
Beyond Basic Diversity Estimates—Analytical Tools for Mechanistic Interpretations of Amplicon Sequencing Data
 , , ,
, , , 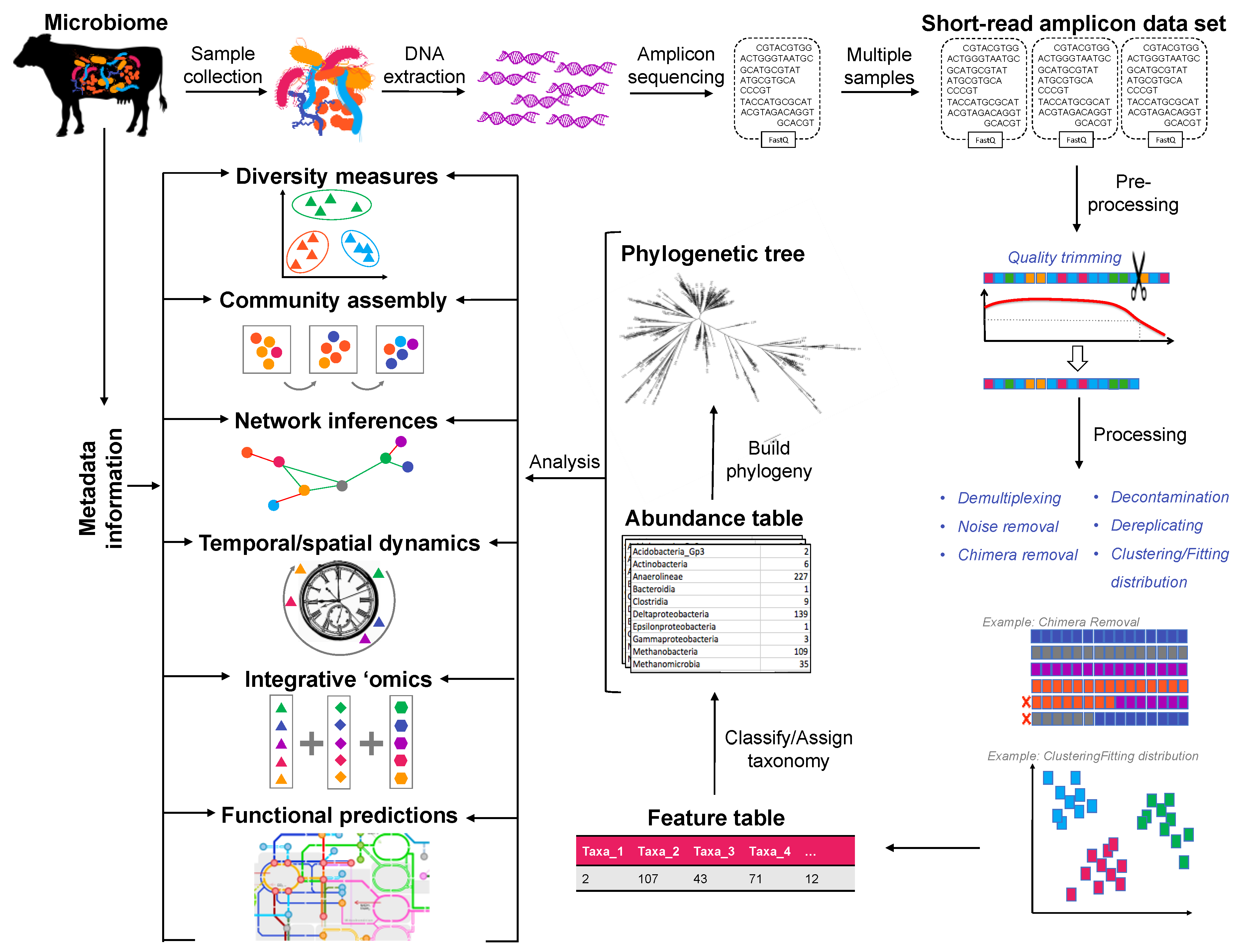{kind=link}
Abstract
1. High-Throughput Sequencing: Widely Used, but Under-Explored
2. Study Design Considerations: Planning for Statistics
3. Current Trends in Data Processing
4. Traditional Measures of Diversity: Revisiting the Basics
5. Identifying Mechanisms Driving Microbial Community Assembly
6. Network Inferences: Identifying Relationships and Revealing Complexity
7. Over Space and Time: Measuring Temporal/Spatial Dynamics
8. Integrative ‘Omics: Combining Multiple Datasets
9. Meta-Data Integration: Using Regressions to Identify Key Taxa
10. Predictive Functional Modelling: Using Structure to Infer Function
11. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Shendure, J.; Balasubramanian, S.; Church, G.M.; Gilbert, W.; Rogers, J.; Schloss, J.A.; Waterston, R.H. DNA sequencing at 40: Past, present and future. Nature 2017, 550, 345–353. [Google Scholar] [CrossRef] [PubMed]
- Shendure, J.; Ji, H. Next-generation DNA sequencing. Nat. Biotechnol. 2008, 26, 1135. [Google Scholar] [CrossRef] [PubMed]
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; FitzHugh, W.; et al. Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921. [Google Scholar] [CrossRef] [PubMed]
- Tyson, G.W.; Chapman, J.; Hugenholtz, P.; Allen, E.E.; Ram, R.J.; Richardson, P.M.; Solovyev, V.V.; Rubin, E.M.; Rokhsar, D.S.; Banfield, J.F. Community structure and metabolism through reconstruction of microbial genomes from the environment. Nature 2004, 428, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Venter, J.C.; Remington, K.; Heidelberg, J.F.; Halpern, A.L.; Rusch, D.; Eisen, J.A.; Wu, D.; Paulsen, I.; Nelson, K.E.; Nelson, W. Environmental genome shotgun sequencing of the Sargasso Sea. Science 2004, 304, 66–74. [Google Scholar] [CrossRef]
- Schloss, P.D.; Handelsman, J. Metagenomics for studying unculturable microorganisms: Cutting the Gordian knot. Genome Biol. 2005, 6, 229. [Google Scholar] [CrossRef]
- Liu, S.; Moon, C.D.; Zheng, N.; Huws, S.; Zhao, S.; Wang, J. Opportunities and challenges of using metagenomic data to bring uncultured microbes into cultivation. Microbiome 2022, 10, 76. [Google Scholar] [CrossRef]
- Lightbody, G.; Haberland, V.; Browne, F.; Taggart, L.; Zheng, H.; Parkes, E.; Blayney, J.K. Review of applications of high-throughput sequencing in personalized medicine: Barriers and facilitators of future progress in research and clinical application. Brief. Bioinform. 2019, 20, 1795–1811. [Google Scholar] [CrossRef]
- Ley, T.J.; Mardis, E.R.; Ding, L.; Fulton, B.; McLellan, M.D.; Chen, K.; Dooling, D.; Dunford-Shore, B.H.; McGrath, S.; Hickenbotham, M.; et al. DNA sequencing of a cytogenetically normal acute myeloid leukaemia genome. Nature 2008, 456, 66–72. [Google Scholar] [CrossRef]
- Miga, K.H.; Koren, S.; Rhie, A.; Vollger, M.R.; Gershman, A.; Bzikadze, A.; Brooks, S.; Howe, E.; Porubsky, D.; Logsdon, G.A. Telomere-to-telomere assembly of a complete human X chromosome. Nature 2020, 585, 79–84. [Google Scholar] [CrossRef]
- Lam, H.Y.K.; Clark, M.J.; Chen, R.; Chen, R.; Natsoulis, G.; O’Huallachain, M.; Dewey, F.E.; Habegger, L.; Ashley, E.A.; Gerstein, M.B.; et al. Performance comparison of whole-genome sequencing platforms. Nat. Biotechnol. 2012, 30, 78–82. [Google Scholar] [CrossRef] [PubMed]
- Quince, C.; Walker, A.W.; Simpson, J.T.; Loman, N.J.; Segata, N. Shotgun metagenomics, from sampling to analysis. Nat. Biotechnol. 2017, 35, 833–844. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Marguerat, S.; Bähler, J. RNA-seq: From technology to biology. Cell. Mol. Life Sci. 2010, 67, 569–579. [Google Scholar] [CrossRef] [PubMed]
- Amarasinghe, S.L.; Su, S.; Dong, X.; Zappia, L.; Ritchie, M.E.; Gouil, Q. Opportunities and challenges in long-read sequencing data analysis. Genome Biol. 2020, 21, 30. [Google Scholar] [CrossRef]
- Saliba, A.-E.; Westermann, A.J.; Gorski, S.A.; Vogel, J. Single-cell RNA-seq: Advances and future challenges. Nucleic Acids Res. 2014, 42, 8845–8860. [Google Scholar] [CrossRef]
- McKenna, A.; Ijaz, U.Z.; Kelly, C.; Linton, M.; Sloan, W.T.; Green, B.D.; Lavery, U.; Dorrell, N.; Wren, B.W.; Richmond, A.; et al. Impact of industrial production system parameters on chicken microbiomes: Mechanisms to improve performance and reduce Campylobacter. Microbiome 2020, 8, 128. [Google Scholar] [CrossRef]
- Trego, A.C.; McAteer, P.G.; Nzeteu, C.; Mahony, T.; Abram, F.; Ijaz, U.Z.; O’Flaherty, V. Combined Stochastic and Deterministic Processes Drive Community Assembly of Anaerobic Microbiomes during Granule Flotation. Front. Microbiol. 2021, 12, 1165. [Google Scholar] [CrossRef]
- Nikolova, C.; Ijaz, U.Z.; Gutierrez, T. Exploration of marine bacterioplankton community assembly mechanisms during chemical dispersant and surfactant-assisted oil biodegradation. Ecol. Evol. 2021, 11, 13862–13874. [Google Scholar] [CrossRef]
- Liu, R.; Wei, X.; Song, W.; Wang, L.; Cao, J.; Wu, J.; Thomas, T.; Jin, T.; Wang, Z.; Wei, W.; et al. Novel Chloroflexi genomes from the deepest ocean reveal metabolic strategies for the adaptation to deep-sea habitats. Microbiome 2022, 10, 75. [Google Scholar] [CrossRef]
- Meek, M.H.; Larson, W.A. The future is now: Amplicon sequencing and sequence capture usher in the conservation genomics era. Mol. Ecol. Resour. 2019, 19, 795–803. [Google Scholar] [CrossRef] [PubMed]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2013, 41, D590–D596. [Google Scholar] [CrossRef] [PubMed]
- Yilmaz, P.; Parfrey, L.W.; Yarza, P.; Gerken, J.; Pruesse, E.; Quast, C.; Schweer, T.; Peplies, J.; Ludwig, W.; Glöckner, F.O. The SILVA and “All-species Living Tree Project (LTP)” taxonomic frameworks. Nucleic Acids Res. 2014, 42, D643–D648. [Google Scholar] [CrossRef] [PubMed]
- Dueholm, M.K.D.; Nierychlo, M.; Andersen, K.S.; Rudkjøbing, V.; Knutsson, S.; Albertsen, M.; Nielsen, P.H. MiDAS 4: A global catalogue of full-length 16S rRNA gene sequences and taxonomy for studies of bacterial communities in wastewater treatment plants. Nat. Commun. 2022, 13, 1908. [Google Scholar] [CrossRef] [PubMed]
- Rohwer, R.; Hamilton, J.; Newton, R.; McMahon, K.; Rodrigues, J. TaxAss: Leveraging a Custom Freshwater Database Achieves Fine-Scale Taxonomic Resolution. mSphere 2022, 3, e00327-18. [Google Scholar] [CrossRef] [PubMed]
- Prodan, A.; Tremaroli, V.; Brolin, H.; Zwinderman, A.H.; Nieuwdorp, M.; Levin, E. Comparing bioinformatic pipelines for microbial 16S rRNA amplicon sequencing. PLoS ONE 2020, 15, e0227434. [Google Scholar] [CrossRef]
- van Dijk, E.L.; Jaszczyszyn, Y.; Naquin, D.; Thermes, C. The Third Revolution in Sequencing Technology. Trends Genet. 2018, 34, 666–681. [Google Scholar] [CrossRef]
- Poretsky, R.; Rodriguez-R, L.M.; Luo, C.; Tsementzi, D.; Konstantinidis, K.T. Strengths and Limitations of 16S rRNA Gene Amplicon Sequencing in Revealing Temporal Microbial Community Dynamics. PLoS ONE 2014, 9, e93827. [Google Scholar] [CrossRef]
- Verster, A.J.; Borenstein, E. Competitive lottery-based assembly of selected clades in the human gut microbiome. Microbiome 2018, 6, 186. [Google Scholar] [CrossRef]
- Eng, A.; Borenstein, E. Taxa-function robustness in microbial communities. Microbiome 2018, 6, 45. [Google Scholar] [CrossRef]
- Darcy, J.L.; Amend, A.S.; Swift, S.O.I.; Sommers, P.S.; Lozupone, C.A. specificity: An R package for analysis of feature specificity to environmental and higher dimensional variables, applied to microbiome species data. Environ. Microbiome 2022, 17, 34. [Google Scholar] [CrossRef] [PubMed]
- Calle, M.L.; Susin, A. coda4microbiome: Compositional data analysis for microbiome studies. bioRxiv 2022. [Google Scholar] [CrossRef]
- Niku, J.; Hui, F.K.C.; Taskinen, S.; Warton, D.I. gllvm: Fast analysis of multivariate abundance data with generalized linear latent variable models in R. Methods Ecol. Evol. 2019, 10, 2173–2182. [Google Scholar] [CrossRef]
- Bharti, R.; Grimm, D.G. Current challenges and best-practice protocols for microbiome analysis. Brief. Bioinform. 2021, 22, 178–193. [Google Scholar] [CrossRef] [PubMed]
- Maki, K.A.; Diallo, A.F.; Lockwood, M.B.; Franks, A.T.; Green, S.J.; Joseph, P. V Considerations when designing a microbiome study: Implications for nursing science. Biol. Res. Nurs. 2019, 21, 125–141. [Google Scholar] [CrossRef]
- Goodrich, J.K.; Di Rienzi, S.C.; Poole, A.C.; Koren, O.; Walters, W.A.; Caporaso, J.G.; Knight, R.; Ley, R.E. Conducting a Microbiome Study. Cell 2014, 158, 250–262. [Google Scholar] [CrossRef]
- Salter, S.J.; Cox, M.J.; Turek, E.M.; Calus, S.T.; Cookson, W.O.; Moffatt, M.F.; Turner, P.; Parkhill, J.; Loman, N.J.; Walker, A.W. Reagent and laboratory contamination can critically impact sequence-based microbiome analyses. BMC Biol. 2014, 12, 87. [Google Scholar] [CrossRef]
- D’Amore, R.; Ijaz, U.Z.; Schirmer, M.; Kenny, J.G.; Gregory, R.; Darby, A.C.; Shakya, M.; Podar, M.; Quince, C.; Hall, N. A comprehensive benchmarking study of protocols and sequencing platforms for 16S rRNA community profiling. BMC Genom. 2016, 17, 55. [Google Scholar] [CrossRef]
- Schirmer, M.; Ijaz, U.Z.; D’Amore, R.; Hall, N.; Sloan, W.T.; Quince, C. Insight into biases and sequencing errors for amplicon sequencing with the Illumina MiSeq platform. Nucleic Acids Res. 2015, 43, gku1341. [Google Scholar] [CrossRef]
- Quince, C.; Ijaz, U.Z.; Loman, N.; Eren, A.M.; Saulnier, D.; Russell, J.; Haig, S.J.; Calus, S.T.; Quick, J.; Barclay, A. Extensive modulation of the fecal metagenome in children with Crohn’s disease during exclusive enteral nutrition. Am. J. Gastroenterol. 2015, 110, 1718. [Google Scholar] [CrossRef]
- Schielzeth, H.; Nakagawa, S. Nested by design: Model fitting and interpretation in a mixed model era. Methods Ecol. Evol. 2013, 4, 14–24. [Google Scholar] [CrossRef]
- Rahman, G.; McDonald, D.; Gonzalez, A.; Vázquez-Baeza, Y.; Jiang, L.; Casals-Pascual, C.; Peddada, S.; Hakim, D.; Dilmore, A.H.; Nowinski, B.; et al. Scalable power analysis and effect size exploration of microbiome community differences with Evident. bioRxiv 2022. [Google Scholar] [CrossRef]
- Trego, A.C.; O’Sullivan, S.; Quince, C.; Mills, S.; Ijaz, U.Z.; Collins, G. Size Shapes the Active Microbiome of the Methanogenic Granules, Corroborating a Biofilm Life Cycle. mSystems 2020, 5, e00323-20. [Google Scholar] [CrossRef] [PubMed]
- Trego, A.C.; Holohan, B.C.; Keating, C.; Graham, A.; O’Connor, S.; Gerardo, M.; Hughes, D.; Zeeshan Ijaz, U.; O’Flaherty, V. First Proof of Concept for Full-Scale, Direct, Low-Temperature Anaerobic Treatment of Municipal Wastewater. Bioresour. Technol. 2021, 341, 125786. [Google Scholar] [CrossRef]
- Keating, C.; Bolton-Warberg, M.; Hinchcliffe, J.; Davies, R.; Whelan, S.; Wan, A.H.L.; Fitzgerald, R.D.; Davies, S.J.; Smith, C.J.; Ijaz, U.Z. Key Drivers of Ecological Assembly in the Hindgut of Atlantic Cod (Gadus morhua) when Fed with a Macroalgal Supplemented diet—How Robust Is the Gut to Taxonomic Perturbation? bioRxiv 2021. [Google Scholar] [CrossRef]
- Thom, C.; Smith, C.J.; Moore, G.; Weir, P.; Ijaz, U.Z. Microbiomes in drinking water treatment and distribution: A meta-analysis from source to tap. Water Res. 2022, 212, 118106. [Google Scholar] [CrossRef]
- Yonatan, Y.; Amit, G.; Friedman, J.; Bashan, A. Complexity–stability trade-off in empirical microbial ecosystems. Nat. Ecol. Evol. 2022, 6, 693–700. [Google Scholar] [CrossRef]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Lu, J.; Salzberg, S.L. Ultrafast and accurate 16S rRNA microbial community analysis using Kraken 2. Microbiome 2020, 8, 124. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Holmes, S.P. Exact sequence variants should replace operational taxonomic units in marker-gene data analysis. ISME J. 2017, 11, 2639–2643. [Google Scholar] [CrossRef] [PubMed]
- Glassman, S.I.; Martiny, J.B.H. Broadscale ecological patterns are robust to use of exact sequence variants versus operational taxonomic units. mSphere 2018, 3, e00148-18. [Google Scholar] [CrossRef] [PubMed]
- Schloss, P.D.; McMahon, K. Amplicon Sequence Variants Artificially Split Bacterial Genomes into Separate Clusters. mSphere 2021, 6, e00191-21. [Google Scholar] [CrossRef] [PubMed]
- Cholet, F.; Lisik, A.; Agogue, H.; Ijaz, U.Z.; Pineau, P.; Lachaussée, N.; Smith, C.J. Ecological Observations Based on Functional Gene Sequencing Are Sensitive to the Amplicon Processing Method. bioRxiv 2022. [Google Scholar] [CrossRef] [PubMed]
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahé, F. VSEARCH: A versatile open source tool for metagenomics. PeerJ 2016, 2016, 2584. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. UNOISE2: Improved error-correction for Illumina 16S and ITS amplicon sequencing. bioRxiv 2016. [Google Scholar] [CrossRef]
- Abellan-Schneyder, I.; Matchado, M.S.; Reitmeier, S.; Sommer, A.; Sewald, Z.; Baumbach, J.; List, M.; Neuhaus, K. Primer, Pipelines, Parameters: Issues in 16S rRNA Gene Sequencing. mSphere 2021, 6, e01202-20. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef]
- Amir, A.; McDonald, D.; Navas-Molina, J.A.; Kopylova, E.; Morton, J.T.; Zech Xu, Z.; Kightley, E.P.; Thompson, L.R.; Hyde, E.R.; Gonzalez, A.; et al. Deblur Rapidly Resolves Single-Nucleotide Community Sequence Patterns. mSystems 2017, 2, e00191-16. [Google Scholar] [CrossRef]
- Gao, X.; Lin, H.; Revanna, K.; Dong, Q. A Bayesian taxonomic classification method for 16S rRNA gene sequences with improved species-level accuracy. BMC Bioinformatics 2017, 18, 247. [Google Scholar] [CrossRef] [PubMed]
- McDonald, D.; Price, M.N.; Goodrich, J.; Nawrocki, E.P.; DeSantis, T.Z.; Probst, A.; Andersen, G.L.; Knight, R.; Hugenholtz, P. An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J. 2012, 6, 610–618. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef] [PubMed]
- Simonsen, D.M.; Skytte, A.K.; Jon, M.S.; Munk, K.J.; Erika, Y.; Michael, K.S.; Mads, A.; Halkjær, N.P.; Nicole, D. Generation of Comprehensive Ecosystem-Specific Reference Databases with Species-Level Resolution by High-Throughput Full-Length 16S rRNA Gene Sequencing and Automated Taxonomy Assignment (AutoTax). MBio 2020, 11, e01557-20. [Google Scholar] [CrossRef]
- Choi, J.; Yang, F.; Stepanauskas, R.; Cardenas, E.; Garoutte, A.; Williams, R.; Flater, J.; Tiedje, J.M.; Hofmockel, K.S.; Gelder, B.; et al. Strategies to improve reference databases for soil microbiomes. ISME J. 2017, 11, 829–834. [Google Scholar] [CrossRef] [PubMed]
- Madi, N.; Vos, M.; Murall, C.L.; Legendre, P.; Shapiro, B.J. Does diversity beget diversity in microbiomes? eLife 2020, 9, e58999. [Google Scholar] [CrossRef] [PubMed]
- Oksanen, J.; Blanchet, F.G.; Friendly, M.; Kindt, R.; Legendre, P.; McGlinn, D.; Minchin, P.R.; O’hara, R.B.; Simpson, G.L.; Solymos, P. Vegan: Community Ecology Package. R package, v. 2.4–6; R Core Team: Vienna, Austria, 2018. [Google Scholar]
- McMurdie, P.J.; Holmes, S. Phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef]
- Lande, R. Statistics and Partitioning of Species Diversity, and Similarity among Multiple Communities. Oikos 1996, 76, 5–13. [Google Scholar] [CrossRef]
- Shannon, C.E.; Weaver, W. The Mathematical Theory of Communication; University of Illinois: Champaign, IL, USA, 1949. [Google Scholar]
- Fisher, R.A.; Corbet, A.S.; Williams, C.B. The relation between the number of species and the number of individuals in a random sample of an animal population. J. Anim. Ecol. 1943, 12, 42–58. [Google Scholar] [CrossRef]
- Simpson, E.H. Measurement of Diversity. Nature 1949, 163, 688. [Google Scholar] [CrossRef]
- Hill, M.O. Diversity and Evenness: A Unifying Notation and Its Consequences. Ecology 1973, 54, 427–432. [Google Scholar] [CrossRef]
- Willis, A.D. Rarefaction, Alpha Diversity, and Statistics. Front. Microbiol. 2019. [CrossRef] [PubMed]
- Lozupone, C.; Lladser, M.E.; Knights, D.; Stombaugh, J.; Knight, R. UniFrac: An effective distance metric for microbial community comparison. ISME J. 2011, 5, 169–172. [Google Scholar] [CrossRef] [PubMed]
- Lozupone, C.A.; Micah, H.; Kelley, S.T.; Knight, R. Quantitative and Qualitative β Diversity Measures Lead to Different Insights into Factors That Structure Microbial Communities. Appl. Environ. Microbiol. 2007, 73, 1576–1585. [Google Scholar] [CrossRef] [PubMed]
- Bray, J.R.; Curtis, J.T. An ordination of the upland forest communities of southern Wisconsin. Ecol. Monogr. 1957, 27, 326–349. [Google Scholar] [CrossRef]
- Beals, E.W. Bray-Curtis Ordination: An Effective Strategy for Analysis of Multivariate Ecological Data. In Advances in Ecological Research; MacFadyen, A., Ford, E.D., Eds.; Academic Press: Cambridge, MA, USA, 1984; Volume 14, pp. 1–55. ISBN 0065-2504. [Google Scholar]
- Ramette, A. Multivariate analyses in microbial ecology. FEMS Microbiol. Ecol. 2007, 62, 142–160. [Google Scholar] [CrossRef]
- Paliy, O.; Shankar, V. Application of multivariate statistical techniques in microbial ecology. Mol. Ecol. 2016, 25, 1032–1057. [Google Scholar] [CrossRef]
- Pierre, L.; Miquel, C.; Hélène, M. Beta diversity as the variance of community data: Dissimilarity coefficients and partitioning. Ecol. Lett. 2013, 16, 951–963. [Google Scholar] [CrossRef]
- Dray, S.; Blanchet, G.; Borcard, D.; Guenard, G.; Jombart, T.; Larocque, G.; Legendre, P.; Madi, N.; Wagner, H.H. Package ‘ adespatial’. 2017. [Google Scholar]
- Keating, C.; Bolton-Warberg, M.; Hinchcliffe, J.; Davies, R.; Whelan, S.; Wan, A.H.L.; Fitzgerald, R.D.; Davies, S.J.; Ijaz, U.Z.; Smith, C.J. Temporal changes in the gut microbiota in farmed Atlantic cod (Gadus morhua) outweigh the response to diet supplementation with macroalgae. Anim. Microbiome 2021, 3, 7. [Google Scholar] [CrossRef]
- Rohart, F.; Gautier, B.; Singh, A.; Lê Cao, K.-A. mixOmics: An R package for ‘omics feature selection and multiple data integration. PLOS Comput. Biol. 2017, 13, e1005752. [Google Scholar] [CrossRef]
- Jalanka-Tuovinen, J.; Salonen, A.; Nikkilä, J.; Immonen, O.; Kekkonen, R.; Lahti, L.; Palva, A.; de Vos, W.M. Intestinal Microbiota in Healthy Adults: Temporal Analysis Reveals Individual and Common Core and Relation to Intestinal Symptoms. PLoS ONE 2011, 6, e23035. [Google Scholar] [CrossRef] [PubMed]
- Dai, T.; Zhang, Y.; Tang, Y.; Bai, Y.; Tao, Y.; Huang, B.; Wen, D. Identifying the key taxonomic categories that characterize microbial community diversity using full-scale classification: A case study of microbial communities in the sediments of Hangzhou Bay. FEMS Microbiol. Ecol. 2016, 92, fiw150. [Google Scholar] [CrossRef] [PubMed]
- De Vrieze, J.; Pinto, A.J.; Sloan, W.T.; Ijaz, U.Z. The active microbial community more accurately reflects the anaerobic digestion process: 16S rRNA (gene) sequencing as a predictive tool. Microbiome 2018, 6, 63. [Google Scholar] [CrossRef]
- Props, R.; Kerckhof, F.-M.; Rubbens, P.; De Vrieze, J.; Hernandez Sanabria, E.; Waegeman, W.; Monsieurs, P.; Hammes, F.; Boon, N. Absolute quantification of microbial taxon abundances. ISME J. 2017, 11, 584–587. [Google Scholar] [CrossRef] [PubMed]
- Furman, O.; Shenhav, L.; Sasson, G.; Kokou, F.; Honig, H.; Jacoby, S.; Hertz, T.; Cordero, O.X.; Halperin, E.; Mizrahi, I. Stochasticity constrained by deterministic effects of diet and age drive rumen microbiome assembly dynamics. Nat. Commun. 2020, 11, 1904. [Google Scholar] [CrossRef] [PubMed]
- Leibold, M.A.; Mikkelson, G.M. Coherence, species turnover, and boundary clumping: Elements of meta-community structure. Oikos 2002, 97, 237–250. [Google Scholar] [CrossRef]
- Vass, M.; Székely, A.J.; Lindström, E.S.; Langenheder, S. Using null models to compare bacterial and microeukaryotic metacommunity assembly under shifting environmental conditions. Sci. Rep. 2020, 10, 2455. [Google Scholar] [CrossRef] [PubMed]
- Presley, S.J.; Higgins, C.L.; Willig, M.R. A comprehensive framework for the evaluation of metacommunity structure. Oikos 2010, 119, 908–917. [Google Scholar] [CrossRef]
- Zhou, J.; Ning, D. Stochastic Community Assembly: Does It Matter in Microbial Ecology? Microbiol. Mol. Biol. Rev. 2017, 81, e00002-17. [Google Scholar] [CrossRef]
- Stegen, J.C.; Lin, X.; Fredrickson, J.K.; Chen, X.; Kennedy, D.W.; Murray, C.J.; Rockhold, M.L.; Konopka, A. Quantifying community assembly processes and identifying features that impose them. ISME J. 2013, 7, 2069–2079. [Google Scholar] [CrossRef]
- Stegen, J.C.; Lin, X.; Fredrickson, J.K.; Konopka, A.E. Estimating and mapping ecological processes influencing microbial community assembly. Front. Microbiol. 2015, 6, 370. [Google Scholar] [CrossRef] [PubMed]
- Ning, D.; Yuan, M.; Wu, L.; Zhang, Y.; Guo, X.; Zhou, X.; Yang, Y.; Arkin, A.P.; Firestone, M.K.; Zhou, J. A quantitative framework reveals ecological drivers of grassland microbial community assembly in response to warming. Nat. Commun. 2020, 11, 4717. [Google Scholar] [CrossRef] [PubMed]
- Ning, D.; Deng, Y.; Tiedje, J.M.; Zhou, J. A general framework for quantitatively assessing ecological stochasticity. Proc. Natl. Acad. Sci. 2019, 116, 16892–16898. [Google Scholar] [CrossRef]
- Modin, O.; Liébana, R.; Saheb-Alam, S.; Wilén, B.-M.; Suarez, C.; Hermansson, M.; Persson, F. Hill-based dissimilarity indices and null models for analysis of microbial community assembly. Microbiome 2020, 8, 132. [Google Scholar] [CrossRef]
- Hubbell, S.P. The unified neutral theory of biodiversity and biogeography (MPB-32). In The Unified Neutral Theory of Biodiversity and Biogeography (MPB-32); Princeton University Press: Princeton, NJ, USA, 2011; ISBN 1400837529. [Google Scholar]
- Sloan, W.T.; Lunn, M.; Woodcock, S.; Head, I.M.; Nee, S.; Curtis, T.P. Quantifying the roles of immigration and chance in shaping prokaryote community structure. Environ. Microbiol. 2006, 8, 732–740. [Google Scholar] [CrossRef] [PubMed]
- Tucker, C.M.; Shoemaker, L.G.; Davies, K.F.; Nemergut, D.R.; Melbourne, B.A. Differentiating between niche and neutral assembly in metacommunities using null models of β-diversity. Oikos 2016, 125, 778–789. [Google Scholar] [CrossRef]
- Kokou, F.; Sasson, G.; Friedman, J.; Eyal, S.; Ovadia, O.; Harpaz, S.; Cnaani, A.; Mizrahi, I. Core gut microbial communities are maintained by beneficial interactions and strain variability in fish. Nat. Microbiol. 2019, 4, 2456–2465. [Google Scholar] [CrossRef]
- Finn, D.R.; Yu, J.; Ilhan, Z.E.; Fernandes, V.M.C.; Penton, C.R.; Krajmalnik-Brown, R.; Garcia-Pichel, F.; Vogel, T.M. MicroNiche: An R package for assessing microbial niche breadth and overlap from amplicon sequencing data. FEMS Microbiol. Ecol. 2020, 96, fiaa131. [Google Scholar] [CrossRef] [PubMed]
- Golovko, G.; Kamil, K.; Albayrak, L.; Nia, A.M.; Duarte, R.S.A.; Chumakov, S.; Fofanov, Y. Identification of multidimensional Boolean patterns in microbial communities. Microbiome 2020, 8, 131. [Google Scholar] [CrossRef]
- Durán, C.; Ciucci, S.; Palladini, A.; Ijaz, U.Z.; Zippo, A.G.; Sterbini, F.P.; Masucci, L.; Cammarota, G.; Ianiro, G.; Spuul, P.; et al. Nonlinear machine learning pattern recognition and bacteria-metabolite multilayer network analysis of perturbed gastric microbiome. Nat. Commun. 2021, 12, 1926. [Google Scholar] [CrossRef]
- Röttjers, L.; Faust, K. From hairballs to hypotheses–biological insights from microbial networks. FEMS Microbiol. Rev. 2018, 42, 761–780. [Google Scholar] [CrossRef] [PubMed]
- Willis, A.D.; Martin, B.D. Estimating diversity in networked ecological communities. Biostatistics 2022, 23, 207–222. [Google Scholar] [CrossRef] [PubMed]
- Oulas, A.; Zachariou, M.; Chasapis, C.T.; Tomazou, M.; Ijaz, U.Z.; Schmartz, G.P.; Spyrou, G.M.; Vlamis-Gardikas, A. Putative Antimicrobial Peptides within Bacterial Proteomes Affect Bacterial Predominance: A Network Analysis Perspective. Front. Microbiol. 2021, 12, 752674. [Google Scholar] [CrossRef]
- Kumar, M.; Ji, B.; Zengler, K.; Nielsen, J. Modelling approaches for studying the microbiome. Nat. Microbiol. 2019, 4, 1253–1267. [Google Scholar] [CrossRef] [PubMed]
- Kurtz, Z.D.; Müller, C.L.; Miraldi, E.R.; Littman, D.R.; Blaser, M.J.; Bonneau, R.A. Sparse and Compositionally Robust Inference of Microbial Ecological Networks. PLOS Comput. Biol. 2015, 11, e1004226. [Google Scholar] [CrossRef] [PubMed]
- Donohue, I.; Hillebrand, H.; Montoya, J.M.; Petchey, O.L.; Pimm, S.L.; Fowler, M.S.; Healy, K.; Jackson, A.L.; Lurgi, M.; McClean, D.; et al. Navigating the complexity of ecological stability. Ecol. Lett. 2016, 19, 1172–1185. [Google Scholar] [CrossRef] [PubMed]
- Kéfi, S.; Domínguez-García, V.; Donohue, I.; Fontaine, C.; Thébault, E.; Dakos, V. Advancing our understanding of ecological stability. Ecol. Lett. 2019, 22, 1349–1356. [Google Scholar] [CrossRef] [PubMed]
- May, R.M. Will a Large Complex System be Stable? Nature 1972, 238, 413–414. [Google Scholar] [CrossRef] [PubMed]
- Becraft, E.D.; Vetter, M.C.Y.L.; Bezuidt, O.K.I.; Brown, J.M.; Labonté, J.M.; Kauneckaite-Griguole, K.; Salkauskaite, R.; Alzbutas, G.; Sackett, J.D.; Kruger, B.R. Evolutionary stasis of a deep subsurface microbial lineage. ISME J. 2021, 15, 2830–2842. [Google Scholar] [CrossRef]
- Hamdan, L.J.; Hampel, J.J.; Moseley, R.D.; Mugge, R.L.; Ray, A.; Salerno, J.L.; Damour, M. Deep-sea shipwrecks represent island-like ecosystems for marine microbiomes. ISME J. 2021, 1–9. [Google Scholar] [CrossRef]
- Ramirez, K.S.; Knight, C.G.; De Hollander, M.; Brearley, F.Q.; Constantinides, B.; Cotton, A.; Creer, S.; Crowther, T.W.; Davison, J.; Delgado-Baquerizo, M. Detecting macroecological patterns in bacterial communities across independent studies of global soils. Nat. Microbiol. 2018, 3, 189–196. [Google Scholar] [CrossRef] [PubMed]
- McAteer, P.G.; Trego, A.C.; Thorn, C.; Mahony, T.; Abram, F.; O’Flaherty, V. Reactor configuration influences microbial community structure during high-rate, low-temperature anaerobic treatment of dairy wastewater. Bioresour. Technol. 2020, 307, 123221. [Google Scholar] [CrossRef] [PubMed]
- Bokulich, N.A.; Dillon, M.R.; Yilong, Z.; Ram, R.J.; Evan, B.; Huilin, L.; Albert, P.S.; Gregory, C.J.; Mani, A. q2-longitudinal: Longitudinal and Paired-Sample Analyses of Microbiome Data. mSystems 2021, 3, e00219-18. [Google Scholar] [CrossRef] [PubMed]
- Riva, F.; Mammola, S. Rarity facets of biodiversity: Integrating Zeta diversity and Dark diversity to understand the nature of commonness and rarity. Ecol. Evol. 2021, 11, 13912–13919. [Google Scholar] [CrossRef]
- Buckley, H.L.; Day, N.J.; Case, B.S.; Lear, G. Measuring change in biological communities: Multivariate analysis approaches for temporal datasets with low sample size. PeerJ 2021, 9, e11096. [Google Scholar] [CrossRef]
- Darcy, J.L.; Washburne, A.D.; Robeson, M.S.; Prest, T.; Schmidt, S.K.; Lozupone, C.A. A phylogenetic model for the recruitment of species into microbial communities and application to studies of the human microbiome. ISME J. 2020, 14, 1359–1368. [Google Scholar] [CrossRef]
- Shields-Cutler, R.R.; Al-Ghalith, G.A.; Yassour, M.; Knights, D. SplinectomeR Enables Group Comparisons in Longitudinal Microbiome Studies. Front. Microbiol. 2018, 9, 785. [Google Scholar] [CrossRef]
- Bodein, A.; Chapleur, O.; Droit, A.; Lê Cao, K.-A. A Generic Multivariate Framework for the Integration of Microbiome Longitudinal Studies With Other Data Types. Front. Genet. 2019, 10, 963. [Google Scholar] [CrossRef]
- Shenhav, L.; Furman, O.; Briscoe, L.; Thompson, M.; Silverman, J.D.; Mizrahi, I.; Halperin, E. Modeling the temporal dynamics of the gut microbial community in adults and infants. PLOS Comput. Biol. 2019, 15, e1006960. [Google Scholar] [CrossRef]
- McGlinn, D.J.; Xiao, X.; May, F.; Gotelli, N.J.; Engel, T.; Blowes, S.A.; Knight, T.M.; Purschke, O.; Chase, J.M.; McGill, B.J. Measurement of Biodiversity (MoB): A method to separate the scale-dependent effects of species abundance distribution, density, and aggregation on diversity change. Methods Ecol. Evol. 2019, 10, 258–269. [Google Scholar] [CrossRef]
- Karczewski, K.J.; Snyder, M.P. Integrative omics for health and disease. Nat. Rev. Genet. 2018, 19, 299–310. [Google Scholar] [CrossRef] [PubMed]
- Planell, N.; Lagani, V.; Sebastian-Leon, P.; van der Kloet, F.; Ewing, E.; Karathanasis, N.; Urdangarin, A.; Arozarena, I.; Jagodic, M.; Tsamardinos, I.; et al. STATegra: Multi-Omics Data Integration—A Conceptual Scheme with a Bioinformatics Pipeline. Front. Genet. 2021, 12, 620453. [Google Scholar] [CrossRef] [PubMed]
- Mills, S.; Trego, A.C.; Lens, P.N.L.; Ijaz, U.Z.; Collins, G. A Distinct, Flocculent, Acidogenic Microbial Community Accompanies Methanogenic Granules in Anaerobic Digesters. Microbiol. Spectr. 2021, 9, e00784-21. [Google Scholar] [CrossRef] [PubMed]
- Frau, A.; Ijaz, U.Z.; Slater, R.; Jonkers, D.; Penders, J.; Campbell, B.J.; Kenny, J.G.; Hall, N.; Lenzi, L.; Burkitt, M.D.; et al. Inter-kingdom relationships in Crohn’s disease explored using a multi-omics approach. Gut Microbes 2021, 13, 1930871. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Shi, P.; Li, H. Generalized linear models with linear constraints for microbiome compositional data. Biometrics 2019, 75, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Ovaskainen, O.; Tikhonov, G.; Norberg, A.; Guillaume Blanchet, F.; Duan, L.; Dunson, D.; Roslin, T.; Abrego, N. How to make more out of community data? A conceptual framework and its implementation as models and software. Ecol. Lett. 2017, 20, 561–576. [Google Scholar] [CrossRef]
- Ovaskainen, O.; Abrego, N. Joint Species Distribution Modelling: With Applications in R; Cambridge University Press: Cambridge, UK, 2020; ISBN 1108674151. [Google Scholar]
- Tikhonov, G.; Opedal, Ø.H.; Abrego, N.; Lehikoinen, A.; de Jonge, M.M.J.; Oksanen, J.; Ovaskainen, O. Joint species distribution modelling with the r-package Hmsc. Methods Ecol. Evol. 2020, 11, 442–447. [Google Scholar] [CrossRef]
- Skrondal, A.; Rabe-Hesketh, S. Generalized Latent Variable Modeling: Multilevel, Longitudinal, and Structural Equation Models; Chapman and Hall/CRC: Boca Raton, FL, USA, 2004; ISBN 042920549X. [Google Scholar]
- Warton, D.I.; Blanchet, F.G.; O’Hara, R.B.; Ovaskainen, O.; Taskinen, S.; Walker, S.C.; Hui, F.K.C. So many variables: Joint modeling in community ecology. Trends Ecol. Evol. 2015, 30, 766–779. [Google Scholar] [CrossRef]
- Niku, J.; Warton, D.I.; Hui, F.K.C.; Taskinen, S. Generalized linear latent variable models for multivariate count and biomass data in ecology. J. Agric. Biol. Environ. Stat. 2017, 22, 498–522. [Google Scholar] [CrossRef]
- Aitchison, J. The statistical analysis of compositional data. J. R. Stat. Soc. Ser. B 1982, 44, 139–160. [Google Scholar] [CrossRef]
- Susin, A.; Wang, Y.; Lê Cao, K.-A.; Calle, M.L. Variable selection in microbiome compositional data analysis. NAR Genom. Bioinform. 2020, 2, lqaa029. [Google Scholar] [CrossRef] [PubMed]
- Rivera-Pinto, J.; Egozcue, J.J.; Pawlowsky-Glahn, V.; Paredes, R.; Noguera-Julian, M.; Calle, M.L. Balances: A new perspective for microbiome analysis. mSystems 2018, 3, e00053-18. [Google Scholar] [CrossRef]
- le Cessie, S.; van Houwelingen, J.C. Ridge Estimators in Logistic Regression. J. R. Stat. Soc. Ser. C Appl. Stat. 1992, 41, 191–201. [Google Scholar] [CrossRef]
- Zou, H.; Hastie, T. Regularization and variable selection via the elastic net. J. R. Stat. Soc. Ser. B Stat. Methodol. 2005, 67, 301–320. [Google Scholar] [CrossRef]
- Tibshirani, R. Regression Shrinkage and Selection via the Lasso. J. R. Stat. Soc. Ser. B 1996, 58, 267–288. [Google Scholar] [CrossRef]
- Knight, R.; Vrbanac, A.; Taylor, B.C.; Aksenov, A.; Callewaert, C.; Debelius, J.; Gonzalez, A.; Kosciolek, T.; McCall, L.-I.; McDonald, D. Best practices for analysing microbiomes. Nat. Rev. Microbiol. 2018, 16, 410–422. [Google Scholar] [CrossRef]
- Langille, M.G.I. Exploring linkages between taxonomic and functional profiles of the human microbiome. mSystems 2018, 3, e00163-17. [Google Scholar] [CrossRef]
- Huttenhower, C.; Gevers, D.; Knight, R.; Abubucker, S.; Badger, J.H.; Chinwalla, A.T.; Giglio, M.G. Structure, function and diversity of the healthy human microbiome. Nature 2012, 486, 207. [Google Scholar]
- Jing, G.; Zhang, Y.; Cui, W.; Liu, L.; Xu, J.; Su, X. Meta-Apo improves accuracy of 16S-amplicon-based prediction of microbiome function. BMC Genom. 2021, 22, 9. [Google Scholar] [CrossRef]
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.R.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Huttenhower, C.; Langille, M.G.I. PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol. 2020, 38, 685–688. [Google Scholar] [CrossRef]
- Wemheuer, F.; Taylor, J.A.; Daniel, R.; Johnston, E.; Meinicke, P.; Thomas, T.; Wemheuer, B. Tax4Fun2: Prediction of habitat-specific functional profiles and functional redundancy based on 16S rRNA gene sequences. Environ. Microbiome 2020, 15, 11. [Google Scholar] [CrossRef] [PubMed]
- Louca, S.; Parfrey, L.W.; Doebeli, M. Decoupling function and taxonomy in the global ocean microbiome. Science 2016, 353, 1272–1277. [Google Scholar] [CrossRef] [PubMed]
- Ward, T.; Larson, J.; Meulemans, J.; Hillmann, B.; Lynch, J.; Sidiropoulos, D.; Spear, J.R.; Caporaso, G.; Blekhman, R.; Knight, R.; et al. BugBase predicts organism-level microbiome phenotypes. bioRxiv 2017. [Google Scholar] [CrossRef]
- Zhang, Y.; Jing, G.; Chen, Y.; Li, J.; Su, X. Hierarchical Meta-Storms enables comprehensive and rapid comparison of microbiome functional profiles on a large scale using hierarchical dissimilarity metrics and parallel computing. Bioinforma. Adv. 2021, 1, vbab003. [Google Scholar] [CrossRef]
- Campanaro, S.; Treu, L.; Rodriguez-R, L.M.; Kovalovszki, A.; Ziels, R.M.; Maus, I.; Zhu, X.; Kougias, P.G.; Basile, A.; Luo, G.; et al. The anaerobic digestion microbiome: A collection of 1600 metagenome-assembled genomes shows high species diversity related to methane production. bioRxiv 2019. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Trego, A.; Keating, C.; Nzeteu, C.; Graham, A.; O’Flaherty, V.; Ijaz, U.Z. Beyond Basic Diversity Estimates—Analytical Tools for Mechanistic Interpretations of Amplicon Sequencing Data. Microorganisms 2022, 10, 1961. https://doi.org/10.3390/microorganisms10101961
Trego A, Keating C, Nzeteu C, Graham A, O’Flaherty V, Ijaz UZ. Beyond Basic Diversity Estimates—Analytical Tools for Mechanistic Interpretations of Amplicon Sequencing Data. Microorganisms. 2022; 10(10):1961. https://doi.org/10.3390/microorganisms10101961
Chicago/Turabian StyleTrego, Anna, Ciara Keating, Corine Nzeteu, Alison Graham, Vincent O’Flaherty, and Umer Zeeshan Ijaz. 2022. "Beyond Basic Diversity Estimates—Analytical Tools for Mechanistic Interpretations of Amplicon Sequencing Data" Microorganisms 10, no. 10: 1961. https://doi.org/10.3390/microorganisms10101961
APA StyleTrego, A., Keating, C., Nzeteu, C., Graham, A., O’Flaherty, V., & Ijaz, U. Z. (2022). Beyond Basic Diversity Estimates—Analytical Tools for Mechanistic Interpretations of Amplicon Sequencing Data. Microorganisms, 10(10), 1961. https://doi.org/10.3390/microorganisms10101961