
Real-Time PCR Protocol for Detection and Quantification of Three Pathogenic Members of the Vibrionaceae Family



Abstract
1. Introduction
2. Materials and Methods
2.1. Absolute Microbial Enumeration
2.2. DNA Extraction Methods
2.3. Primer Pair Design and PCR Conditions Optimisation
2.4. Absolute qPCR Calibration Curves
2.5. The Influence of a Complex Matrix in Quantification of Artificially Spiked Microalgae Samples
3. Results
3.1. The Standard Curves
3.2. The qPCR Efficiency (E%)
3.3. The Influence of Complex Matrices
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Parte, A.C.; Carbasse, J.S.; Meier-Kolthoff, J.P.; Reimer, L.C.; Göker, M. List of Prokaryotic names with Standing in Nomenclature (LPSN) moves to the DSMZ. Int. J. Syst. Evol. Microbiol. 2020, 70, 5607–5612. [Google Scholar] [CrossRef]
- Gomez-Gil, B.; Thompson, C.; Matsumura, Y.; Sawabe, T.; Iida, T.; Christen, R.; Thompson, F.; Sawabe, T. The family Vibrionaceae. In The prokaryotes—Gammaproteobacteria; Springer: Berlin/Heidelberg, Germany, 2014; Volume 9, pp. 659–747. [Google Scholar]
- Baker-Austin, C.; Trinanes, J.; Gonzalez-Escalona, N.; Martinez-Urtaza, J. Non-Cholera Vibrios: The Microbial Barometer of Climate Change. Trends Microbiol. 2017, 25, 76–84. [Google Scholar] [CrossRef]
- Damir, K.; Irena, V.S.; Damir, V.; Emin, T. Occurrence, characterization and antimicrobial susceptibility of Vibrio alginolyticus in the Eastern Adriatic Sea. Mar. Pollut. Bull. 2013, 75, 46–52. [Google Scholar] [CrossRef]
- Thompson, F.L.; Lida, T.; Swings, S. Biodiversity of Vibrios. MMBR 2004, 68, 403–431. [Google Scholar] [CrossRef]
- Austin, B. Vibrios as causal agents of zoonoses. Vet. Microbiol. 2010, 140, 310–317. [Google Scholar] [CrossRef]
- Robert-Pillot, A.; Copin, S.; Himber, C.; Gay, M.; Quilici, M.-L. Occurrence of the three major Vibrio species pathogenic for human in seafood products consumed in France using real-time PCR. Int. J. Food Microbiol. 2014, 189, 75–81. [Google Scholar] [CrossRef]
- Baker-Austin, C.; Oliver, J.D.; Alam, M.; Ali, A.; Waldor, M.K.; Qadri, F.; Martinez-Urtaza, J. Vibrio spp. infections. Nat. Rev. Dis. Primers 2018, 4, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Finkelstein, R.; Edelstein, S.; Mahamid, G. Fulminant wound infections due to Vibrio vulnificus. Isr. Med. Assoc. J. 2002, 4, 654–655. [Google Scholar]
- CDC. Chapter 6—Isolation and Identification of Vibrio cholerae. In Laboratory Methods for the Diagnosis of Epidemic Dysentery and Cholera.; CDC: Atlanta, GA, USA, 1999. [Google Scholar]
- Borrego, J.J.; Castro, D.; Luque, A.; Paillard, C.; Maes, P.; Garcia, M.T.; Ventosa, A. Vibrio tapetis sp. nov., the Causative Agent of the Brown Ring Disease Affecting Cultured Clams. Int. J. Syst. Evol. Microbiol. 1996, 46, 480–484. [Google Scholar] [CrossRef]
- Diggles, B.K.; Carson, J.; Hine, P.M.; Hickman, R.W.; Tait, M.J. Vibrio species associated with mortalities in hatchery-reared turbot (Colistium nudipinnis) and brill (C. guntheri) in New Zealand. Aquaculture 2000, 183, 1–12. [Google Scholar] [CrossRef]
- Hansen, G.H.; Olafsen, J.A. Bacterial Interactions in Early Life Stages of Marine Cold Water Fish. Microb. Ecol. 1999, 38, 1–26. [Google Scholar] [CrossRef]
- Ishimaru, K.; Akagawa-Matsushita, M.; Muroga, K. Vibrio penaeicida sp. nov., a Pathogen of Kuruma Prawns (Penaeus japonicus). Int. J. Syst. Evol. Microbiol. 1995, 45, 134–138. [Google Scholar] [CrossRef]
- Toranzo, A.E.; Magariños, B.; Avendaño-Herrera, R. Vibriosis: Vibrio anguillarum, V. ordalii and Aliivibrio salmonicida. In Fish Viruses and Bacteria: Pathobiology and Protection; Woo, P.T., Cipriano, R.C., Eds.; CABI: Bikaner, India, 2017; pp. 314–333. [Google Scholar]
- Frans, I.; Michiels, C.W.; Bossier, P.; Willems, K.; Lievens, B.; Rediers, H. Vibrio anguillarum as a fish pathogen: Virulence factors, diagnosis and prevention. J. Fish Dis. 2011, 34, 643–661. [Google Scholar] [CrossRef]
- Prayitno, S.; Latchford, J. Experimental infections of crustaceans with luminous bacteria related to Photobacterium and Vibrio. Effect of salinity and pH on infectiosity. Aquaculture 1995, 132, 105–112. [Google Scholar] [CrossRef]
- Zhang, X.-H.; He, X.; Austin, B. Vibrio harveyi: A serious pathogen of fish and invertebrates in mariculture. Mar. Life Sci. Technol. 2020, 2, 231–245. [Google Scholar] [CrossRef]
- Balebona, M.C.; Andreu, M.J.; Bordas, M.A.; Zorrilla, I.; Moriñigo, M.A.; Borrego, J.J. Pathogenicity of Vibrio alginolyticus for cultured gilt-head sea bream (Sparus aurata L.). Appl. Environ. Microbiol. 1998, 64, 4269–4275. [Google Scholar] [CrossRef]
- Samad, A.P.A.; Santoso, U.; Lee, M.-C.; Nan, F.-H. Effects of dietary katuk (Sauropus androgynus L. Merr.) on growth, non-specific immune and diseases resistance against Vibrio alginolyticus infection in grouper Epinephelus coioides. Fish Shellfish Immunol. 2014, 36, 582–589. [Google Scholar] [CrossRef]
- Arunkumar, M.; LewisOscar, F.; Thajuddin, N.; Pugazhendhi, A.; Nithya, C. In vitro and in vivo biofilm forming Vibrio spp: A significant threat in aquaculture. Process Biochem. 2020, 94, 213–223. [Google Scholar] [CrossRef]
- Defoirdt, T.; Bossier, P.; Sorgeloos, P.; Verstraete, W. The impact of mutations in the quorum sensing systems of Aeromonas hydrophila, Vibrio anguillarum and Vibrio harveyi on their virulence towards gnotobiotically cultured Artemia franciscana. Environ. Microbiol. 2005, 7, 1239–1247. [Google Scholar] [CrossRef]
- Gram, L.; Løvold, T.; Nielsen, J.; Melchiorsen, J.; Spanggaard, B. In vitro antagonism of the probiont Pseudomonas fluorescens strain AH2 against Aeromonas salmonicida does not confer protection of salmon against furunculosis. Aquaculture 2001, 199, 1–11. [Google Scholar] [CrossRef]
- Mohamed, S.; Nagaraj, G.; Chua, F.H.C.; Wang, Y.G. The use of chemicals in aquaculture in Malaysia and Singapore. In Use of Chemicals in Aquaculture in Asia, Proceedings of the Meeting on the Use of Chemicals in Aquaculture in Asia, Tigbauan, Iloilo, Philippines, 20–22 May 1996; SEAFDEC: Tigbauan, Philippines, 2000; pp. 127–140. [Google Scholar]
- Duff, D.C.B. The Oral Immunization of Trout Against Bacterium Salmonicida. J. Immunol. 1942, 44, 87–94. [Google Scholar]
- Prol, M.; Bruhn, J.B.; Pintado, J.; Gram, L. Real-time PCR detection and quantification of fish probiotic Phaeobacter strain 27-4 and fish pathogenic Vibrio in microalgae, rotifer, Artemia and first feeding turbot (Psetta maxima) larvae. J. Appl. Microbiol. 2009, 106, 1292–1303. [Google Scholar] [CrossRef]
- Munro, P.D.; Birkbeck, T.H.; Barbour, A. Influence of rate of bacterial colonisation of the gut of turbot larvae on larval survival. In Fish Farming Technology; CRC Press: Boca Raton, FL, USA, 2020; pp. 85–92. [Google Scholar]
- Noble, R.T.; Weisberg, S.B. A review of technologies for rapid detection of bacteria in recreational waters. J. Water Health 2005, 3, 381–392. [Google Scholar] [CrossRef]
- Castillo, M.; Martín-Orúe, S.M.; Manzanilla, E.G.; Badiola, I.; Martín, M.; Gasa, J. Quantification of total bacteria, enterobacteria and lactobacilli populations in pig digesta by real-time PCR. Vet. Microbiol. 2006, 114, 165–170. [Google Scholar] [CrossRef]
- Zhang, Z.; Qu, Y.; Li, S.; Feng, K.; Wang, S.; Cai, W.; Liang, Y.; Li, H.; Xu, M.; Yin, H. Soil bacterial quantification approaches coupling with relative abundances reflecting the changes of taxa. Sci. Rep. 2017, 7, 4837. [Google Scholar] [CrossRef]
- Head, I.M.; Saunders, J.R.; Pickup, R.W. Microbial Evolution, Diversity, and Ecology: A Decade of Ribosomal RNA Analysis of Uncultivated Microorganisms. Microb. Ecol. 1998, 35, 1–21. [Google Scholar] [CrossRef]
- Matsuki, T.; Watanabe, K.; Tanaka, R. Genus-and species-specific PCR primers for the detection and identification of bifidobacteria. Curr. Issues Intest. Microbiol. 2003, 4, 61–69. [Google Scholar]
- Hossain, M.; Kim, E.Y.; Kim, Y.R.; Kim, D.G.; Kong, I.S. Application of groEL gene for the species-specific detection of Vibrio parahaemolyticus by PCR. Lett. Appl. Microbiol. 2012, 54, 67–72. [Google Scholar] [CrossRef]
- Paillard, C.; Gausson, S.; Nicolas, J.L.; Le Pennec, J.P.; Haras, D. Molecular identification of Vibrio tapetis, the causative agent of the brown ring disease of Ruditapes philippinarum. Aquaculture 2006, 253, 25–38. [Google Scholar] [CrossRef][Green Version]
- Pang, L.; Zhang, X.H.; Zhong, Y.; Chen, J.; Li, Y.; Austin, B. Identification of Vibrio harveyi using PCR amplification of the toxR gene. Lett. Appl. Microbiol. 2006, 43, 249–255. [Google Scholar] [CrossRef]
- Saulnier, D.; Avarre, J.C.; Le Moullac, G.; Ansquer, D.; Levy, P.; Vonau, V. Rapid and sensitive PCR detection of Vibrio penaeicida, the putative etiological agent of syndrome 93 in New Caledonia. Dis. Aquat. Organ. 2000, 40, 109–115. [Google Scholar] [CrossRef]
- Mackay, I.M.; Arden, K.E.; Nitsche, A. Real-time PCR in virology. Nucleic Acids Res. 2002, 30, 1292–1305. [Google Scholar] [CrossRef]
- Nagy, A.; Vitásková, E.; Černíková, L.; Křivda, V.; Jiřincová, H.; Sedlák, K.; Horníčková, J.; Havlíčková, M. Evaluation of TaqMan qPCR system integrating two identically labelled hydrolysis probes in single assay. Sci. Rep. 2017, 7, 41392. [Google Scholar] [CrossRef]
- Ward, L.N.; Bej, A.K. Detection of Vibrio parahaemolyticus in shellfish by use of multiplexed real-time PCR with TaqMan fluorescent probes. Appl. Environ. Microbiol. 2006, 72, 2031–2042. [Google Scholar] [CrossRef]
- Nordstrom, J.L.; Vickery, M.C.; Blackstone, G.M.; Murray, S.L.; DePaola, A. Development of a multiplex real-time PCR assay with an internal amplification control for the detection of total and pathogenic Vibrio parahaemolyticus bacteria in oysters. Appl. Environ. Microbiol. 2007, 73, 5840–5847. [Google Scholar] [CrossRef]
- Greig, D.R.; Hickey, T.J.; Boxall, M.D.; Begum, H.; Gentle, A.; Jenkins, C.; Chattaway, M.A. A real-time multiplex PCR for the identification and typing of Vibrio cholerae. Diagn. Microbiol. Infect. Dis. 2018, 90, 171–176. [Google Scholar] [CrossRef]
- Campbell, M.S.; Wright, A.C. Real-time PCR analysis of Vibrio vulnificus from oysters. Appl. Environ. Microbiol. 2003, 69, 7137–7144. [Google Scholar] [CrossRef]
- Azinheiro, S.; Ghimire, D.; Carvalho, J.; Prado, M.; Garrido-Maestu, A. Next-day detection of viable Listeria monocytogenes by multiplex reverse transcriptase real-time PCR. Food Control 2022, 133, 108593. [Google Scholar] [CrossRef]
- Lebonah, D.; Dileep, A.; Chandrasekhar, K.; Sreevani, S.; Sreedevi, B.; Pramoda Kumari, J. DNA barcoding on bacteria: A review. Adv. Biol. 2014, 2014, 541787. [Google Scholar] [CrossRef]
- Montieri, S.; Suffredini, E.; Ciccozzi, M.; Croci, L. Phylogenetic and evolutionary analysis of Vibrio parahaemolyticus and Vibrio alginolyticus isolates based on toxR gene sequence. New Microbiol. 2010, 33, 359–372. [Google Scholar]
- Blaiotta, G.; Fusco, V.; Ercolini, D.; Aponte, M.; Pepe, O.; Villani, F. Lactobacillus strain diversity based on partial hsp60 gene sequences and design of PCR-restriction fragment length polymorphism assays for species identification and differentiation. Appl. Environ. Microbiol. 2008, 74, 208–215. [Google Scholar] [CrossRef]
- Yushan, H.; Lei, L.; Weijia, L.; Xiaoguang, C. Sequence analysis of the groEL gene and its potential application in identification of pathogenic bacteria. Afr. J. Microbiol. Res. 2017, 4, 1733–1741. [Google Scholar]
- Nishibuchi, M. Molecular Identification. In The Biology of Vibrios; Thompson, F.L., Austin, B., Swings, J., Eds.; ASM Press: Washington, DC, USA, 2006. [Google Scholar]
- Ahmed, R.; Rafiquzaman, S.; Hossain, M.T.; Lee, J.-M.; Kong, I.-S. Species-specific detection of Vibrio alginolyticus in shellfish and shrimp by real-time PCR using the groEL gene. Aquac. Int. 2016, 24, 157–170. [Google Scholar] [CrossRef]
- Liu, B.; Gao, Q.; Zhang, X.; Chen, H.; Zhang, Y.; Sun, Y.; Yang, S.; Chen, C. CsrA Regulates Swarming Motility and Carbohydrate and Amino Acid Metabolism in Vibrio alginolyticus. Microorganisms 2021, 9, 2383. [Google Scholar] [CrossRef]
- Barbosa, C.; Nogueira, S.; Gadanho, M.; Chaves, S. DNA extraction: Finding the most suitable method. In Molecular Microbial Diagnostic Methods; Elsevier: Amsterdam, The Netherlands, 2016; pp. 135–154. [Google Scholar] [CrossRef]
- Kralik, P.; Ricchi, M. A basic guide to real time PCR in microbial diagnostics: Definitions, parameters, and everything. Front. Microbiol. 2017, 8, 108. [Google Scholar] [CrossRef]
- Mygind, T.; Birkelund, S.; Birkebæk, N.H.; Østergaard, L.; Jensen, J.S.; Christiansen, G. Determination of PCR efficiency in chelex-100 purified clinical samples and comparison of real-time quantitative PCR and conventional PCR for detection of Chlamydia pneumoniae. BMC Microbiol. 2002, 2, 17. [Google Scholar] [CrossRef]
- Thomas, T.; Gilbert, J.; Meyer, F. Metagenomics-a guide from sampling to data analysis. Microb. Inform. Exp. 2012, 2, 3. [Google Scholar] [CrossRef]
- Dashti, A.A.; Jadaon, M.M.; Abdulsamad, A.M.; Dashti, H.M. Heat treatment of bacteria: A simple method of DNA extraction for molecular techniques. Kuwait Med. J. 2009, 41, 117–122. [Google Scholar]
- Peng, X.; Yu, K.-Q.; Deng, G.-H.; Jiang, Y.-X.; Wang, Y.; Zhang, G.-X.; Zhou, H.-W. Comparison of direct boiling method with commercial kits for extracting fecal microbiome DNA by Illumina sequencing of 16S rRNA tags. J. Microbiol. Methods 2013, 95, 455–462. [Google Scholar] [CrossRef]
- Abdelhai, M.H.; Hassanin, H.A.; Sun, X. Comparative study of rapid DNA extraction methods of pathogenic bacteria. Am. J. Biosci. Bioeng. 2016, 4, 1–8. [Google Scholar] [CrossRef]
- Johnson, G.; Nolan, T.; Bustin, S.A. Real-time quantitative PCR, pathogen detection and MIQE. In PCR Detection of Microbial Pathogens; Springer: Berlin/Heidelberg, Germany, 2013; pp. 1–16. [Google Scholar] [CrossRef]
- Junior, J.C.R.; Tamanini, R.; Soares, B.F.; de Oliveira, A.M.; de Godoi Silva, F.; da Silva, F.F.; Augusto, N.A.; Beloti, V. Efficiency of boiling and four other methods for genomic DNA extraction of deteriorating spore-forming bacteria from milk. Semin. Ciências Agrárias 2016, 37, 3069–3078. [Google Scholar] [CrossRef]
- Campos, M.J.; Quesada, A. Strategies to Improve Efficiency and Specificity of Degenerate Primers in PCR. Methods Mol. Biol. 2017, 1620, 75–85. [Google Scholar] [CrossRef]
- Chuang, L.-Y.; Cheng, Y.-H.; Yang, C.-H. Specific primer design for the polymerase chain reaction. Biotechnol. Lett. 2013, 35, 1541–1549. [Google Scholar] [CrossRef]
- Mitsuhashi, M. Technical report: Part 2. Basic requirements for designing optimal PCR primers. J. Clin. Lab. Anal. 1996, 10, 285–293. [Google Scholar] [CrossRef]
- Slana, I.; Kralik, P.; Kralova, A.; Pavlik, I. On-farm spread of Mycobacterium avium subsp. paratuberculosis in raw milk studied by IS900 and F57 competitive real time quantitative PCR and culture examination. Int. J. Food Microbiol. 2008, 128, 250–257. [Google Scholar] [CrossRef]
- Kralik, P.; Slana, I.; Kralova, A.; Babak, V.; Whitlock, R.H.; Pavlik, I. Development of a predictive model for detection of Mycobacterium avium subsp. paratuberculosis in faeces by quantitative real time PCR. Vet. Microbiol. 2011, 149, 133–138. [Google Scholar] [CrossRef]
- Ricchi, M.; Savi, R.; Bolzoni, L.; Pongolini, S.; Grant, I.R.; De Cicco, C.; Cerutti, G.; Cammi, G.; Garbarino, C.A.; Arrigoni, N. Estimation of Mycobacterium avium subsp. paratuberculosis load in raw bulk tank milk in Emilia-Romagna Region (Italy) by qPCR. Microbiol. Open 2016, 5, 551–559. [Google Scholar] [CrossRef]
- Blackstone, G.M.; Nordstrom, J.L.; Vickery, M.C.; Bowen, M.D.; Meyer, R.F.; DePaola, A. Detection of pathogenic Vibrio parahaemolyticus in oyster enrichments by real time PCR. J. Microbiol. Methods 2003, 53, 149–155. [Google Scholar] [CrossRef]
- Kaufman, G.; Blackstone, G.; Vickery, M.; Bej, A.; Bowers, J.; Bowen, M.D.; Meyer, R.F.; DePaola, A. Real-time PCR quantification of Vibrio parahaemolyticus in oysters using an alternative matrix. J. Food Prot. 2004, 67, 2424–2429. [Google Scholar] [CrossRef]
- Ferrero, E.M.; de Godos, I.; Rodríguez, E.M.; García-Encina, P.A.; Muñoz, R.; Bécares, E. Molecular Characterization of Bacterial Communities in Algal–Bacterial Photobioreactors Treating Piggery Wastewaters; Elsevier: Amsterdam, The Netherlands, 2012; Volume 40, pp. 121–130. [Google Scholar]
- Lakaniemi, A.M.; Intihar, V.M.; Tuovinen, O.H.; Puhakka, J.A. Growth of Chlorella vulgaris and associated bacteria in photobioreactors. Microb. Biotechnol. 2012, 5, 69–78. [Google Scholar] [CrossRef]
- Lee, J.; Lee, J.; Lee, T.K.; Woo, S.-G.; Baek, G.S.; Park, J. In-depth characterization of wastewater bacterial community in response to algal growth using pyrosequencing. J. Microbiol. Biotechnol. 2013, 23, 1472–1477. [Google Scholar] [CrossRef] [PubMed]
- EC. GMO Method Validation. Available online: https://ec.europa.eu/jrc/en/research-topic/gmos (accessed on 28 January 2022).
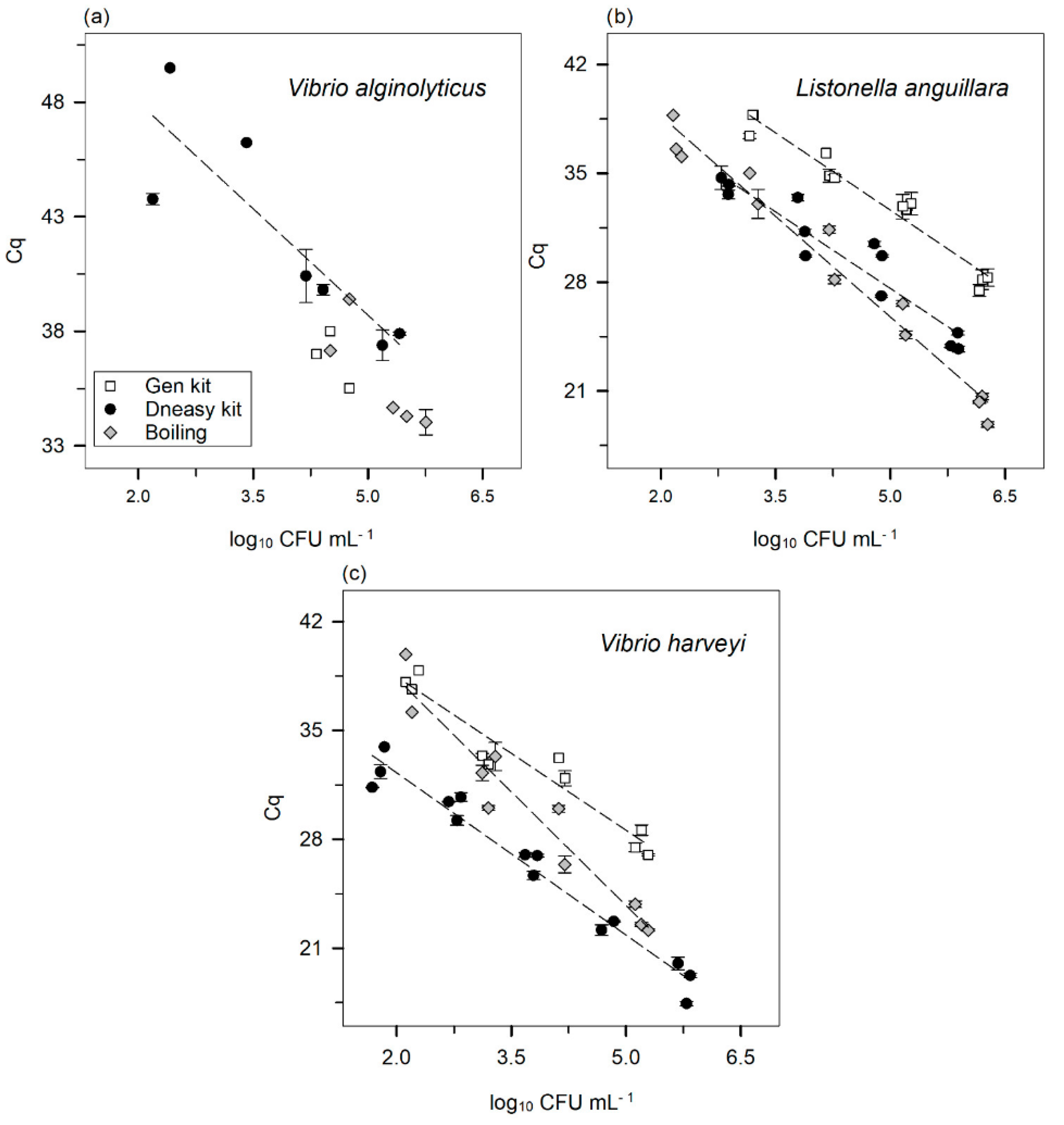
 —Gen kit,
—Gen kit,  —DNeasy kit, and
—DNeasy kit, and  —boiling method. Error bars ± standard error. The Cq was always obtained with a threshold of 160 RFU.
—Gen kit, —DNeasy kit, and —boiling method. Error bars ± standard error. The Cq was always obtained with a threshold of 160 RFU.
—boiling method. Error bars ± standard error. The Cq was always obtained with a threshold of 160 RFU.
—Gen kit, —DNeasy kit, and —boiling method. Error bars ± standard error. The Cq was always obtained with a threshold of 160 RFU.
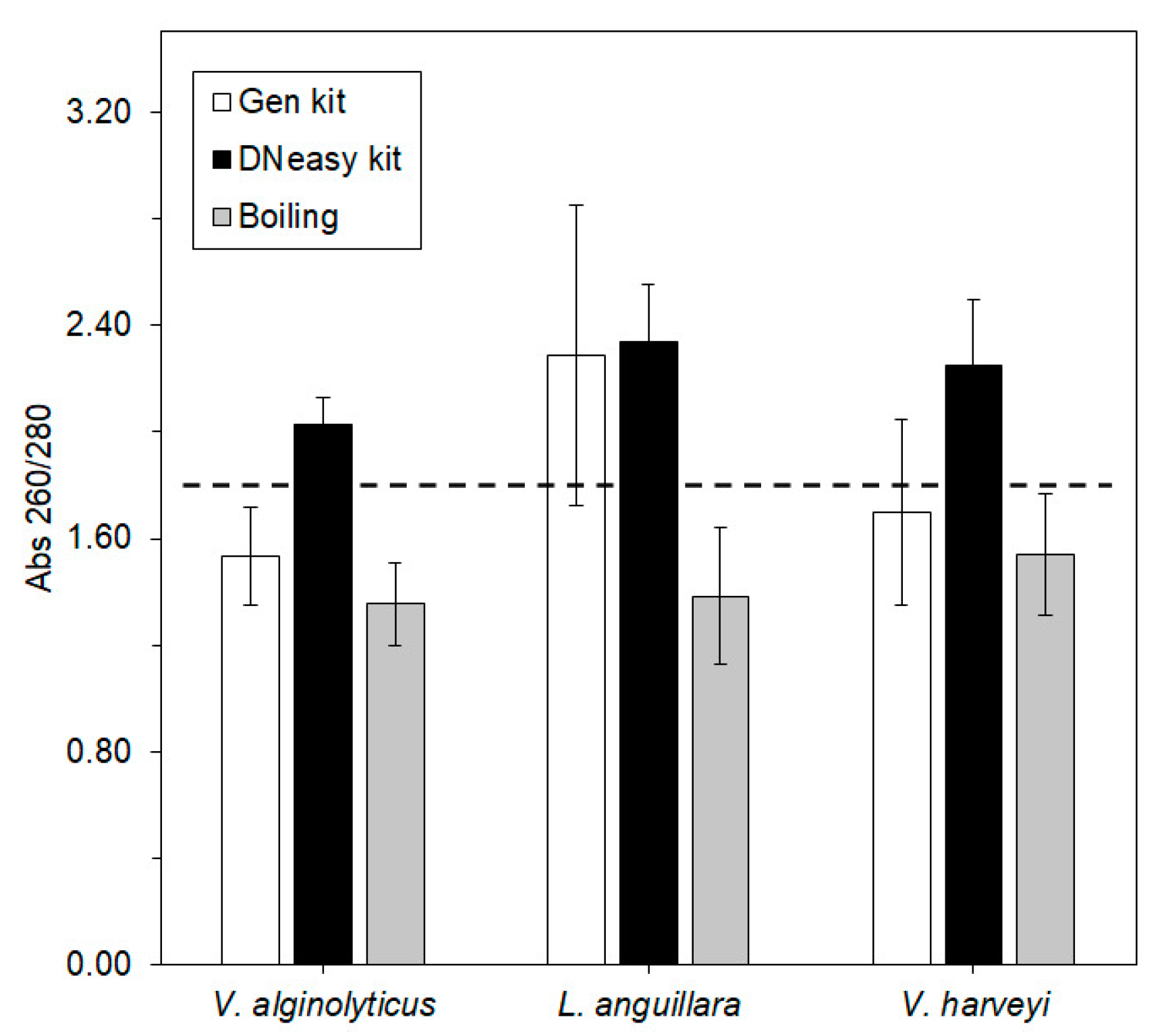
 —Gen kit,
—Gen kit,  —DNeasy kit, and
—DNeasy kit, and  —boiling. The dashed line denotes the threshold ratio of 1.80, above which DNA is considered pure. Error bars ± standard error.
—Gen kit, —DNeasy kit, and —boiling. The dashed line denotes the threshold ratio of 1.80, above which DNA is considered pure. Error bars ± standard error.
—boiling. The dashed line denotes the threshold ratio of 1.80, above which DNA is considered pure. Error bars ± standard error.
—Gen kit, —DNeasy kit, and —boiling. The dashed line denotes the threshold ratio of 1.80, above which DNA is considered pure. Error bars ± standard error.
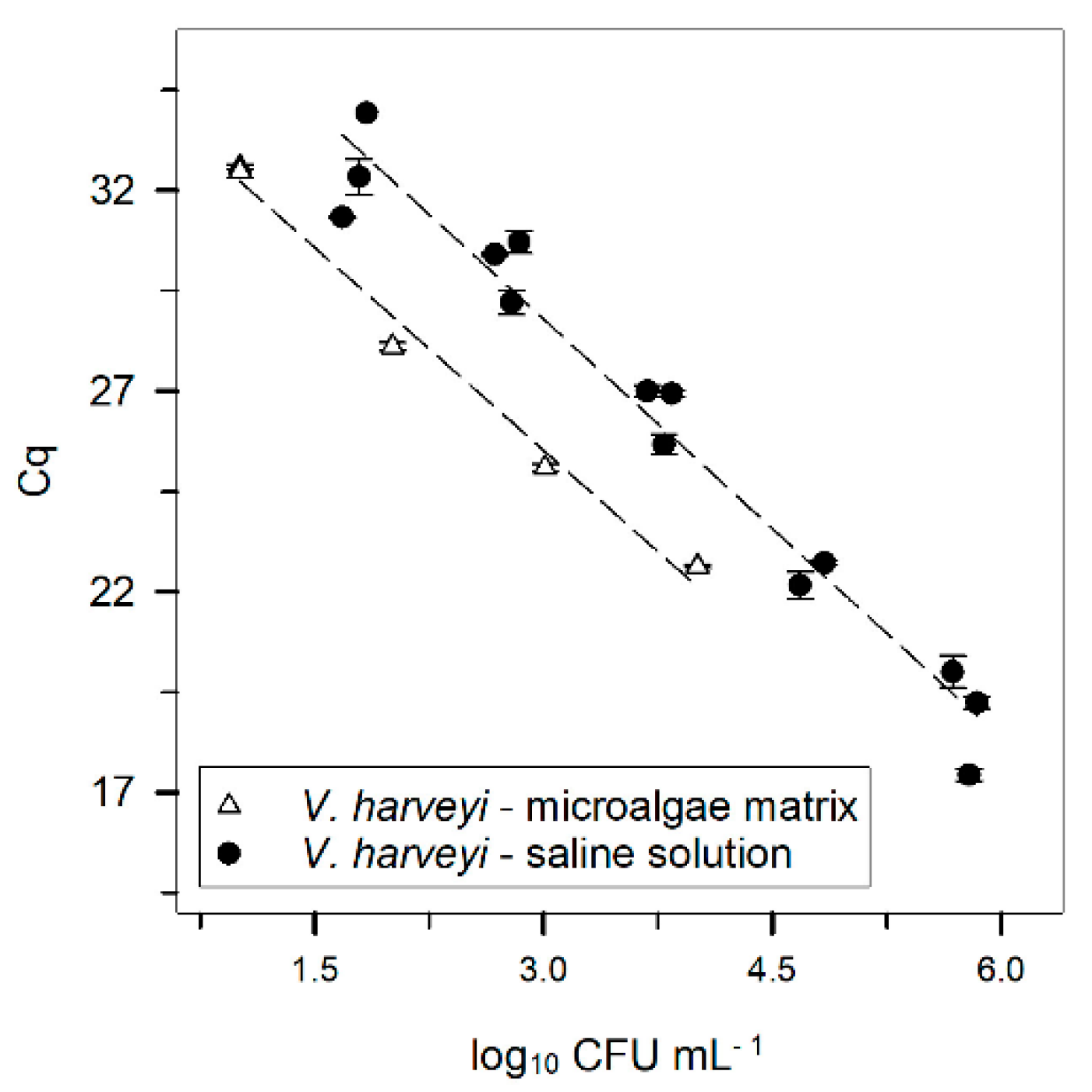
 saline solution (data as shown in Figure 1), and
saline solution (data as shown in Figure 1), and  microalgae matrix. Both results were obtained after DNA extraction using the DNeasy kit. Error bars ± standard error. The Cq was always obtained with a threshold of 160 RFU.
saline solution (data as shown in Figure 1), and microalgae matrix. Both results were obtained after DNA extraction using the DNeasy kit. Error bars ± standard error. The Cq was always obtained with a threshold of 160 RFU.
microalgae matrix. Both results were obtained after DNA extraction using the DNeasy kit. Error bars ± standard error. The Cq was always obtained with a threshold of 160 RFU.
saline solution (data as shown in Figure 1), and microalgae matrix. Both results were obtained after DNA extraction using the DNeasy kit. Error bars ± standard error. The Cq was always obtained with a threshold of 160 RFU.

{kind=link}
{kind=link}
{kind=link}
| Species Strain | Accession Number |
|---|---|
| Vibrio alginolyticus ATCC 17749 | CP006719.1 |
| Listonella anguillara 775 | CP002284.1 |
| Vibrio harveyi FDAARGOS_107 | CP014038.2 |
| Vibrio atlanticus LGP32 | FM954972.2 |
| Vibrio campbellii ATCC BAA-1116 | CP006605.1 |
| Vibrio cholerae N16961 | CP047297.1 |
| Vibrio fluvialis ATCC 33809 | CP014035.2 |
| Vibrio mediterranei 117-T6 | CP033577.1 |
| Vibrio metschnikovii CIP 69.14 | NZ_ACZO01000005.1 |
| Vibrio nigripulchritudo Snf1 | FO203526.1 |
| Vibrio parahaemolyticus RIMD 2210633 | BA000032.2 |
| Vibrio toranzoniae Vb 10.8 | LMXU01000002.1 |
| Vibrio tubiashii ATCC 19109 | CP009354.1 |
| Vibrio vulnificus CECT 4999 | CP014637.1 |
| Escherichia coli pAR060302 | HQ023864.1 |
| Pseudomonas aeruginosa PAO1 | CP053028.1 |
| Staphylococcus aureus NCTC 8325 | CP000253.1 |
| Salmonella enterica CMCST_CEPR_1 | CP053702.1 |
| DNA extraction methods | V. alginolyticus | |||
| Equation | R2 | E (%) | Amplification factor | |
| Gen kit | ------- | ------- | ------- | ------- |
| Boiling | ------ | ------ | ------ | ------ |
| DNeasy kit | y = −3.095x + 54.176 | R2 = 0.76 | 110 | 2.10 |
| DNA extraction methods | L. anguillara | |||
| Equation | R2 | E (%) | Amplification factor | |
| Gen kit | y = −3.288x + 49.057 | R2 = 0.95 | 101 | 2.01 |
| Boiling | y = −4.310x + 47.310 | R2 = 0.97 | 71 | 1.71 |
| DNeasy kit | y = −3.284x + 44.015 | R2 = 0.90 | 102 | 2.02 |
| DNA extraction methods | V. harveyi | |||
| Equation | R2 | E (%) | Amplification factor | |
| Gen kit | y = −3.296x + 45.049 | R2 = 0.93 | 101 | 2.01 |
| Boiling | y = −4.868x + 48.075 | R2 = 0.94 | 60 | 1.60 |
| DNeasy kit | y = −3.478x + 39.227 | R2 = 0.96 | 94 | 1.94 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Costa, C.; Ferreira, G.D.; Simões, M.; Silva, J.L.; Campos, M.J. Real-Time PCR Protocol for Detection and Quantification of Three Pathogenic Members of the Vibrionaceae Family. Microorganisms 2022, 10, 2060. https://doi.org/10.3390/microorganisms10102060
Costa C, Ferreira GD, Simões M, Silva JL, Campos MJ. Real-Time PCR Protocol for Detection and Quantification of Three Pathogenic Members of the Vibrionaceae Family. Microorganisms. 2022; 10(10):2060. https://doi.org/10.3390/microorganisms10102060
Chicago/Turabian StyleCosta, Cátia, Guilherme D. Ferreira, Marco Simões, Joana L. Silva, and Maria J. Campos. 2022. "Real-Time PCR Protocol for Detection and Quantification of Three Pathogenic Members of the Vibrionaceae Family" Microorganisms 10, no. 10: 2060. https://doi.org/10.3390/microorganisms10102060
APA StyleCosta, C., Ferreira, G. D., Simões, M., Silva, J. L., & Campos, M. J. (2022). Real-Time PCR Protocol for Detection and Quantification of Three Pathogenic Members of the Vibrionaceae Family. Microorganisms, 10(10), 2060. https://doi.org/10.3390/microorganisms10102060