
Species-Specific Duplication of Surface Antigen Genes in Paramecium


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Identification of the SAg Dataset
2.2. Alignments and Phylogenetic Analyses
2.3. Prediction of Protein Characteristics and Motifs
3. Results
3.1. Identification of Putative SAgs in Different Paramecium Species
3.2. Internal Tandem Repeats Are Not Present in All SAgs
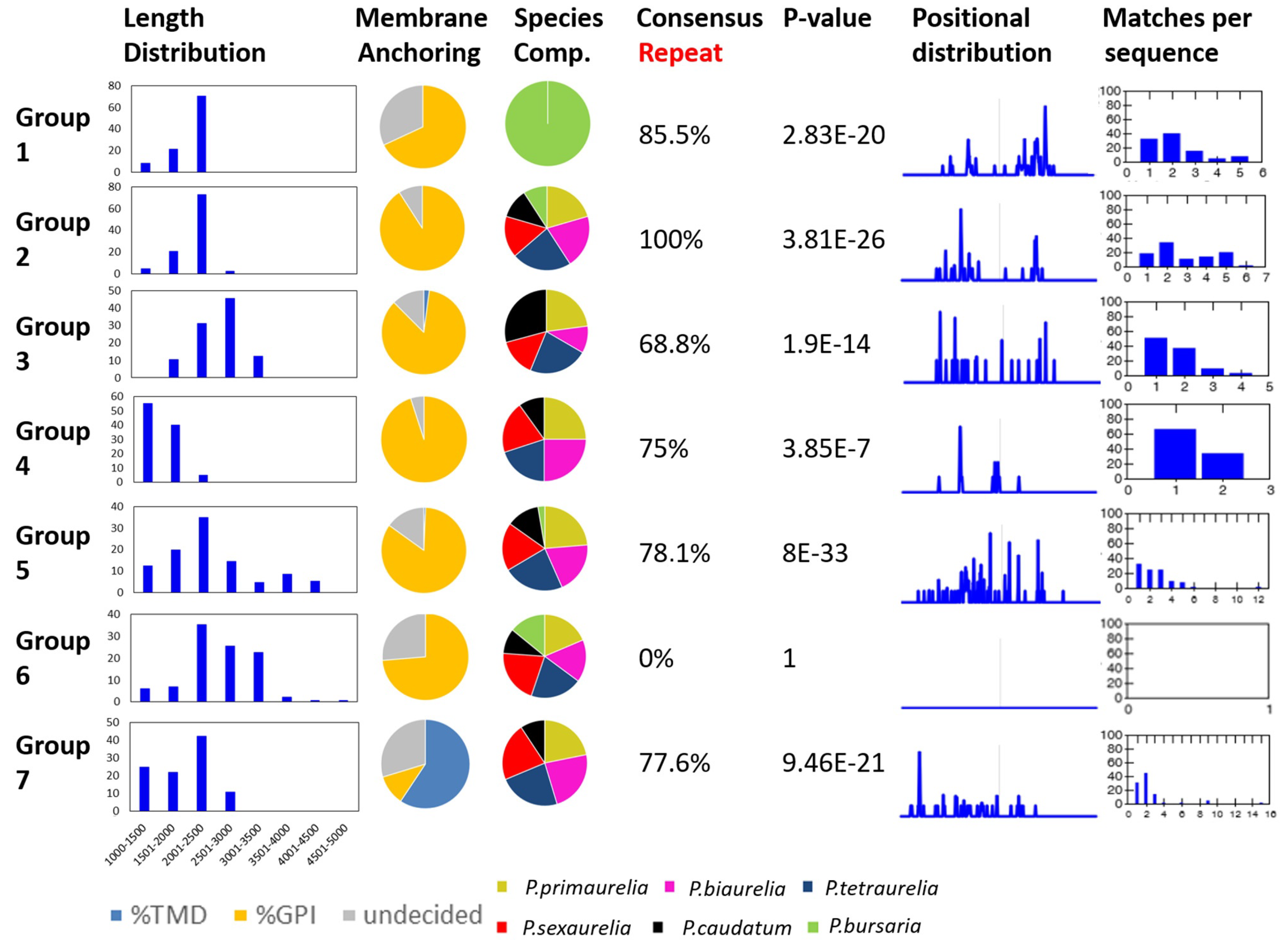
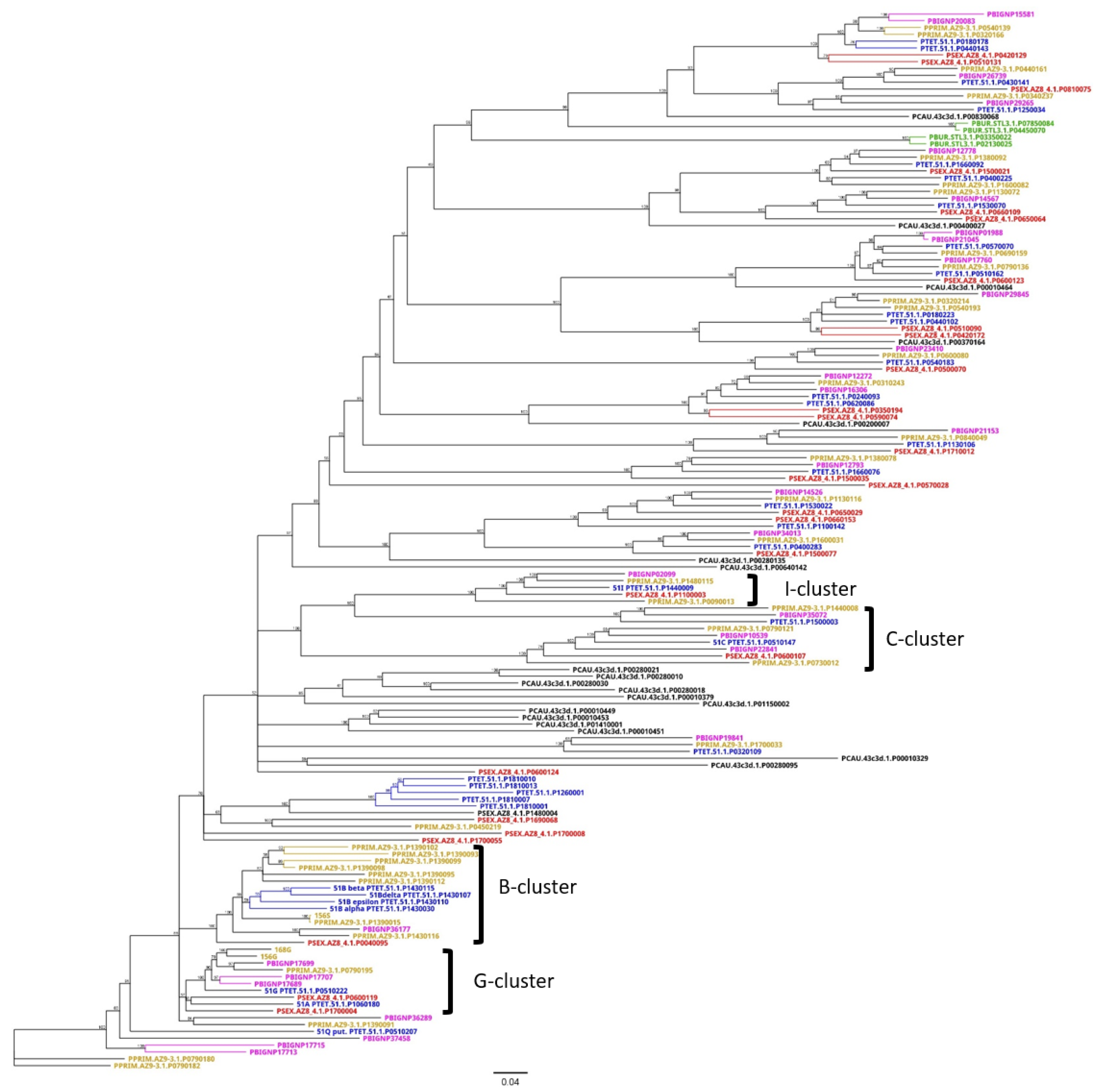
3.3. An Attempt at the Categorization of SAg Groups
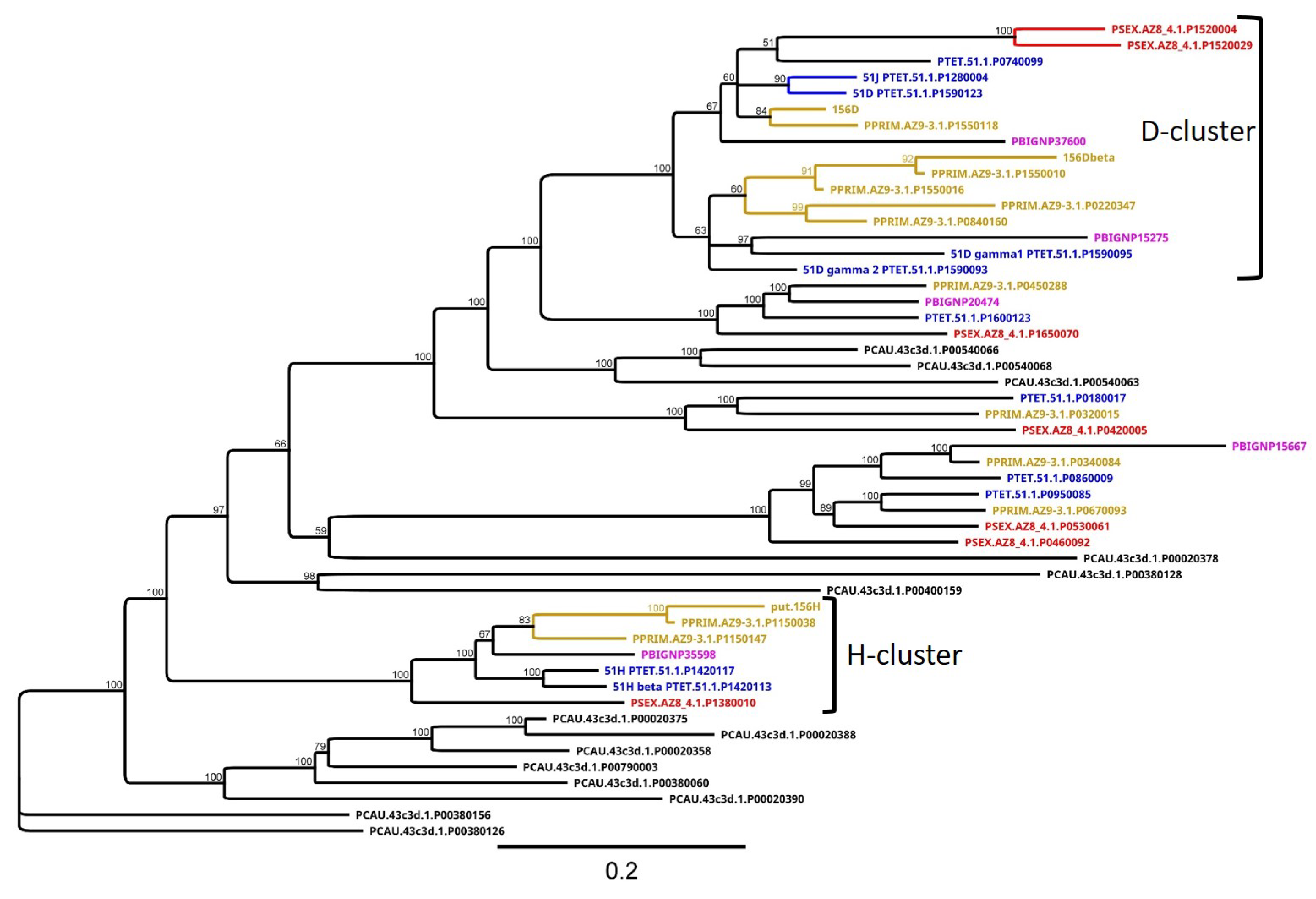
3.4. Group 3 Contains the D and H SAg Isogene Families
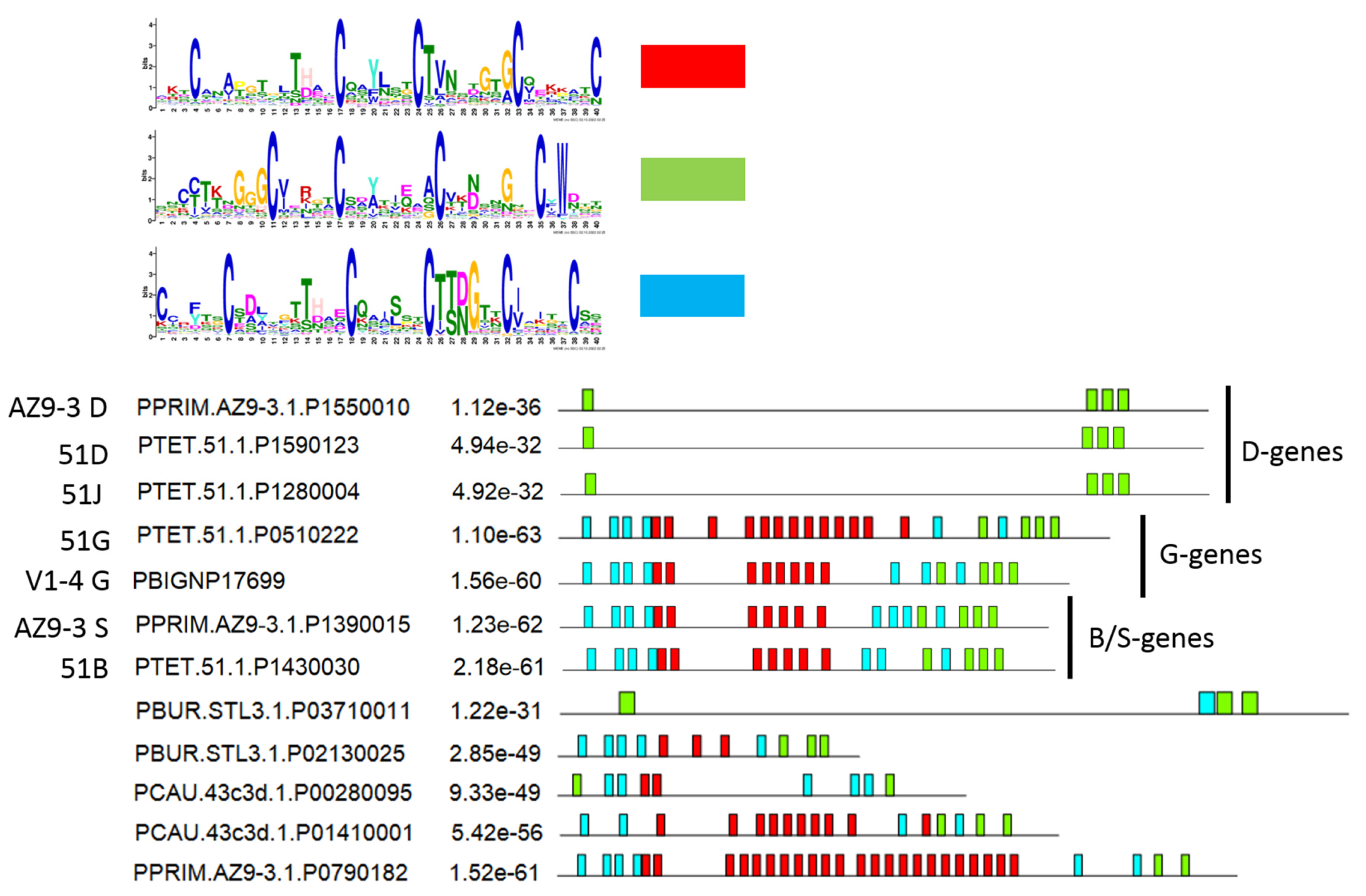
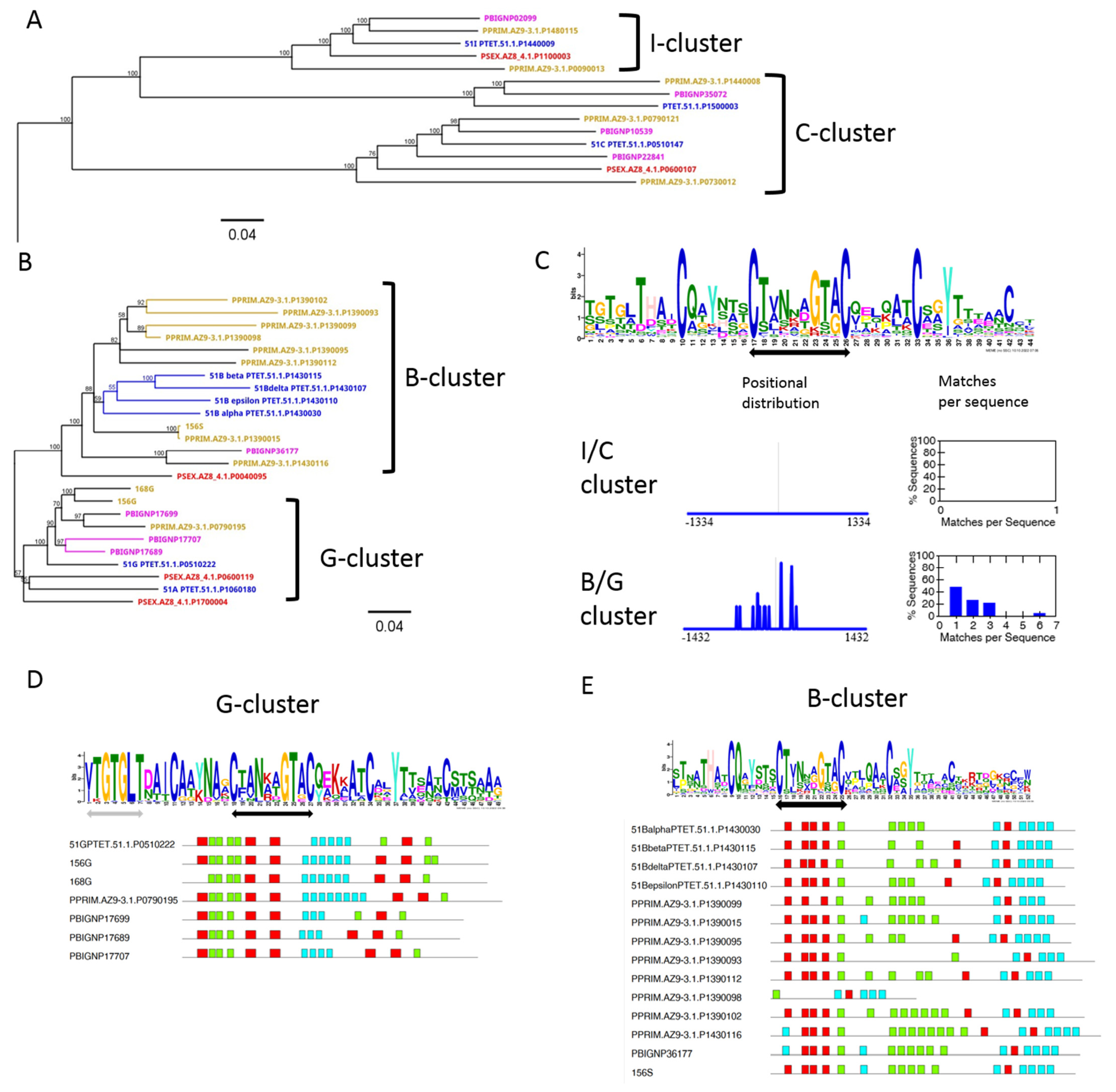
3.5. Diversification of Internal Repeats in SAg Subfamilies
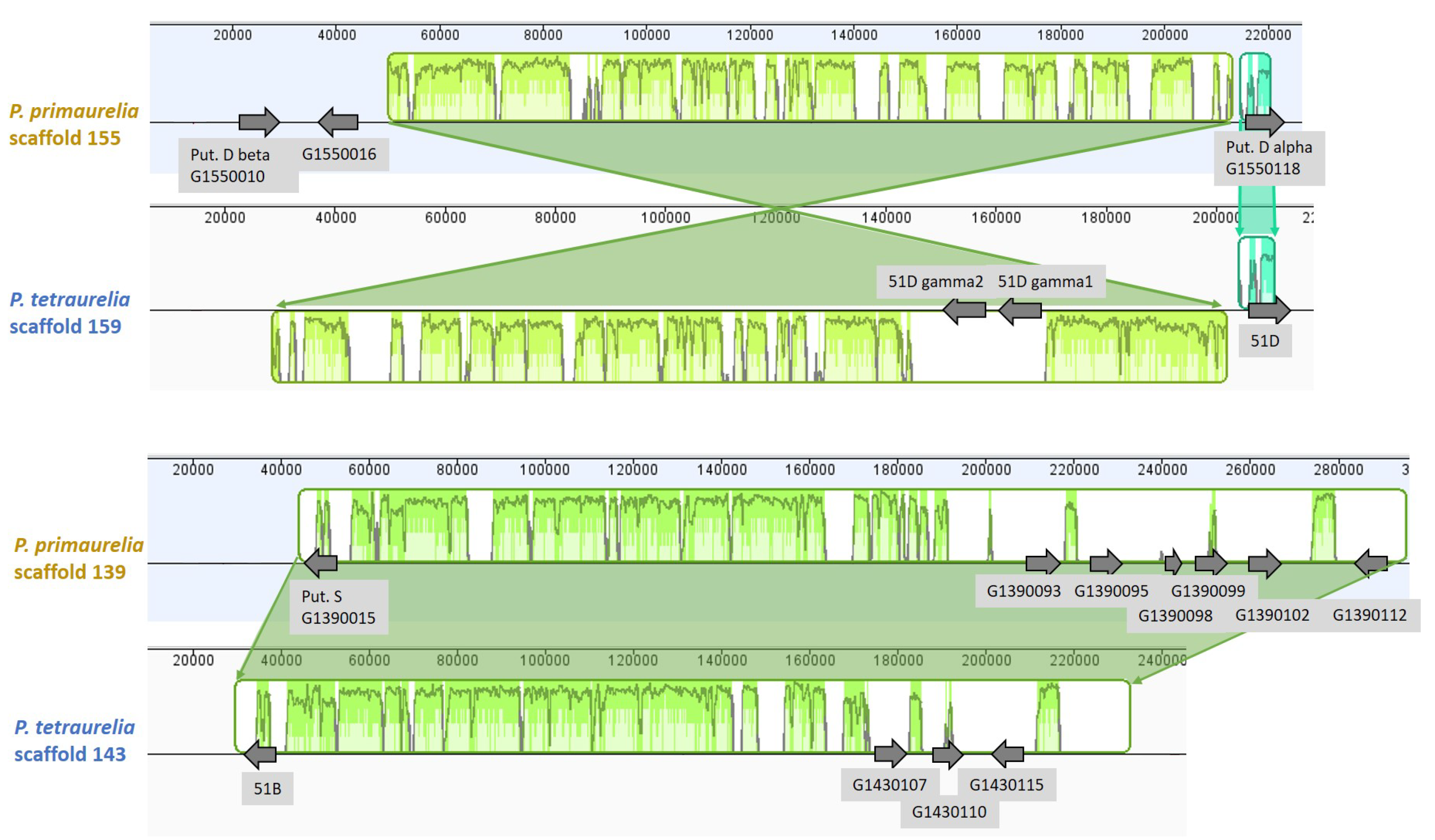
3.6. Chromosome Synteny: 156S Is the Ortholog of 51B
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A

References
- Deitsch, K.W.; Lukehart, S.A.; Stringer, J.R. Common strategies for antigenic variation by bacterial, fungal and protozoan pathogens. Nat. Rev. Microbiol. 2009, 7, 493–503. [Google Scholar] [CrossRef]
- Taylor, J.E.; Rudenko, G. Switching trypanosome coats: What’s in the wardrobe? Trends Genet. 2006, 22, 614–620. [Google Scholar] [CrossRef]
- Kaviratne, M.; Fernandez, V.; Jarra, W.; Cunningham, D.; Galinski, M.R.; Wahlgren, M.; Preiser, P.R. Antigenic variation in Plasmodium falciparum and other Plasmodium species. In Antigenic Variation; Elsevier: Amsterdam, The Netherlands, 2003; pp. 291–318. [Google Scholar]
- Deitsch, K.W.; Chitnis, C.E. Molecular basis of severe malaria. Proc. Natl. Acad. Sci. USA 2012, 109, 10130–10131. [Google Scholar] [CrossRef] [PubMed]
- Avril, M.; Tripathi, A.K.; Brazier, A.J.; Andisi, C.; Janes, J.H.; Soma, V.L.; Sullivan, D.J., Jr.; Bull, P.C.; Stins, M.F.; Smith, J.D. A restricted subset of var genes mediates adherence of Plasmodium falciparum-infected erythrocytes to brain endothelial cells. Proc. Natl. Acad. Sci. USA 2012, 109, E1782–E1790. [Google Scholar] [CrossRef] [PubMed]
- Claessens, A.; Adams, Y.; Ghumra, A.; Lindergard, G.; Buchan, C.C.; Andisi, C.; Bull, P.C.; Mok, S.; Gupta, A.P.; Wang, C.W.; et al. A subset of group A-like var genes encodes the malaria parasite ligands for binding to human brain endothelial cells. Proc. Natl. Acad. Sci. USA 2012, 109, E1772–E1781. [Google Scholar] [CrossRef] [PubMed]
- Lavstsen, T.; Turner, L.; Saguti, F.; Magistrado, P.; Rask, T.S.; Jespersen, J.S.; Wang, C.W.; Berger, S.S.; Baraka, V.; Marquard, A.M.; et al. Plasmodium falciparum erythrocyte membrane protein 1 domain cassettes 8 and 13 are associated with severe malaria in children. Proc. Natl. Acad. Sci. USA 2012, 109, E1791–E1800. [Google Scholar] [CrossRef] [PubMed]
- Simon, M.C.; Schmidt, H.J. Antigenic Variation in Ciliates: Antigen Structure, Function, Expression 1. J. Eukaryot. Microbiol. 2007, 54, 1–7. [Google Scholar] [CrossRef]
- Simon, M.; Plattner, H. Unicellular eukaryotes as models in cell and molecular biology: Critical appraisal of their past and future value. Int. Rev. Cell Mol. Biol. 2014, 309, 141–198. [Google Scholar]
- Marker, S.; Le Mouel, A.; Meyer, E.; Simon, M. Distinct RNA-dependent RNA polymerases are required for RNAi triggered by double-stranded RNA versus truncated transgenes in Paramecium tetraurelia. Nucleic Acids Res. 2010, 38, 4092–4107. [Google Scholar] [CrossRef]
- Karunanithi, S.; Oruganti, V.; Marker, S.; Rodriguez-Viana, A.M.; Drews, F.; Pirritano, M.; Nordström, K.; Simon, M.; Schulz, M.H. Exogenous RNAi mechanisms contribute to transcriptome adaptation by phased siRNA clusters in Paramecium. Nucleic Acids Res. 2019, 47, 8036–8049. [Google Scholar] [CrossRef]
- Capdeville, Y.; de Almeida, M.L.C.; Deregnaucourt, C. The membrane-anchor of Paramecium temperature-specific surface antigens is a glycosylinositol phospholipid. Biochem. Biophys. Res. Commun. 1987, 147, 1219–1225. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, E.; You, Y.; Forney, J. Cysteine residue periodicity is a conserved structural feature of variable surface proteins from Paramecium tetraurelia. J. Mol. Biol. 1991, 222, 835–841. [Google Scholar] [CrossRef] [PubMed]
- Prat, A. Conserved sequences flank variable tandem repeats in two alleles of the G surface protein of Paramecium primaurelia. J. Mol. Biol. 1990, 211, 521–535. [Google Scholar] [CrossRef] [PubMed]
- Capdeville, Y.; Deregnaucourt, C.; Keller, A.M. Surface antigens of Paramecium primaurelia: Membrane-bound and soluble forms. Exp. Cell Res. 1985, 161, 495–508. [Google Scholar] [CrossRef] [PubMed]
- Meyer, E.; Caron, F.; Baroin, A. Macronuclear structure of the G surface antigen gene of Paramecium primaurelia and direct expression of its repeated epitopes in Escherichia coli. Mol. Cell. Biol. 1985, 5, 2414–2422. [Google Scholar]
- Simon, M.C.; Kusch, J. Communicative functions of GPI-anchored surface proteins in unicellular eukaryotes. Crit. Rev. Microbiol. 2013, 39, 70–78. [Google Scholar] [CrossRef]
- Harumoto, T.; Miyake, A. Possible participation of surface antigens of Paramecium in predator-prey interaction. J. Eukaryot. Microbiol. 1993, 40, 27A. [Google Scholar]
- Klöppel, C.; Müller, A.; Marker, S.; Simon, M. Two isoforms of eukaryotic phospholipase C in Paramecium affecting transport and release of GPI-anchored proteins in vivo. Eur. J. Cell Biol. 2009, 88, 577–592. [Google Scholar] [CrossRef]
- Müller, A.; Klöppel, C.; Smith-Valentine, M.; Van Houten, J.; Simon, M. Selective and programmed cleavage of GPI-anchored proteins from the surface membrane by phospholipase C. Biochim. Biophys. Acta BBA Biomembr. 2012, 1818, 117–124. [Google Scholar] [CrossRef]
- Staudt, E.; Ramasamy, P.; Plattner, H.; Simon, M. Differential subcellular distribution of four phospholipase C isoforms and secretion of GPI-PLC activity. Biochim. Biophys. Acta BBA Biomembr. 2016, 1858, 3157–3168. [Google Scholar] [CrossRef]
- Preer, J.R., Jr. Surface antigens of Paramecium. In The Molecular Biology of Ciliated Protozoa; Gall, J., Ed.; Elsevier: New York, NY, USA, 1986; pp. 301–336. [Google Scholar]
- Tenaglia, A.H.; Luján, L.A.; Ríos, D.N.; Midlej, V.; Iribarren, P.A.; Molina, C.R.; Berazategui, M.A.; Torri, A.; Saura, A.; Peralta, D.O.; et al. Antibodies to protozoan variable surface antigens induce antigenic variation. bioRxiv 2022. [Google Scholar] [CrossRef]
- Paquette, C.; Villa, R.; Bush, A.; van Houten, J.L. Glycophosphatidylinositol-anchored proteins in Paramecium tetraurelia: Possible role in chemoresponse. J. Exp. Biol. 2001, 204, 2899–2910. [Google Scholar] [CrossRef] [PubMed]
- Aury, J.M.; Jaillon, O.; Duret, L.; Noel, B.; Jubin, C.; Porcel, B.M.; Ségurens, B.; Daubin, V.; Anthouard, V.; Aiach, N.; et al. Global trends of whole-genome duplications revealed by the ciliate Paramecium tetraurelia. Nature 2006, 444, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Baranasic, D.; Oppermann, T.; Cheaib, M.; Cullum, J.; Schmidt, H.; Simon, M. Genomic characterization of variable surface antigens reveals a telomere position effect as a prerequisite for RNA interference-mediated silencing in Paramecium tetraurelia. MBio 2014, 5, e01328-14. [Google Scholar] [CrossRef]
- Breuer, M.; Schulte, G.; Sschwegmann, K.J.; Schmidt, H.J. Molecular characterization of the D surface protein gene subfamily in Paramecium tetraurelia. J. Eukaryot. Microbiol. 1996, 43, 314–322. [Google Scholar] [CrossRef]
- Bourgain-Guglielmetti, F.M.; Caron, F.M. Molecular characterization of the D surface protein gene subfamily in Paramecium primaurelia. J. Eukaryot. Microbiol. 1996, 43, 303–313. [Google Scholar] [CrossRef]
- Simon, M.C.; Schmidt, H.J. Variety of serotypes of Paramecium primaurelia: Single epitopes are responsible for immunological differentiation. J. Eukaryot. Microbiol. 2005, 52, 319–327. [Google Scholar] [CrossRef]
- Preer, J.; Preer, L.; Rudman, B.; Barnett, A. Deviation from the universal code shown by the gene for surface protein 51A in Paramecium. Nature 1985, 314, 188–190. [Google Scholar] [CrossRef]
- Scott, J.; Leeck, C.; Forney, J. Molecular and genetic analyses of the B type surface protein gene from Paramecium tetraurelia. Genetics 1993, 134, 189–198. [Google Scholar] [CrossRef]
- Duharcourt, S.; Keller, A.M.; Meyer, E. Homology-dependent maternal inhibition of developmental excision of internal eliminated sequences in Paramecium tetraurelia. Mol. Cell. Biol. 1998, 18, 7075–7085. [Google Scholar] [CrossRef]
- Godiska, R. Structure and sequence of the H surface protein gene of Paramecium and comparison with related genes. Mol. Gen. Genet. 1987, 208, 529–536. [Google Scholar] [CrossRef] [PubMed]
- Arnaiz, O.; Meyer, E.; Sperling, L. Paramecium DB 2019: Integrating genomic data across the genus for functional and evolutionary biology. Nucleic Acids Res. 2020, 48, D599–D605. [Google Scholar] [PubMed]
- Dereeper, A.; Guignon, V.; Blanc, G.; Audic, S.; Buffet, S.; Chevenet, F.; Dufayard, J.F.; Guindon, S.; Lefort, V.; Lescot, M.; et al. Phylogeny. fr: Robust phylogenetic analysis for the non-specialist. Nucleic Acids Res. 2008, 36, W465–W469. [Google Scholar] [CrossRef] [PubMed]
- Darling, A.C.; Mau, B.; Blattner, F.R.; Perna, N.T. Mauve: Multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 2004, 14, 1394–1403. [Google Scholar] [CrossRef]
- Krogh, A.; Larsson, B.; Von Heijne, G.; Sonnhammer, E.L. Predicting transmembrane protein topology with a hidden Markov model: Application to complete genomes. J. Mol. Biol. 2001, 305, 567–580. [Google Scholar] [CrossRef] [PubMed]
- Fankhauser, N.; Mäser, P. Identification of GPI anchor attachment signals by a Kohonen self-organizing map. Bioinformatics 2005, 21, 1846–1852. [Google Scholar] [CrossRef] [PubMed]
- Bailey, T.L.; Johnson, J.; Grant, C.E.; Noble, W.S. The MEME suite. Nucleic Acids Res. 2015, 43, W39–W49. [Google Scholar] [CrossRef]
- Bailey, T.L.; Elkan, C. Fitting a mixture model by expectation maximization to discover motifs in bipolymers. Proc. Int. Conf. Intell. Syst. Mol. Biol. 1994, 2, 28–36. [Google Scholar]
- Bailey, T.L.; Grant, C.E. SEA: Simple Enrichment Analysis of motifs. bioRxiv 2021. [Google Scholar] [CrossRef]
- Przybos, E.; Rautian, M.; Potekhin, A. First European record of Paramecium septaurelia and the discovery of new European habitats of P. pentaurelia and P. sexaurelia in Russia (Astrakhan and Vologograd regions). Folia Biol. Krakow 2004, 52, 87–90. [Google Scholar]
- Potekhin, A.; Nekrasova, I.; Przyboś, E.; Rautian, M. Comparative description of macronuclear electrophoretic karyotypes of Paramecium primaurelia and Paramecium novaurelia sibling species. Cell Tissue Biol. 2008, 2, 614–624. [Google Scholar] [CrossRef]
- Nekrasova, I.V.; Przyboś, E.; Rautian, M.S.; Potekhin, A.A. Electrophoretic karyotype polymorphism of sibling species of the Paramecium aurelia complex. J. Eukaryot. Microbiol. 2010, 57, 494–507. [Google Scholar] [CrossRef] [PubMed]
- Prat, A.; Katinka, M.; Caron, F.; Meyer, E. Nucleotide sequence of the Paramecium primaurelia G surface protein: A huge protein with a highly periodic structure. J. Mol. Biol. 1986, 189, 47–60. [Google Scholar] [CrossRef] [PubMed]
- Beale, G.H. The Genetics of Paramecium Aurelia; Camebridge University Press: Cambridge, UK, 1954. [Google Scholar]
- Cheng, Y.H.; Liu, C.F.J.; Yu, Y.H.; Jhou, Y.T.; Fujishima, M.; Tsai, I.J.; Leu, J.Y. Genome plasticity in Paramecium bursaria revealed by population genomics. BMC Biol. 2020, 18, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Landis, W. Ecology. In Paramecium; Spring: Berlin/Heidelberg, Germany, 1988; Volume 36, pp. 402–408. [Google Scholar]
- Drews, F.; Boenigk, J.; Simon, M. Paramecium epigenetics in development and proliferation. J. Eukaryot. Microbiol. 2022, 69, e12914. [Google Scholar] [CrossRef] [PubMed]
- Duret, L.; Cohen, J.; Jubin, C.; Dessen, P.; Goût, J.F.; Mousset, S.; Aury, J.M.; Jaillon, O.; Noël, B.; Arnaiz, O.; et al. Analysis of sequence variability in the macronuclear DNA of Paramecium tetraurelia: A somatic view of the germline. Genome Res. 2008, 18, 585–596. [Google Scholar] [CrossRef]
- Sellis, D.; Guérin, F.; Arnaiz, O.; Pett, W.; Lerat, E.; Boggetto, N.; Krenek, S.; Berendonk, T.; Couloux, A.; Aury, J.M.; et al. Massive colonization of protein-coding exons by selfish genetic elements in Paramecium germline genomes. PLoS Biol. 2021, 19, e3001309. [Google Scholar] [CrossRef]
- Dubrana, K.; Amar, L. Programmed DNA under-amplification in Paramecium primaurelia. Chromosoma 2000, 109, 460–466. [Google Scholar] [CrossRef]
- Forney, J.D.; Blackburn, E.H. Developmentally controlled telomere addition in wild-type and mutant paramecia. Mol. Cell. Biol. 1988, 8, 251–258. [Google Scholar]
- Epstein, L.M.; Forney, J.D. Mendelian and non-Mendelian mutations affecting surface antigen expression in Paramecium tetraurelia. Mol. Cell. Biol. 1984, 4, 1583–1590. [Google Scholar]
- Phan, H.L.; Forney, J.; Blackburn, E.H. Analysis of Paramecium macronuclear DNA using pulsed field gel electrophoresis. J. Protozool. 1989, 36, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Capdeville, Y.; Caron, F.; Antony, C.; Deregnaucourt, C.; Keller, A.M. Allelic antigen and membrane-anchor epitopes of Paramecium primaurelia surface antigens. J. Cell Sci. 1987, 88, 553–562. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pirritano, M.; Yakovleva, Y.; Potekhin, A.; Simon, M. Species-Specific Duplication of Surface Antigen Genes in Paramecium. Microorganisms 2022, 10, 2378. https://doi.org/10.3390/microorganisms10122378
Pirritano M, Yakovleva Y, Potekhin A, Simon M. Species-Specific Duplication of Surface Antigen Genes in Paramecium. Microorganisms. 2022; 10(12):2378. https://doi.org/10.3390/microorganisms10122378
Chicago/Turabian StylePirritano, Marcello, Yulia Yakovleva, Alexey Potekhin, and Martin Simon. 2022. "Species-Specific Duplication of Surface Antigen Genes in Paramecium" Microorganisms 10, no. 12: 2378. https://doi.org/10.3390/microorganisms10122378
APA StylePirritano, M., Yakovleva, Y., Potekhin, A., & Simon, M. (2022). Species-Specific Duplication of Surface Antigen Genes in Paramecium. Microorganisms, 10(12), 2378. https://doi.org/10.3390/microorganisms10122378