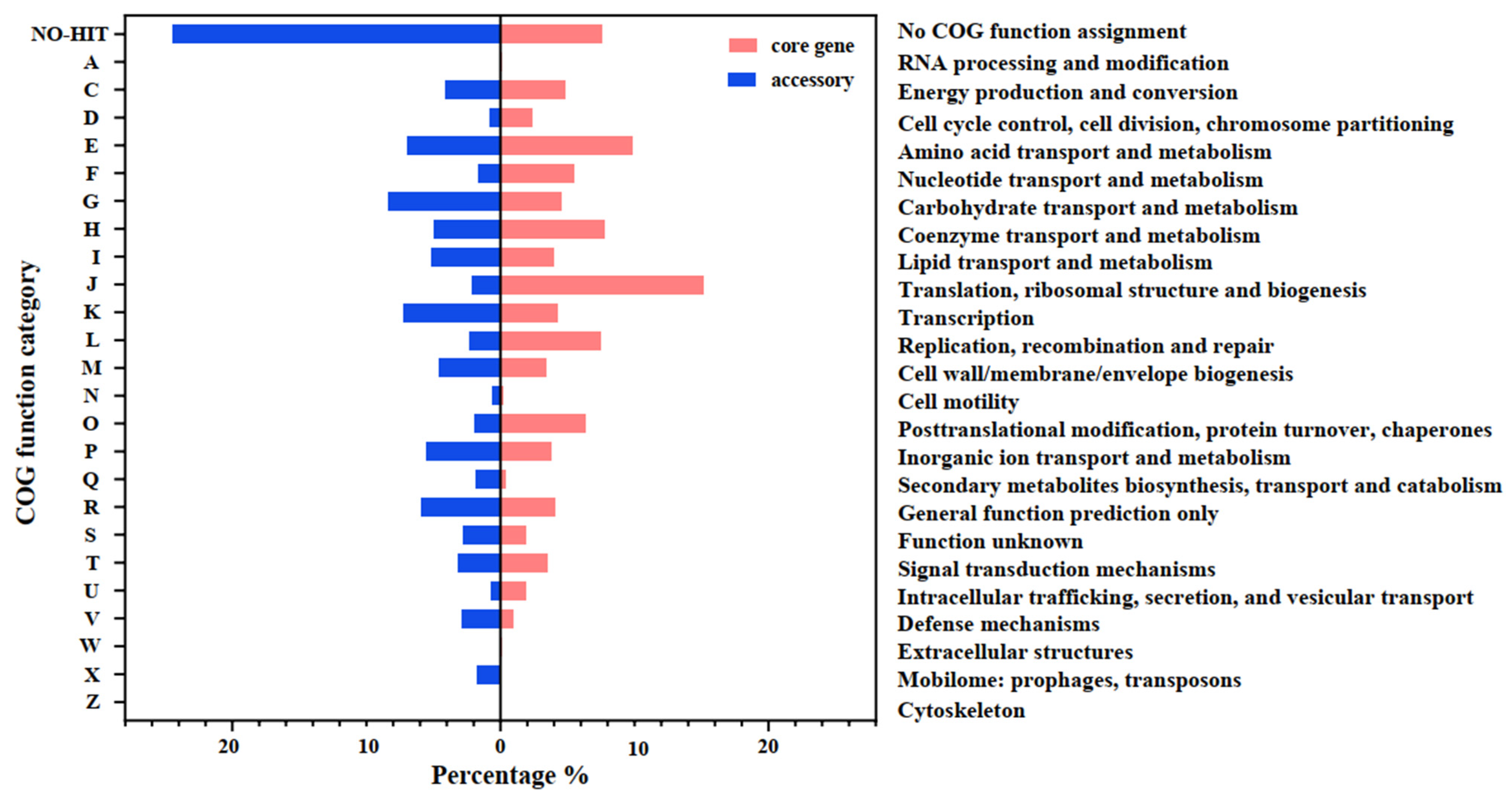
3.2. Comparative Genomic Analysis
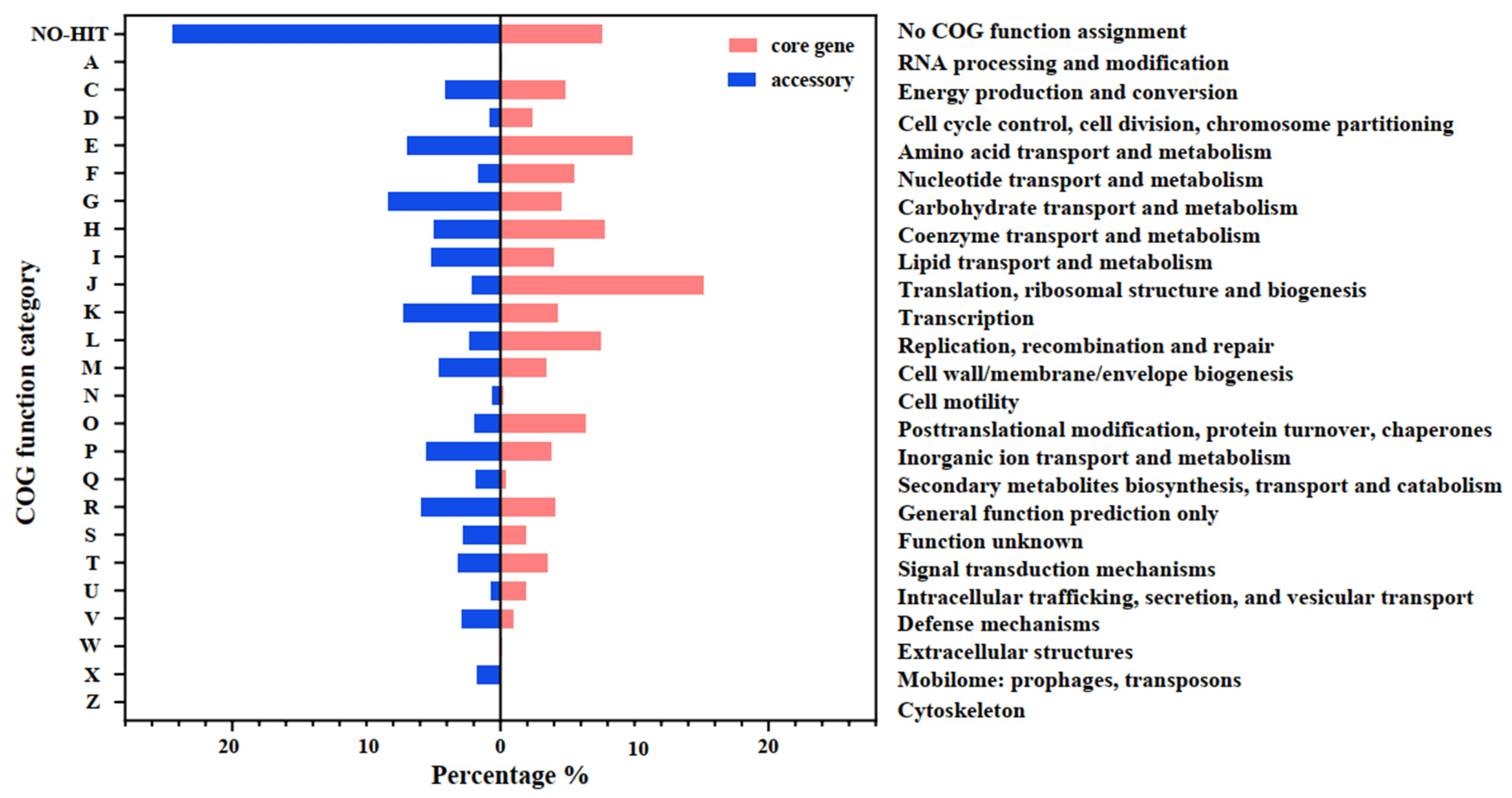
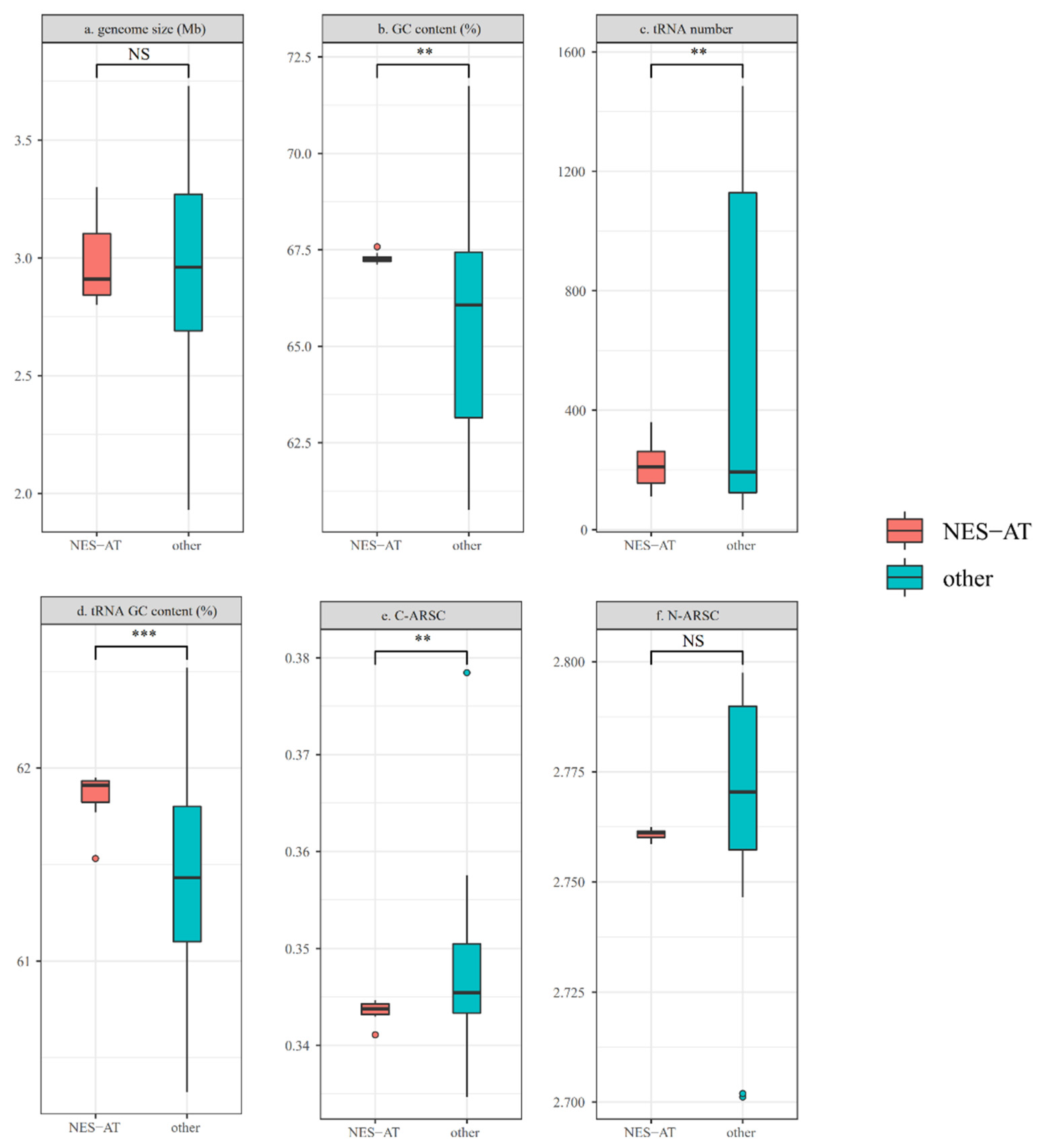
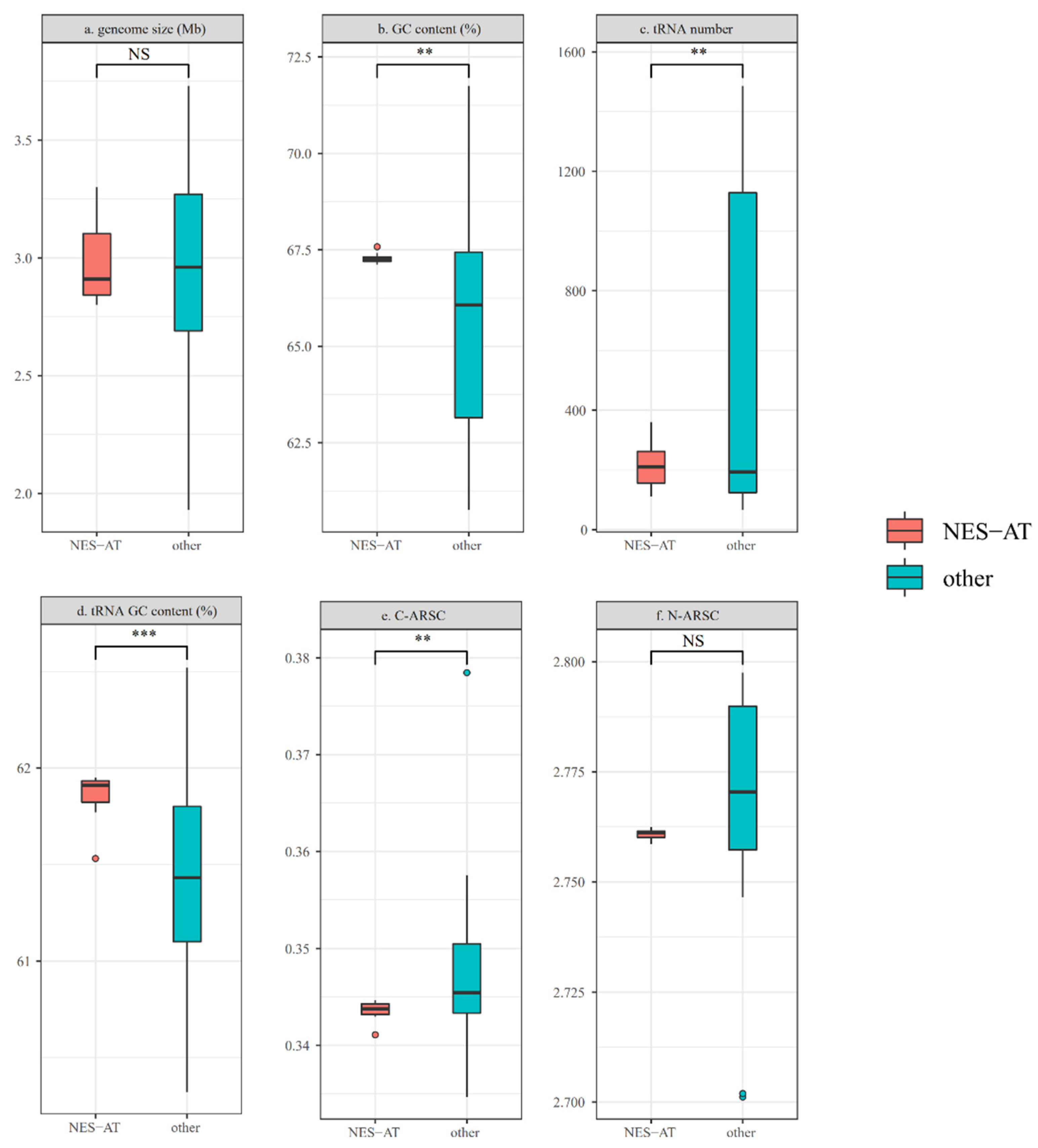
3.3. Genomic Feature Comparison between NES-AT Isolates and Other Nesterenkonia Isolates
3.4. Functional Genes Related to the “Polar” Environmental Adaption
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Yao, T.; Thompson, L.G.; Mosbrugger, V.; Zhang, F.; Ma, Y.; Luo, T.; Xu, B.; Yang, X.; Joswiak, D.R.; Wang, W.; et al. Third Pole Environment (TPE). Environ. Dev. 2012, 3, 52–64. [Google Scholar] [CrossRef]
- Williams, T.J.; Allen, M.A.; DeMaere, M.; Kyrpides, N.; Tringe, S.; Woyke, T.; Cavicchioli, R. Microbial ecology of an Antarctic hypersaline lake: Genomic assessment of ecophysiology among dominant haloarchaea. ISME J. 2014, 8, 1645–1658. [Google Scholar] [CrossRef] [Green Version]
- Sheridan, P.O.; Thames Consortium; Raguideau, S.; Quince, C.; Holden, J.; Zhang, L.; Williams, T.A.; Gubry-Rangin, C. Gene duplication drives genome expansion in a major lineage of Thaumarchaeota. Nat. Commun. 2020, 11, 5494. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Liu, Y.; Allen, M.A.; Xu, B.; Wang, N.; Williams, T.J.; Wang, F.; Zhou, Y.; Liu, Q.; Cavicchioli, R. Linking genomic and physiological characteristics of psychrophilic Arthrobacter to metagenomic data to explain global environmental distribution. Microbiome 2021, 9, 136. [Google Scholar] [CrossRef]
- Teufel, A.G.; Li, W.; Kiss, A.J.; Morgan-Kiss, R.M. Impact of nitrogen and phosphorus on phytoplankton production and bacterial community structure in two stratified Antarctic lakes: A bioassay approach. Polar Biol. 2017, 40, 1007–1022. [Google Scholar] [CrossRef]
- Liu, Y.; Yao, T.; Zhu, L.; Jiao, N.; Liu, X.; Zeng, Y.; Jiang, H. Bacterial Diversity of Freshwater Alpine Lake Puma Yumco on the Tibetan Plateau. Geomicrobiol. J. 2009, 26, 131–145. [Google Scholar] [CrossRef]
- Møller, A.K.; Søborg, D.A.; Abu Al-Soud, W.; Sørensen, S.; Kroer, N. Bacterial community structure in High-Arctic snow and freshwater as revealed by pyrosequencing of 16S rRNA genes and cultivation. Polar Res. 2013, 32, 17390. [Google Scholar] [CrossRef]
- Liu, K.; Yao, T.; Pearce, D.A.; Jiao, N.; Zeng, Y.; Guo, B.; Liu, Y. Bacteria in the lakes of the Tibetan Plateau and polar regions. Sci. Total. Environ. 2021, 754, 142248. [Google Scholar] [CrossRef] [PubMed]
- Liao, L.; Su, S.; Zhao, B.; Fan, C.; Zhang, J.; Li, H.; Chen, B. Biosynthetic Potential of a Novel Antarctic Actinobacterium Marisediminicola antarctica ZS314T Revealed by Genomic Data Mining and Pigment Characterization. Mar. Drugs 2019, 17, 388. [Google Scholar] [CrossRef]
- Silva, T.R.E.; Silva, L.C.F.; de Queiroz, A.C.; Moreira, M.S.A.; Fraga, C.A.D.C.; de Menezes, G.C.A.; Rosa, L.H.; Bicas, J.; de Oliveira, V.M.; Duarte, A.W.F. Pigments from Antarctic bacteria and their biotechnological applications. Crit. Rev. Biotechnol. 2021, 41, 809–826. [Google Scholar] [CrossRef] [PubMed]
- Benaud, N.; Edwards, R.J.; Amos, T.G.; D’Agostino, P.M.; Gutiérrez-Chávez, C.; Montgomery, K.; Nicetic, I.; Ferrari, B.C. Antarctic desert soil bacteria exhibit high novel natural product potential, evaluated through long-read genome sequencing and comparative genomics. Environ. Microbiol. 2021, 23, 3646–3664. [Google Scholar] [CrossRef] [PubMed]
- Dsouza, M.; Taylor, M.W.; Turner, S.J.; Aislabie, J. Genomic and phenotypic insights into the ecology of Arthrobacter from Antarctic soils. BMC Genom. 2015, 16, 36. [Google Scholar] [CrossRef] [Green Version]
- Li, W.J.; Chen, H.H.; Zhang, Y.Q.; Schumann, P.; Stackebrandt, E.; Xu, L.H.; Jiang, C.L. Nesterenkonia halotolerans sp. nov.; Nesterenkonia xinjiangensis sp. nov.; actinobacteria from saline soils in the west of China. Int. J. Syst. Evol. Microbiol. 2004, 54, 837–841. [Google Scholar] [CrossRef]
- Delgado, O.; Quillaguaman, J.; Bakhtiar, S.; Mattiasson, B.; Gessesse, A.; Hatti-Kaul, R. Nesterenkonia aethiopica sp. nov.; an alkaliphilic, moderate halophile isolated from an Ethiopian soda lake. Int. J. Syst. Evol. Microbiol. 2006, 56, 1229–1232. [Google Scholar] [CrossRef] [Green Version]
- Finore, I.; Orlando, P.; Di Donato, P.; Leone, L.; Nicolaus, B.; Poli, A. Nesterenkonia aurantiaca sp. nov.; an alkaliphilic actinobacterium isolated from Antarctica. Int. J. Syst. Evol. Microbiol. 2016, 66, 1554–1560. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.M.; Tuo, L.; Habden, X.; Guo, L.; Jiang, Z.K.; Liu, X.F.; Chen, L.; Zhang, Y.Q.; Sun, C.H. Nesterenkonia populi sp. nov.; an actinobacterium isolated from Populus euphratica. Int. J. Syst. Evol. Microbiol. 2015, 65 Pt 5, 1474–1479. [Google Scholar] [CrossRef] [Green Version]
- Chander, A.; Nair, R.G.; Kaur, G.; Kochhar, R.; Dhawan, D.K.; Bhadada, S.K.; Mayilraj, S. Genome Insight and Comparative Pathogenomic Analysis of Nesterenkonia jeotgali Strain CD08_7 Isolated from Duodenal Mucosa of Celiac Disease Patient. Front. Microbiol. 2017, 8, 129. [Google Scholar] [CrossRef] [Green Version]
- Edouard, S.; Sankar, S.; Dangui, N.P.M.; Lagier, J.-C.; Michelle, C.; Raoult, D.; Fournier, P.-E. Genome sequence and description of Nesterenkonia massiliensis sp. nov. strain NP1T. Stand. Genom. Sci. 2014, 9, 866–882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, P.; Kapse, N.; Gowdaman, V.; Tsuji, M.; Singh, S.; Dhakephalkar, P. Comparative Genomic Analysis of Arctic Permafrost Bacterium Nesterenkonia sp. PF2B19 to Gain Insights into Its Cold Adaptation Tactic and Diverse Biotechnological Potential. Sustainability 2021, 13, 4590. [Google Scholar] [CrossRef]
- Aliyu, H.; De Maayer, P.; Cowan, D. The genome of the Antarctic polyextremophile Nesterenkonia sp. AN1 reveals adaptive strategies for survival under multiple stress conditions. FEMS Microbiol. Ecol. 2016, 92, fiw032. [Google Scholar] [CrossRef] [Green Version]
- Alonso-Reyes, D.G.; Galván, F.S.; Portero, L.R.; Alvarado, N.N.; Farías, M.E.; Vazquez, M.P.; Albarracín, V.H. Genomic insights into an andean multiresistant soil actinobacterium of biotechnological interest. World J. Microbiol. Biotechnol. 2021, 37, 116. [Google Scholar] [CrossRef] [PubMed]
- Vernikos, G.; Medini, D.; Riley, D.R.; Tettelin, H. Ten years of pan-genome analyses. Curr. Opin. Microbiol. 2015, 23, 148–154. [Google Scholar] [CrossRef]
- Sugawara, M.; Epstein, B.; Badgley, B.D.; Unno, T.; Xu, L.; Reese, J.; Gyaneshwar, P.; Denny, R.; Mudge, J.; Bharti, A.K.; et al. Comparative genomics of the core and accessory genomes of 48 Sinorhizobium strains comprising five genospecies. Genome Biol. 2013, 14, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Zhong, C.; Wang, L.; Ning, K. Pan-genome study of Thermococcales reveals extensive genetic diversity and genetic evidence of thermophilic adaption. Environ. Microbiol. 2021, 23, 3599–3613. [Google Scholar] [CrossRef]
- Lefébure, T.; Stanhope, M.J. Evolution of the core and pan-genome of Streptococcus: Positive selection, recombination, and genome composition. Genome Biol. 2007, 8, 1–17. [Google Scholar] [CrossRef] [Green Version]
- D’Auria, G.; Hernández, N.J.; Peris-Bondia, F.; Moya, A.; Latorre, A. Legionella pneumophila pangenome reveals strain-specific virulence factors. BMC Genom. 2010, 11, 181. [Google Scholar] [CrossRef] [Green Version]
- Sehgal, S.N.; Gibbons, N.E. Effect of Some Metal Ions on the Growth of Halobacterium Cutirubrum. Can. J. Microbiol. 1960, 6, 165–169. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.-H.; Ha, S.-M.; Kwon, S.; Lim, J.; Kim, Y.; Seo, H.; Chun, J. Introducing EzBioCloud: A taxonomically united database of 16S rRNA gene sequences and whole-genome assemblies. Int. J. Syst. Evol. Microbiol. 2017, 67, 1613. [Google Scholar] [CrossRef]
- Federation, W.E.; Association, A. Standard Methods for the Examination of Water and Wastewater; American Public Health Association (APHA): Washington, DC, USA, 2005. [Google Scholar]
- Lim, H.J.; Lee, E.; Yoon, Y.; Chua, B.; Son, A. Portable lysis apparatus for rapid single-step DNA extraction of Bacillus subtilis. J. Appl. Microbiol. 2016, 120, 379–387. [Google Scholar] [CrossRef] [Green Version]
- Bushnell, B.; Rood, J.; Singer, E. BBMerge–Accurate paired shotgun read merging via overlap. PLoS ONE 2017, 12, e0185056. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parks, D.H.; Imelfort, M.; Skennerton, C.T.; Hugenholtz, P.; Tyson, G.W. CheckM: Assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015, 25, 1043–1055. [Google Scholar] [CrossRef] [Green Version]
- Shen, W.; Le, S.; Li, Y.; Hu, F. SeqKit: A Cross-Platform and Ultrafast Toolkit for FASTA/Q File Manipulation. PLoS ONE 2016, 11, e0163962. [Google Scholar] [CrossRef] [PubMed]
- Seemann, T. Prokka: Rapid Prokaryotic Genome Annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef]
- Chan, P.P.; Lin, B.Y.; Mak, A.J.; Lowe, T.M. tRNAscan-SE 2.0: Improved detection and functional classification of transfer RNA genes. Nucleic Acids Res. 2021, 49, 9077–9096. [Google Scholar] [CrossRef]
- Kanehisa, M.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016, 44, D457–D462. [Google Scholar] [CrossRef] [Green Version]
- Galperin, M.Y.; Wolf, Y.I.; Makarova, K.S.; Alvarez, R.V.; Landsman, D.; Koonin, E.V. COG database update: Focus on microbial diversity, model organisms, and widespread pathogens. Nucleic Acids Res. 2021, 49, D274–D281. [Google Scholar] [CrossRef]
- Eddy, S.R. A new generation of homology search tools based on probabilistic inference. In Genome Informatics 2009; Imperial College Press and Distributed by World Scientific Publishing Co.: London, UK, 2009; pp. 205–211. [Google Scholar]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, L.-T.; Schmidt, H.A.; Von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Emms, D.M.; Kelly, S. OrthoFinder: Solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biol. 2015, 16, 157. [Google Scholar] [CrossRef] [Green Version]
- Capella-Gutiérrez, S.; Silla-Martínez, J.M.; Gabaldón, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef]
- Jain, C.; Rodriguez, R.L.M.; Phillippy, A.M.; Konstantinidis, K.T.; Aluru, S. High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 2018, 9, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Rodriguez, R.L.M.; Konstantinidis, K.T. The Enveomics Collection: A Toolbox for Specialized Analyses of Microbial Genomes and Metagenomes; PeerJ Preprints: London, UK, 2016. [Google Scholar]
- R Core Team. R: A Language and Environment for Statistical Computing; R Core Team: Vienna, Austria, 2013. [Google Scholar]
- Eren, A.M.; Kiefl, E.; Shaiber, A.; Veseli, I.; Miller, S.E.; Schechter, M.S.; Fink, I.; Pan, J.N.; Yousef, M.; Fogarty, E.C. Community-led, integrated, reproducible multi-omics with anvi’o. Nat. Microbiol. 2021, 6, 3–6. [Google Scholar] [CrossRef]
- Enright, A.J.; van Dongen, S.; Ouzounis, C.A. An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res. 2002, 30, 1575–1584. [Google Scholar] [CrossRef] [PubMed]
- Snipen, L.-G.; Liland, K.H. Micropan: An R-package for microbial pan-genomics. BMC Bioinform. 2015, 16, 1–8. [Google Scholar] [CrossRef]
- Zhao, Y.; Jia, X.; Yang, J.; Ling, Y.; Zhang, Z.; Yu, J.; Wu, J.; Xiao, J. PanGP: A tool for quickly analyzing bacterial pan-genome profile. Bioinformatics 2014, 30, 1297–1299. [Google Scholar] [CrossRef] [Green Version]
- Moreno-Pino, M.; Ugalde, J.A.; Valdés, J.H.; Rodríguez-Marconi, S.; Parada-Pozo, G.; Trefault, N. Bacteria Isolated from the Antarctic Sponge Iophon sp. Reveals Mechanisms of Symbiosis in Sporosarcina, Cellulophaga, and Nesterenkonia. Front. Microbiol. 2021, 12, 660779. [Google Scholar] [CrossRef] [PubMed]
- Gu, S.; Tang, Y.; Du, M.; Kato, T.; Li, Y.; Cui, X.; Zhao, X. Short-term variation of CO2 flux in relation to environmental controls in an alpine meadow on the Qinghai-Tibetan Plateau. J. Geophys. Res. Space Phys. 2003, 108. [Google Scholar] [CrossRef]
- Schloss, I.R.; Abele, D.; Moreau, S.; Demers, S.; Bers, A.; González, O.; Ferreyra, G.A. Response of phytoplankton dynamics to 19-year (1991–2009) climate trends in Potter Cove (Antarctica). J. Mar. Syst. 2012, 92, 53–66. [Google Scholar] [CrossRef]
- Lieth, H. Modeling the Primary Productivity of the World. In Primary Productivity of the Biosphere; Springer: Berlin/Heidelberg, Germany, 1975; pp. 237–263. [Google Scholar]
- Crawford, A.R. The Indus Suture Line, the Himalaya, Tibet and Gondwanaland. Geol. Mag. 1974, 111, 369–383. [Google Scholar] [CrossRef]
- Tettelin, H.; Riley, D.; Cattuto, C.; Medini, D. Comparative genomics: The bacterial pan-genome. Curr. Opin. Microbiol. 2008, 11, 472–477. [Google Scholar] [CrossRef]
- Medini, D.; Donati, C.; Tettelin, H.; Masignani, V.; Rappuoli, R. The microbial pan-genome. Curr. Opin. Genet. Dev. 2005, 15, 589–594. [Google Scholar] [CrossRef] [PubMed]
- Thomas, C.M.; Nielsen, K.M. Mechanisms of, and Barriers to, Horizontal Gene Transfer between Bacteria. Nat. Rev. Microbiol. 2005, 3, 711–721. [Google Scholar] [CrossRef] [PubMed]
- Mende, D.R.; Bryant, J.A.; Aylward, F.; Eppley, J.M.; Nielsen, T.; Karl, D.M.; Delong, E.F. Environmental drivers of a microbial genomic transition zone in the ocean’s interior. Nat. Microbiol. 2017, 2, 1367–1373. [Google Scholar] [CrossRef]
- Weissman, J.L.; Fagan, W.F.; Johnson, P.L.F. Linking high GC content to the repair of double strand breaks in prokaryotic genomes. PLoS Genet. 2019, 15, e1008493. [Google Scholar] [CrossRef] [Green Version]
- Slieman, T.A.; Nicholson, W.L. Artificial and Solar UV Radiation Induces Strand Breaks and Cyclobutane Pyrimidine Dimers in Bacillus subtilis Spore DNA. Appl. Environ. Microbiol. 2000, 66, 199–205. [Google Scholar] [CrossRef] [Green Version]
- Smith, R.C.; Prézelin, B.B.; Baker, K.S.; Bidigare, R.R.; Boucher, N.P.; Coley, T.; Karentz, D.; MacIntyre, S.; Matlick, H.A.; Menzies, D.; et al. Ozone Depletion: Ultraviolet Radiation and Phytoplankton Biology in Antarctic Waters. Science 1992, 255, 952–959. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Liu, J.; Linderholm, H.W.; Chen, D.; Yu, Q.; Wu, D.; Haginoya, S. Observation and calculation of the solar radiation on the Tibetan Plateau. Energy Convers. Manag. 2012, 57, 23–32. [Google Scholar] [CrossRef]
- Su, Z.; Wilson, B.; Kumar, P.; Dutta, A. Noncanonical Roles of tRNAs: tRNA Fragments and Beyond. Annu. Rev. Genet. 2020, 54, 47–69. [Google Scholar] [CrossRef]
- Raina, M.; Ibba, M. tRNAs as regulators of biological processes. Front. Genet. 2014, 5, 171. [Google Scholar] [CrossRef] [Green Version]
- Torrent, M.; Chalancon, G.; de Groot, N.S.; Wuster, A.; Babu, M.M. Cells alter their tRNA abundance to selectively regulate protein synthesis during stress conditions. Sci. Signal. 2018, 11, eaat6409. [Google Scholar] [CrossRef] [Green Version]
- Dutta, A.; Chaudhuri, K. Analysis of tRNA composition and folding in psychrophilic, mesophilic and thermophilic genomes: Indications for thermal adaptation. FEMS Microbiol. Lett. 2010, 305, 100–108. [Google Scholar] [CrossRef] [PubMed]
- Kanaya, S.; Yamada, Y.; Kudo, Y.; Ikemura, T. Studies of codon usage and tRNA genes of 18 unicellular organisms and quantification of Bacillus subtilis tRNAs: Gene expression level and species-specific diversity of codon usage based on multivariate analysis. Gene 1999, 238, 143–155. [Google Scholar] [CrossRef]
- Satapathy, S.S.; Dutta, M.; Ray, S.K. Higher tRNA diversity in thermophilic bacteria: A possible adaptation to growth at high temperature. Microbiol. Res. 2010, 165, 609–616. [Google Scholar] [CrossRef]
- Médigue, C.; Krin, E.; Pascal, G.; Barbe, V.; Bernsel, A.; Bertin, P.N.; Cheung, F.; Cruveiller, S.; D’Amico, S.; Duilio, A.; et al. Coping with cold: The genome of the versatile marine Antarctica bacterium Pseudoalteromonas haloplanktis TAC125. Genome Res. 2005, 15, 1325–1335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saunders, N.F.; Thomas, T.; Curmi, P.M.; Mattick, J.S.; Kuczek, E.; Slade, R.; Davis, J.; Franzmann, P.D.; Boone, D.; Rusterholtz, K.; et al. Mechanisms of Thermal Adaptation Revealed From the Genomes of the Antarctic Archaea Methanogenium frigidum and Methanococcoides burtonii. Genome Res. 2003, 13, 1580–1588. [Google Scholar] [CrossRef] [Green Version]
- Getz, E.W.; Tithi, S.S.; Zhang, L.; Aylward, F.O. Parallel Evolution of Genome Streamlining and Cellular Bioenergetics across the Marine Radiation of a Bacterial Phylum. MBio. 2018, 9, e01089-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, X.; Yu, L.; Yi, Y.; Wang, J.; Wang, S.; Meng, C.; Liu, S.; Hao, Y.; Zhang, Y.; Cao, X.; et al. Phylogenomic analysis reveals a two-stage process of the evolutionary transition of Shewanella from the upper ocean to the hadal zone. Environ. Microbiol. 2021, 23, 744–756. [Google Scholar] [CrossRef] [PubMed]
- Yoshitake, S.; Uchida, M.; Koizumi, H.; Nakatsubo, T. Carbon and nitrogen limitation of soil microbial respiration in a High Arctic successional glacier foreland near Ny-Ålesund, Svalbard. Polar Res. 2007, 26, 22–30. [Google Scholar] [CrossRef]
- Vero, S.; Garmendia, G.; Martínez-Silveira, A.; Cavello, I.; Wisniewski, M. Yeast activities involved in carbon and nitrogen cycles in Antarctica. In The Ecological Role of Micro-Organisms in the Antarctic Environment; Springer: Berlin/Heidelberg, Germany, 2019; pp. 45–64. [Google Scholar]
- Hu, Y.; Wang, Z.; Wang, Q.; Wang, S.; Zhang, Z.; Zhang, Z.; Zhao, Y. Climate change affects soil labile organic carbon fractions in a Tibetan alpine meadow. J. Soils Sediments 2017, 17, 326–339. [Google Scholar] [CrossRef]
- Grzymski, J.J.; Carter, B.J.; DeLong, E.F.; Feldman, R.A.; Ghadiri, A.; Murray, A.E. Comparative Genomics of DNA Fragments from Six Antarctic Marine Planktonic Bacteria. Appl. Environ. Microbiol. 2006, 72, 1532–1541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ayala-Del-Río, H.L.; Chain, P.S.; Grzymski, J.J.; Ponder, M.A.; Ivanova, N.; Bergholz, P.W.; Di Bartolo, G.; Hauser, L.; Land, M.; Bakermans, C.; et al. The Genome Sequence of Psychrobacter arcticus 273-4, a Psychroactive Siberian Permafrost Bacterium, Reveals Mechanisms for Adaptation to Low-Temperature Growth. Appl. Environ. Microbiol. 2010, 76, 2304–2312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DasSarma, S.; Capes, M.D.; Karan, R.; DasSarma, P. Amino Acid Substitutions in Cold-Adapted Proteins from Halorubrum lacusprofundi, an Extremely Halophilic Microbe from Antarctica. PLoS ONE 2013, 8, e58587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ronholm, J.; Raymond-Bouchard, I.; Creskey, M.; Cyr, T.; Cloutis, E.A.; Whyte, L.G. Characterizing the surface-exposed proteome of Planococcus halocryophilus during cryophilic growth. Extremophiles 2015, 19, 619–629. [Google Scholar] [CrossRef] [PubMed]
- Arpigny, J.; Lamotte, J.; Gerday, C. Molecular adaptation to cold of an Antarctic bacterial lipase. J. Mol. Catal. B: Enzym. 1997, 3, 29–35. [Google Scholar] [CrossRef]
- Zhao, J.-S.; Deng, Y.; Manno, D.; Hawari, J. Shewanella spp. Genomic Evolution for a Cold Marine Lifestyle and In-Situ Explosive Biodegradation. PLoS ONE 2010, 5, e9109. [Google Scholar] [CrossRef] [Green Version]
- Raymond-Bouchard, I.; Goordial, J.; Zolotarov, Y.; Ronholm, J.; Stromvik, M.; Bakermans, C.; Whyte, L.G. Conserved genomic and amino acid traits of cold adaptation in subzero-growing Arctic permafrost bacteria. FEMS Microbiol. Ecol. 2018, 94, fiy023. [Google Scholar] [CrossRef] [Green Version]
- Bölter, M. Effects of carbohydrates and leucine on growth of bacteria from Antarctic soils (Casey Station, Wilkes Land). Polar Biol. 1993, 13, 297–306. [Google Scholar] [CrossRef]
- Moura, A.; Savageau, M.A.; Alves, R. Relative Amino Acid Composition Signatures of Organisms and Environments. PLoS ONE 2013, 8, e77319. [Google Scholar] [CrossRef] [Green Version]
- Dutton, R.J.; Boyd, D.; Berkmen, M.; Beckwith, J. Bacterial species exhibit diversity in their mechanisms and capacity for protein disulfide bond formation. Proc. Natl. Acad. Sci. USA 2008, 105, 11933–11938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gromiha, M.M.; Suwa, M. Structural analysis of residues involving cation-π interactions in different folding types of membrane proteins. Int. J. Biol. Macromol. 2005, 35, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Oren, A.; Gunde-Cimerman, N. Mycosporines and mycosporine-like amino acids: UV protectants or multipurpose secondary metabolites? FEMS Microbiol. Lett. 2007, 269, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Metpally, R.P.R.; Reddy, B.V.B. Comparative proteome analysis of psychrophilic versus mesophilic bacterial species: Insights into the molecular basis of cold adaptation of proteins. BMC Genom. 2009, 10, 11. [Google Scholar] [CrossRef] [Green Version]
- Mocali, S.; Chiellini, C.; Fabiani, A.; Decuzzi, S.; De Pascale, D.; Parrilli, E.; Tutino, M.L.; Perrin, E.; Bosi, E.; Fondi, M.; et al. Ecology of cold environments: New insights of bacterial metabolic adaptation through an integrated genomic-phenomic approach. Sci. Rep. 2017, 7, 839. [Google Scholar] [CrossRef] [Green Version]
- Goordial, J.; Raymond-Bouchard, I.; Zolotarov, Y.; de Bethencourt, L.; Ronholm, J.; Shapiro, N.; Woyke, T.; Stromvik, M.; Greer, C.W.; Bakermans, C.; et al. Cold adaptive traits revealed by comparative genomic analysis of the eurypsychrophile Rhodococcus sp. JG3 isolated from high elevation McMurdo Dry Valley permafrost, Antarctica. FEMS Microbiol. Ecol. 2016, 92, fiv154. [Google Scholar]
- Reyes-Lamothe, R.; Sherratt, D.J. The bacterial cell cycle, chromosome inheritance and cell growth. Nat. Rev. Genet. 2019, 17, 467–478. [Google Scholar] [CrossRef]
- Lauro, F.M.; McDougald, D.; Thomas, T.; Williams, T.J.; Egan, S.; Rice, S.; DeMaere, M.; Ting, L.; Ertan, H.; Johnson, J.; et al. The genomic basis of trophic strategy in marine bacteria. Proc. Natl. Acad. Sci. USA 2009, 106, 15527–15533. [Google Scholar] [CrossRef] [Green Version]
- Sharma, A.K.; Zhaxybayeva, O.; Papke, R.T.; Doolittle, W.F. Actinorhodopsins: Proteorhodopsin-like gene sequences found predominantly in non-marine environments. Environ. Microbiol. 2008, 10, 1039–1056. [Google Scholar] [CrossRef]
- Delong, E.F.; Béjà, O. The Light-Driven Proton Pump Proteorhodopsin Enhances Bacterial Survival during Tough Times. PLoS Biol. 2010, 8, e1000359. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.-H.; Xu, J.-L.; Hu, J.; Wang, L.-H.; Ong, S.L.; Leadbetter, J.; Zhang, L.-H. Acyl-homoserine lactone acylase from Ralstonia strain XJ12B represents a novel and potent class of quorum-quenching enzymes. Mol. Microbiol. 2003, 47, 849–860. [Google Scholar] [CrossRef] [PubMed]
- Chambers, J.R.; Liao, J.; Schurr, M.J.; Sauer, K. BrlR from P seudomonas aeruginosa is ac-di-GMP-responsive transcription factor. Mol. Microbiol. 2014, 92, 471–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medie, F.M.; Davies, G.; Drancourt, M.; Henrissat, B. Genome analyses highlight the different biological roles of cellulases. Nat. Rev. Genet. 2012, 10, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Ramasamy, K.P.; Telatin, A.; Mozzicafreddo, M.; Miceli, C.; Pucciarelli, S. Draft Genome Sequence of a New Pseudomonas sp. Strain, ef1, Associated with the Psychrophilic Antarctic Ciliate Euplotes focardii. Microbiol. Resour. Announc. 2019, 8, e00867-19. [Google Scholar] [CrossRef] [Green Version]
- Patel, K.B.; Toh, E.; Fernandez, X.B.; Hanuszkiewicz, A.; Hardy, G.G.; Brun, Y.V.; Bernards, M.A.; Valvano, M.A. Functional Characterization of UDP-Glucose:Undecaprenyl-Phosphate Glucose-1-Phosphate Transferases of Escherichia coli and Caulobacter crescentus. J. Bacteriol. 2012, 194, 2646–2657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becker, A. Challenges and perspectives in combinatorial assembly of novel exopolysaccharide biosynthesis pathways. Front. Microbiol. 2015, 6, 687. [Google Scholar] [CrossRef] [Green Version]
- John, M.S.; Nagoth, J.A.; Ramasamy, K.P.; Ballarini, P.; Mozzicafreddo, M.; Mancini, A.; Telatin, A.; Liò, P.; Giuli, G.; Natalello, A.; et al. Horizontal gene transfer and silver nanoparticles production in a new Marinomonas strain isolated from the Antarctic psychrophilic ciliate Euplotes focardii. Sci. Rep. 2020, 10, 10218. [Google Scholar] [CrossRef]
- Rönsch, H.; Krämer, R.; Morbach, S. Impact of osmotic stress on volume regulation, cytoplasmic solute composition and lysine production in Corynebacterium glutamicum MH20-22B. J. Biotechnol. 2003, 104, 87–97. [Google Scholar] [CrossRef]
- Imanaka, H.; Yamatsu, A.; Fukui, T.; Atomi, H.; Imanaka, T. Phosphoenolpyruvate synthase plays an essential role for glycolysis in the modified Embden-Meyerhof pathway in Thermococcus kodakarensis. Mol. Microbiol. 2006, 61, 898–909. [Google Scholar] [CrossRef]
- Lidbury, I.; Mausz, M.A.; Scanlan, D.; Chen, Y. Identification of dimethylamine monooxygenase in marine bacteria reveals a metabolic bottleneck in the methylated amine degradation pathway. ISME J. 2017, 11, 1592–1601. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain Name | Accession Number | Scaffold Number | Geneome Size (M) | Gene Number | GC% | rRNA Number | tRNA Number | Completeness | Contamination |
|---|---|---|---|---|---|---|---|---|---|
| Nesterenkonia sp. AY15 | JAJOYV000000000 | 20 | 2,801,805 | 2574 | 67.24 | 5 | 234 | 99.01 | 0.46 |
| Nesterenkonia sp. DZ6 | JAJOYW000000000 | 9 | 2,869,920 | 2614 | 67.2 | 5 | 360 | 99.01 | 0.57 |
| Nesterenkonia sp. F | GCA_000220985.2 | 134 | 2,809,541 | 2514 | 71.49 | 3 | 93 | 98.59 | 0.52 |
| Nesterenkonia alba DSM 19423 | GCA_000421745.1 | 36 | 2,591,866 | 2384 | 63.75 | 6 | 140 | 98.38 | 0 |
| Nesterenkonia massiliensis NP1 | GCA_000455245.1 | 19 | 2,672,431 | 2550 | 62.16 | 3 | 261 | 97.73 | 0.46 |
| Nesterenkonia sp. AN1 | GCA_000582475.1 | 42 | 3,040,130 | 2932 | 67.42 | 3 | 167 | 97.15 | 0.69 |
| Nesterenkonia jeotgali strain CD08_7 | GCA_001483765.1 | 8 | 2,925,195 | 2715 | 67.63 | 3 | 364 | 98.51 | 0.98 |
| Nesterenkonia sp. PF2B19 | GCA_001758425.2 | 134 | 3,696,919 | 3701 | 69.49 | 5 | 91 | 96.95 | 0.07 |
| Nesterenkonia sandarakina strain CG 35 | GCA_003003175.1 | 56 | 3,224,976 | 3001 | 67.46 | 9 | 124 | 99.39 | 0.46 |
| Nesterenkonia natronophila strain M8 | GCA_003595215.1 | 5 | 2,520,774 | 2363 | 61.82 | 3 | 402 | 98.34 | 0.07 |
| Nesterenkonia muleiensis strain RB2 | GCA_003600155.1 | 57 | 3,676,111 | 3493 | 63.55 | 2 | 95 | 97.79 | 0.86 |
| Nesterenkonia salmonea strain GY074 | GCA_005771525.1 | 110 | 3,267,177 | 3175 | 61.13 | 3 | 114 | 99.16 | 0.34 |
| Nesterenkonia sphaerica strain GY239 | GCA_005771565.1 | 52 | 2,770,794 | 2633 | 64.28 | 4 | 143 | 99.03 | 1.41 |
| Nesterenkonia sp. NBAIMH1 | GCA_007922635.1 | 1 | 2,691,978 | 2605 | 66.41 | 6 | 1128 | 97.94 | 0.18 |
| Nesterenkonia populi strain DSM 27959 | GCA_007994735.1 | 2 | 2,551,278 | 2414 | 66.85 | 6 | 1177 | 98.17 | 0.88 |
| Nesterenkonia sp. MD2 | GCA_008711175.1 | 41 | 3,733,063 | 3593 | 63.15 | 5 | 142 | 98.41 | 1.41 |
| Nesterenkonia alkaliphila strain F10 | GCA_009758175.1 | 103 | 3,318,774 | 3105 | 64.83 | 2 | 66 | 98.85 | 1.43 |
| Nesterenkonia haasae strain Hz 6-5 | GCA_010119385.1 | 29 | 3,422,101 | 3258 | 60.8 | 7 | 193 | 99.16 | 1.21 |
| Nesterenkonia sp. MY13 | GCA_012641515.1 | 41 | 3,101,056 | 2965 | 63.07 | 3 | 144 | 98.8 | 1.68 |
| Nesterenkonia sandarakina strain DSM 15664 | GCA_013410215.1 | 2 | 3,017,448 | 2780 | 67.51 | 6 | 1128 | 98.58 | 1.05 |
| Nesterenkonia xinjiangensis strain DSM 15475 | GCA_013410745.1 | 1 | 3,569,370 | 3182 | 68.81 | 6 | 1225 | 99.77 | 0.61 |
| Nesterenkonia jeotgali strain DSM 19081 | GCA_014138825.1 | 1 | 3,002,985 | 2767 | 67.44 | 6 | 1275 | 98.51 | 3.28 |
| Nesterenkonia alkaliphila CGMCC 1 | GCA_014639295.1 | 81 | 3,386,621 | 3181 | 64.79 | 4 | 103 | 98.85 | 1.43 |
| Nesterenkonia cremea CGMCC 1 | GCA_014642675.1 | 37 | 3,082,200 | 2850 | 66.86 | 5 | 167 | 99.56 | 0.88 |
| Nesterenkonia lutea strain DSM 15666 | GCA_014873955.1 | 2 | 2,958,123 | 2702 | 66.73 | 6 | 1128 | 99.54 | 0.07 |
| Nesterenkonia halotolerans strain DSM 15474 | GCA_014874065.1 | 3 | 2,966,101 | 2742 | 66.24 | 6 | 648 | 99.16 | 1.28 |
| Nesterenkonia sp. E16_7 | GCA_017347075.1 | 82 | 3,294,162 | 3074 | 67.28 | 3 | 111 | 98.88 | 1.44 |
| Nesterenkonia sp. E16_10 | GCA_017347085.1 | 49 | 3,295,232 | 3071 | 67.28 | 3 | 122 | 98.42 | 1.44 |
| Nesterenkonia lacusekhoensis strain DSM 12544 | GCA_017876395.1 | 2 | 2,742,649 | 2662 | 66.68 | 6 | 1486 | 100 | 0.99 |
| Nesterenkonia sp. Act20 | GCA_019173455.1 | 2 | 2,930,097 | 2732 | 65.93 | 7 | 697 | 99.58 | 0.75 |
| Nesterenkonia massiliensis MGYG-HGUT-01450 | GCA_902375145.1 | 19 | 2,672,431 | 2550 | 62.16 | 3 | 261 | 97.73 | 0.46 |
| Nesterenkonia sp. LB17 | JAJOYX000000000 | 10 | 2,819,602 | 2569 | 67.19 | 4 | 297 | 99.01 | 0 |
| Nesterenkonia aurantiaca strain DSM 27373 | GCA_004364585.1 | 25 | 2,948,026 | 2704 | 67.58 | 4 | 186 | 99.11 | 0 |
| Nesterenkonia sp. YGD6 | JAJOYY000000000 | 11 | 2,853,887 | 2592 | 67.12 | 3 | 250 | 99.01 | 0 |
| Function Class | COG Function | Enrichment Score | Adjusted q Value | Enriched Groups | Accession |
| Energy production | Bacteriorhodopsin | 37 | 0 | AT | COG5524 |
| Transcriptional regulators | Penicillin V acylase or related amidase, Ntn superfamily (YxeI) | 37 | 0 | AT | COG3049 |
| GyrI-like small molecule binding domain (BltR2) | 31.7525 | 0 | AT | COG4978 | |
| HD superfamily phosphodieaserase, includes HD domain of RNase Y (RnaY) | 16.3079 | 0.0061 | AT | COG1418 | |
| Polysaccharides metabolism | Cellulase/cellobiase CelA1 (CelA1) | 27.5549 | 0.0001 | AT | COG5297 |
| O-antigen ligase (RfaL) | 24.1298 | 0.0004 | AT | COG3307 | |
| Phosphoglycerol transferase MdoB/OpgB, AlkP superfamily (MdoB) | 14.9693 | 0.0089 | AT | COG1368 | |
| Glycolysis | Phosphoenolpyruvate synthase/pyruvate phosphate dikinase (PpsA) | 18.8444 | 0.0024 | AT | COG0574 |
| Lysine metabolism | Saccharopine dehydrogenase, NADP-dependent (Lys9) | 18.8444 | 0.0024 | AT | COG1748 |
| Ion transporters | H+/Cl− antiporter ClcA (ClcA) | 18.8444 | 0.0024 | AT | COG0038 |
| Mg2+ and Co2+ transporter CorA (CorA) | 14.9693 | 0.0089 | AT | COG0598 | |
| Electron transfer chain | Flavodoxin (FldA) | 18.8444 | 0.0024 | AT | COG0716 |
| Flavodoxin/ferredoxin-NADP reductase (Fpr) | 18.8444 | 0.0024 | AT | COG1018 | |
| Fe-S cluster carrier ATPase, Mrp/ApbC/NBP35 family (Mrp) | 14.9693 | 0.0089 | AT | COG0489 | |
| Cell motility | Flagellar motor protein MotB (MotB) | 18.8444 | 0.0024 | other | COG1360 |
| Cell surface structure | Sialic acid synthase SpsE, contains C-terminal SAF domain (SpsE) | 14.9693 | 0.0089 | other | COG2089 |
| CDP-glycerol glycerophosphotransferase, TagB/SpsB family | 14.9693 | 0.0089 | other | COG1887 | |
| Murein tripeptide amidase MpaA (MpaA) | 17.1582 | 0.0054 | AT | COG2866 | |
| Thiol:disulfide interchange protein DsbD (DsbD) | 16.7683 | 0.0058 | AT | COG4232 | |
| Unknown | Uncharacterized conserved protein YchJ, contains N- and C-terminal SEC-C domains (YchJ) | 14.9693 | 0.0089 | AT | COG3012 |
| Uncharacterized membrane protein YccF, DUF307 family (YccF) | 14.9693 | 0.0089 | AT | COG3304 | |
| Predicted peptidase | 16.7683 | 0.0058 | AT | COG4099 | |
| Function Class | KOfam | Enrichment Score | Adjusted q Value | Enriched Groups | Accession |
| Polysaccharides metabolism | exopolysaccharide production protein ExoQ | 31.7525 | 0 | AT | K16567 |
| Lysine metabolism | saccharopine dehydrogenase (NAD+, L-lysine forming) | 31.7525 | 0 | AT | K00290 |
| Glycolysis | pyruvate, water dikinase | 21.2667 | 0.0012 | AT | K01007 |
| Solute transporter | solute: Na+ symporter, SSS family | 24.1298 | 0.0006 | other | K03307 |
| ethanolamine permease | 14.9693 | 0.0095 | AT | K16238 | |
| putative amide transporter protein | 18.8444 | 0.0029 | AT | K22112 | |
| Dimethylamine oxidation | dimethylamine monooxygenase subunit B | 21.2667 | 0.0012 | AT | K22343 |
| dimethylamine monooxygenase subunit C | 21.2667 | 0.0012 | AT | K22344 | |
| dimethylamine monooxygenase subunit A | 21.2667 | 0.0012 | AT | K22342 | |
| Cell surface structure | prokaryotic ubiquitin-like protein Pup | 20.925 | 0.0012 | other | K13570 |
| 3-deoxy-manno-octulosonate cytidylyltransferase (CMP-KDO synthetase) | 14.9693 | 0.0095 | other | K00979 | |
| N5-(carboxyethyl)ornithine synthase | 14.9693 | 0.0095 | AT | K00298 | |
| phosphoglycerol transferase | 14.9693 | 0.0095 | AT | K01002 | |
| Stress defense | glyoxylase I family protein | 18.8444 | 0.0029 | AT | K08234 |
| Rhamnose metabolism | rhamnulokinase | 16.7683 | 0.0073 | other | K00848 |
| Methionine biosynthesis | 5-methyltetrahydropteroyltriglutamate-homocysteinmethyltransferase | 16.7683 | 0.0073 | other | K00549 |
| Methanogenesis | formylmethanofuran dehydrogenase subunit E | 16.2576 | 0.0088 | AT | K11261 |
| Antibiotic resistance | fluoroquinolone resistance protein | 14.9693 | 0.0095 | AT | K18555 |
| Transcriptional regulators | MarR family transcriptional regulator, lower aerobic nicotinate degradation pathway regulator | 14.9693 | 0.0095 | AT | K22296 |
| Lipid metabolism | mitochondrial enoyl-[acyl-carrier protein] reductase/trans-2-enoyl-CoA reductase | 14.9693 | 0.0095 | AT | K07512 |
| 4′-phosphopantetheinyl transferase | 14.9693 | 0.0095 | AT | K06133 | |
| sterol 3beta-glucosyltransferase | 14.9693 | 0.0095 | AT | K05841 | |
| Unknown | SEC-C motif domain protein | 14.9693 | 0.0095 | AT | K09858 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dai, D.; Lu, H.; Xing, P.; Wu, Q. Comparative Genomic Analyses of the Genus Nesterenkonia Unravels the Genomic Adaptation to Polar Extreme Environments. Microorganisms 2022, 10, 233. https://doi.org/10.3390/microorganisms10020233
Dai D, Lu H, Xing P, Wu Q. Comparative Genomic Analyses of the Genus Nesterenkonia Unravels the Genomic Adaptation to Polar Extreme Environments. Microorganisms. 2022; 10(2):233. https://doi.org/10.3390/microorganisms10020233
Chicago/Turabian StyleDai, Daoxin, Huibin Lu, Peng Xing, and Qinglong Wu. 2022. "Comparative Genomic Analyses of the Genus Nesterenkonia Unravels the Genomic Adaptation to Polar Extreme Environments" Microorganisms 10, no. 2: 233. https://doi.org/10.3390/microorganisms10020233
APA StyleDai, D., Lu, H., Xing, P., & Wu, Q. (2022). Comparative Genomic Analyses of the Genus Nesterenkonia Unravels the Genomic Adaptation to Polar Extreme Environments. Microorganisms, 10(2), 233. https://doi.org/10.3390/microorganisms10020233