
_Brussaard.png)
The Viral Fraction Metatranscriptomes of Lake Baikal

 , , , , and
, , , , and
Abstract
:1. Introduction
2. Materials and Methods
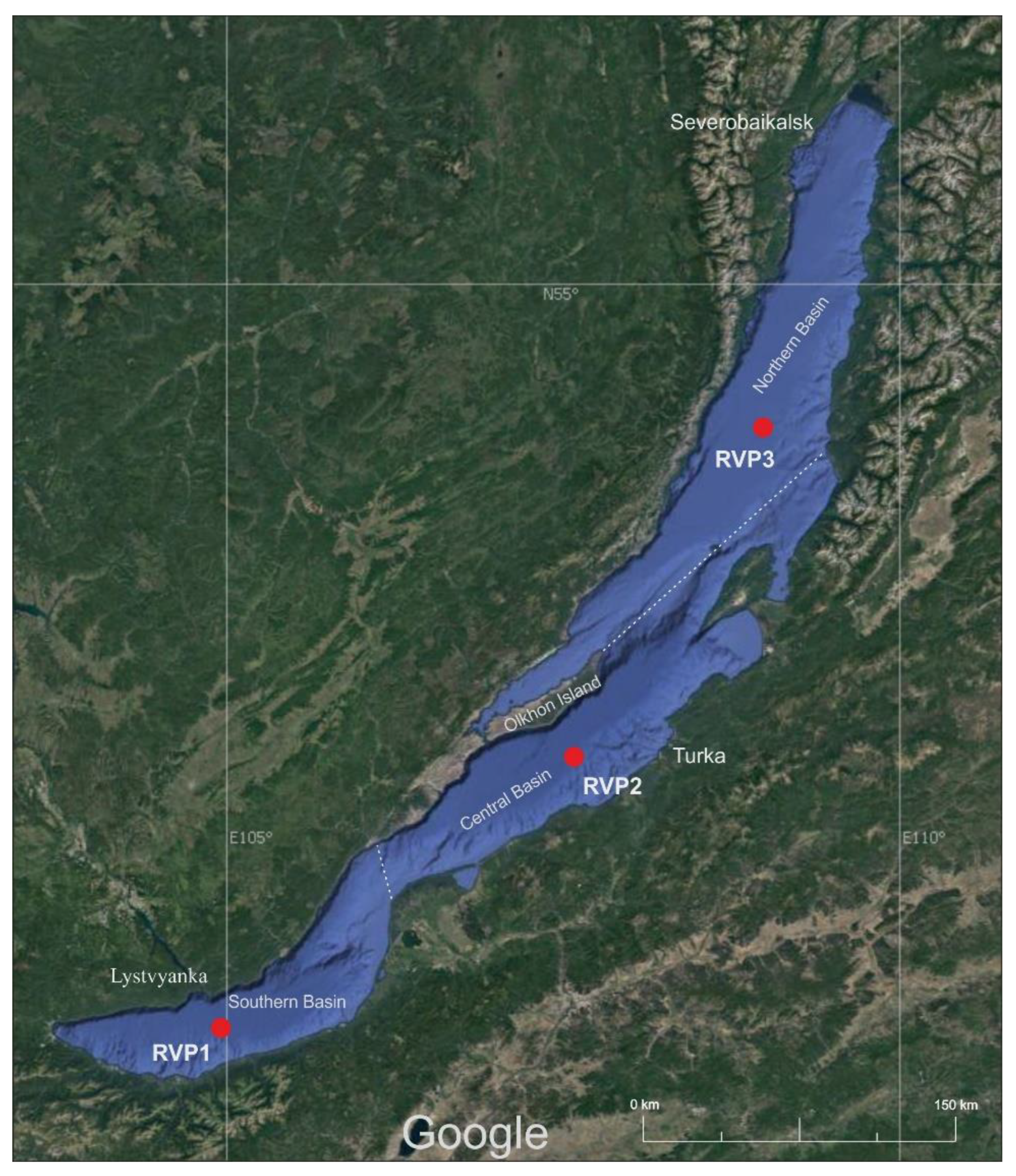
2.1. Sample Preparation and Sequencing
2.2. Bioinformatic Analysis
3. Results
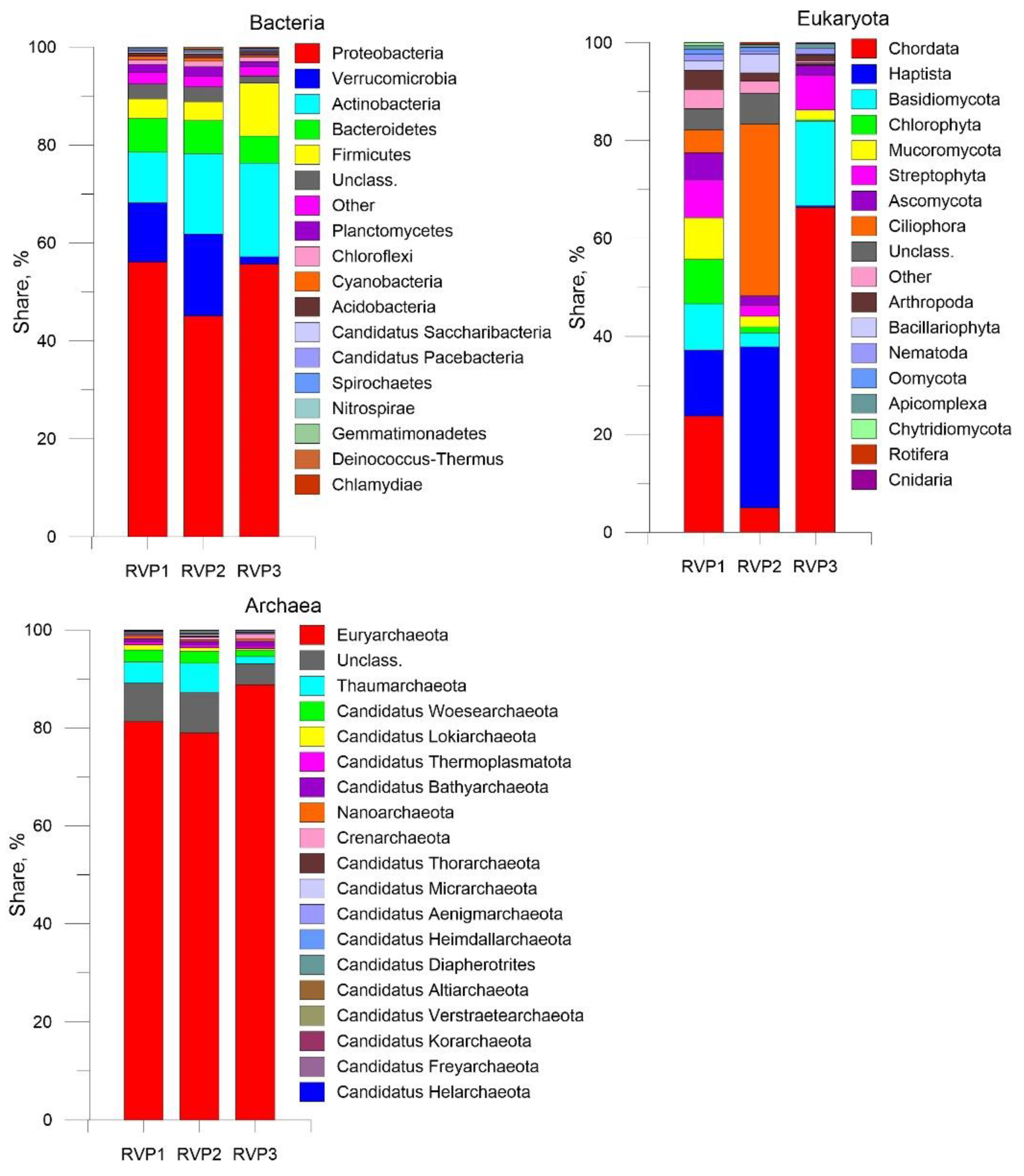
3.1. Taxonomic Annotation of Transcripts
- putative viral DNA polymerase (YP_009243641), identity 50–71%, lowest e-value 2.7 × 10−32, ORF length 147–327 nt, transcript belongs to Bovine retrovirus CH15 (Betaretrovirus) hosted by large cattle;
- pol protein (YP_009513211), identity 46.3–64.7%, lowest e-value 5.0 × 10−22, ORF length 198–231 nt, similar to Koala retrovirus (Gammaretrovirus) according to the annotation;
- ubiquitin-like protein (NP_598374), identity 58.6%, e-value 2.3 × 10−12, closest relative is Murine osteosarcoma virus (Gammaretrovirus), natural hosts are mice;
- gag protein (NP_056901), identity 100%, e-value 3.6 × 10−62, ORF length 231–288 nt, the closest relative is equine infectious anaemia virus (Orthoretrovirinae), infects members of the horse family (Equidae) and others.
3.2. Functional Analysis
3.2.1. PHROG Database
3.2.2. VOG Database
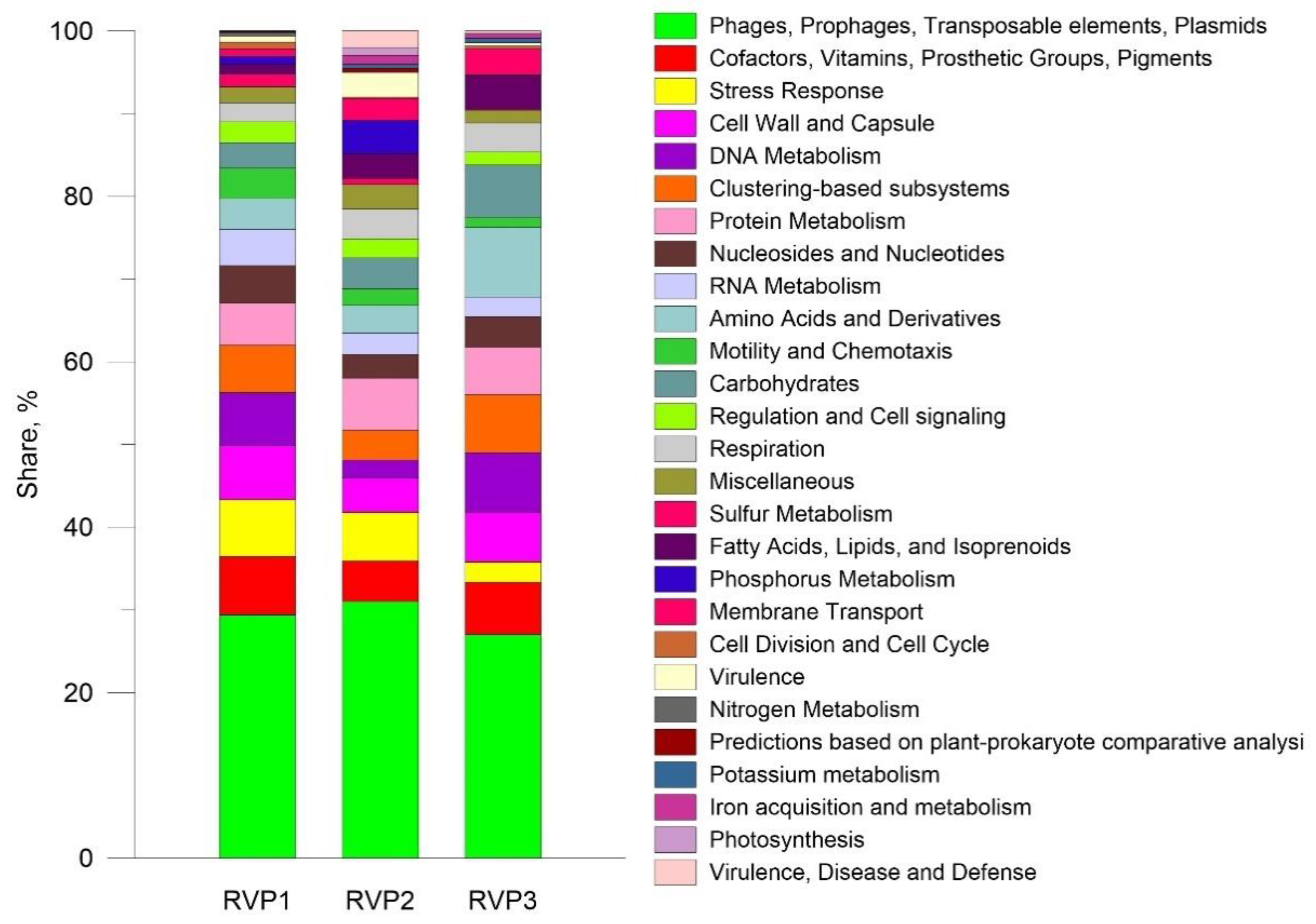
3.2.3. SEED Subsystems
3.2.4. AMG Genes in Metatranscriptomes
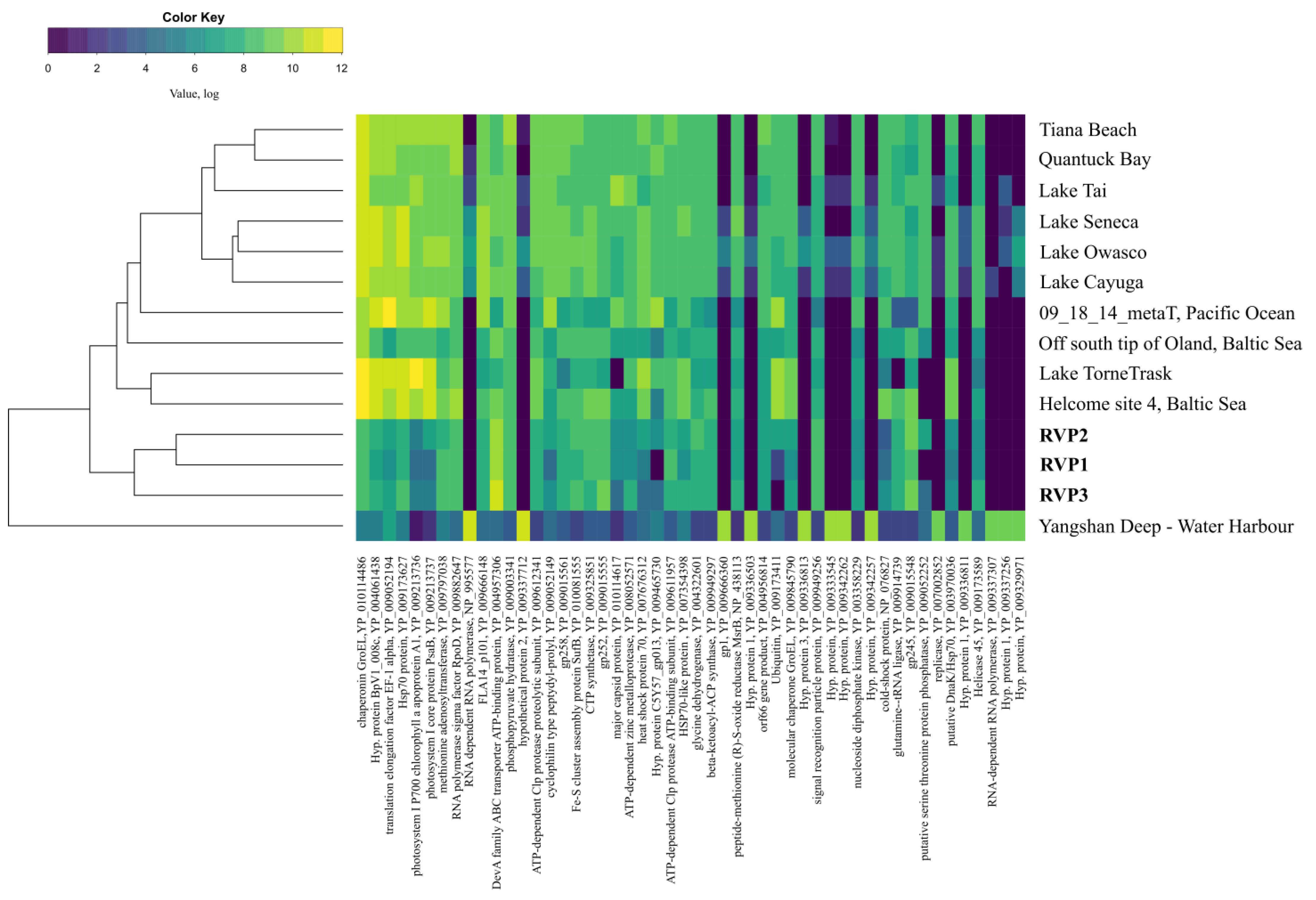
3.3. Comparative Analysis of Transcriptomes
3.4. Transcripts of Putative Hosts
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Steward, G.F.; Culley, A.I.; Mueller, J.A.; Wood-Charlson, E.M.; Belcaid, M.; Poisson, G. Are We Missing Half of the Viruses in the Ocean? ISME J. 2013, 7, 672–679. [Google Scholar] [CrossRef] [PubMed]
- Miranda, J.A.; Culley, A.I.; Schvarcz, C.R.; Steward, G.F. RNA Viruses as Major Contributors to Antarctic Virioplankton. Environ. Microbiol. 2016, 18, 3714–3727. [Google Scholar] [CrossRef] [PubMed]
- Suttle, C.A. Marine Viruses—Major Players in the Global Ecosystem. Nat. Rev. Microbiol. 2007, 5, 801–812. [Google Scholar] [CrossRef] [PubMed]
- Palermo, C.N.; Fulthorpe, R.R.; Saati, R.; Short, S.M. Metagenomic Analysis of Virus Diversity and Relative Abundance in a Eutrophic Freshwater Harbour. Viruses 2019, 11, 792. [Google Scholar] [CrossRef]
- Arkhipova, K.; Skvortsov, T.; Quinn, J.P.; McGrath, J.W.; Allen, C.C.; Dutilh, B.E.; Mcelarney, Y.; Kulakov, L.A. Temporal Dynamics of Uncultured Viruses: A New Dimension in Viral Diversity. ISME J. 2018, 12, 199–211. [Google Scholar] [CrossRef] [PubMed]
- Moon, K.; Kim, S.; Kang, I.; Cho, J.-C. Viral Metagenomes of Lake Soyang, the Largest Freshwater Lake in South Korea. Sci. Data 2020, 7, 349. [Google Scholar] [CrossRef]
- Culley, A.I.; Mueller, J.A.; Belcaid, M.; Wood-Charlson, E.M.; Poisson, G.; Steward, G.F. The Characterization of RNA Viruses in Tropical Seawater Using Targeted PCR and Metagenomics. mBio 2014, 5, e01210-14. [Google Scholar] [CrossRef]
- Djikeng, A.; Kuzmickas, R.; Anderson, N.G.; Spiro, D.J. Metagenomic Analysis of RNA Viruses in a Fresh Water Lake. PLoS ONE 2009, 4, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Moniruzzaman, M.; Wurch, L.L.; Alexander, H.; Dyhrman, S.T.; Gobler, C.J.; Wilhelm, S.W. Virus-Host Relationships of Marine Single-Celled Eukaryotes Resolved from Metatranscriptomics. Nat. Commun. 2017, 8, 16054. [Google Scholar] [CrossRef]
- Zeigler Allen, L.; McCrow, J.P.; Ininbergs, K.; Dupont, C.L.; Badger, J.H.; Hoffman, J.M.; Ekman, M.; Allen, A.E.; Bergman, B.; Venter, J.C. The Baltic Sea Virome: Diversity and Transcriptional Activity of DNA and RNA Viruses. mSystems 2017, 2, e00125-16. [Google Scholar] [CrossRef] [Green Version]
- Hewson, I.; Bistolas, K.S.I.; Button, J.B.; Jackson, E.W. Occurrence and Seasonal Dynamics of RNA Viral Genotypes in Three Contrasting Temperate Lakes. PLoS ONE 2018, 13, e0194419. [Google Scholar] [CrossRef]
- Wolf, Y.I.; Silas, S.; Wang, Y.; Wu, S.; Bocek, M.; Kazlauskas, D.; Krupovic, M.; Fire, A.; Dolja, V.V.; Koonin, E.V. Doubling of the Known Set of RNA Viruses by Metagenomic Analysis of an Aquatic Virome. Nat. Microbiol. 2020, 5, 1262–1270. [Google Scholar] [CrossRef] [PubMed]
- Grachev, M.A.; Kumarev, V.P.; Mamaev, L.V.; Zorin, V.L.; Baranova, L.V.; Denikina, N.N.; Belikov, S.I.; Petrov, E.A.; Kolesnik, V.S.; Kolesnik, R.S.; et al. Distemper Virus in Baikal Seals. Nature 1989, 338, 209–210. [Google Scholar] [CrossRef]
- Likhoshway, Y.V.; Grachev, M.A.; Kumarev, V.P.; Solodun, Y.V.; Goldberg, O.A.; Belykh, O.I.; Nagieva, F.G.; Nikulina, V.G.; Kolesnik, B.S. Baikal Seal Virus. Nature 1989, 339, 266. [Google Scholar] [CrossRef]
- Belykh, O.I.; Goldberg, O.A.; Likhoshway, E.V.; Grachev, M.A. Light, Electron and Immuno-Electron Microscopy of Organs from Seals of Lake Baikal Sampled during the Morbillivirus Infection of 1987–1988. Eur. J. Vet. Pathol. 1997, 3, 133–145. [Google Scholar]
- Butina, T.V.; Belykh, O.I.; Belikov, S.I. Molecular-Genetic Identification of T4 Bacteriophages in Lake Baikal. Dokl. Biochem. Biophys. 2010, 433, 175–178. [Google Scholar] [CrossRef]
- Butina, T.V.; Potapov, S.A.; Belykh, O.I.; Damdinsuren, N.; Choidash, B. Genetic Diversity of the Family Myoviridae Cyanophages in Lake Baikal. Seriya Biologiya. Ekol. Izv. Irkutsk. Gos. Univ. 2012, 5, 17–22. [Google Scholar]
- Potapov, S.A.; Butina, T.V.; Belykh, O.I.; Belikov, S.I. Genetic Diversity of T4-like Bacteriophages in Lake Baikal. Bull. Irkutsk. State Univ. Series Biol. Ecol. 2013, 3, 14–19. [Google Scholar]
- Butina, T.V.; Bukin, Y.S.; Krasnopeev, A.S.; Belykh, O.I.; Tupikin, A.E.; Kabilov, M.R.; Sakirko, M.V.; Belikov, S.I. Estimate of the Diversity of Viral and Bacterial Assemblage in the Coastal Water of Lake Baikal. FEMS Microbiol. Lett. 2019, 366, fnz094. [Google Scholar] [CrossRef]
- Potapov, S.A.; Tikhonova, I.V.; Krasnopeev, A.Y.; Kabilov, M.R.; Tupikin, A.E.; Chebunina, N.S.; Zhuchenko, N.A.; Belykh, O.I. Metagenomic Analysis of Virioplankton from the Pelagic Zone of Lake Baikal. Viruses 2019, 11, 991. [Google Scholar] [CrossRef]
- Sykilinda, N.N.; Bondar, A.A.; Gorshkova, A.S.; Kurochkina, L.P.; Kulikov, E.E.; Shneider, M.M.; Kadykov, V.A.; Solovjeva, N.V.; Kabilov, M.R.; Mesyanzhinov, V.V.; et al. Complete Genome Sequence of the Novel Giant Pseudomonas Phage PaBG. Genome Announc. 2014, 2, e00929-13. [Google Scholar] [CrossRef] [PubMed]
- Butina, T.V.; Bukin, Y.S.; Petrushin, I.S.; Tupikin, A.E.; Kabilov, M.R.; Belikov, S.I. Extended Evaluation of Viral Diversity in Lake Baikal through Metagenomics. Microorganisms 2021, 9, 760. [Google Scholar] [CrossRef]
- Evseev, P.; Lukianova, A.; Sykilinda, N.; Gorshkova, A.; Bondar, A.; Shneider, M.; Kabilov, M.; Drucker, V.; Miroshnikov, K. Pseudomonas Phage MD8: Genetic Mosaicism and Challenges of Taxonomic Classification of Lambdoid Bacteriophages. Int. J. Mol. Sci. 2021, 22, 10350. [Google Scholar] [CrossRef] [PubMed]
- Coutinho, F.H.; Cabello-Yeves, P.J.; Gonzalez-Serrano, R.; Rosselli, R.; López-Pérez, M.; Zemskaya, T.I.; Zakharenko, A.S.; Ivanov, V.G.; Rodriguez-Valera, F. New Viral Biogeochemical Roles Revealed through Metagenomic Analysis of Lake Baikal. Microbiome 2020, 8, 163. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Lin, X.-D.; Tian, J.-H.; Chen, L.-J.; Chen, X.; Li, C.-X.; Qin, X.-C.; Li, J.; Cao, J.-P.; Eden, J.-S.; et al. Redefining the Invertebrate RNA Virosphere. Nature 2016, 540, 539–543. [Google Scholar] [CrossRef] [PubMed]
- Andrews, S. FastQC: A Quality Control Tool for High Throughput Sequence Data. Available online: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 5 September 2022).
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map Format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Laetsch, D.R.; Blaxter, M.L. BlobTools: Interrogation of Genome Assemblies. F1000Research 2017, 6, 1287. [Google Scholar] [CrossRef]
- Besemer, J. GeneMarkS: A Self-Training Method for Prediction of Gene Starts in Microbial Genomes. Implications for Finding Sequence Motifs in Regulatory Regions. Nucleic Acids Res. 2001, 29, 2607–2618. [Google Scholar] [CrossRef]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and Sensitive Protein Alignment Using DIAMOND. Nat. Methods 2014, 12, 59–60. [Google Scholar] [CrossRef]
- Terzian, P.; Olo Ndela, E.; Galiez, C.; Lossouarn, J.; Pérez Bucio, R.E.; Mom, R.; Toussaint, A.; Petit, M.-A.; Enault, F. PHROG: Families of Prokaryotic Virus Proteins Clustered Using Remote Homology. NAR Genom. Bioinform. 2021, 3, lqab067. [Google Scholar] [CrossRef] [PubMed]
- Silva, G.G.Z.; Green, K.T.; Dutilh, B.E.; Edwards, R.A. SUPER-FOCUS: A Tool for Agile Functional Analysis of Shotgun Metagenomic Data. Bioinformatics 2016, 32, 354–361. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Bolduc, B.; Zayed, A.A.; Varsani, A.; Dominguez-Huerta, G.; Delmont, T.O.; Pratama, A.A.; Gazitúa, M.C.; Vik, D.; Sullivan, M.B.; et al. VirSorter2: A Multi-Classifier, Expert-Guided Approach to Detect Diverse DNA and RNA Viruses. Microbiome 2021, 9, 37. [Google Scholar] [CrossRef] [PubMed]
- Merchant, N.; Lyons, E.; Goff, S.; Vaughn, M.; Ware, D.; Micklos, D.; Antin, P. The IPlant Collaborative: Cyberinfrastructure for Enabling Data to Discovery for the Life Sciences. PLoS Biol. 2016, 14, e1002342. [Google Scholar] [CrossRef]
- Kieft, K.; Zhou, Z.; Anantharaman, K. VIBRANT: Automated Recovery, Annotation and Curation of Microbial Viruses, and Evaluation of Virome Function from Genomic Sequences. bioRxiv 2019, 855387. [Google Scholar] [CrossRef]
- Nishimura, Y.; Yoshida, T.; Kuronishi, M.; Uehara, H.; Ogata, H.; Goto, S. ViPTree: The Viral Proteomic Tree Server. Bioinformatics 2017, 33, 2379–2380. [Google Scholar] [CrossRef]
- Pound, H.L.; Gann, E.R.; Tang, X.; Krausfeldt, L.E.; Huff, M.; Staton, M.E.; Talmy, D.; Wilhelm, S.W. The “Neglected Viruses” of Taihu: Abundant Transcripts for Viruses Infecting Eukaryotes and Their Potential Role in Phytoplankton Succession. Front. Microbiol. 2020, 11, 338. [Google Scholar] [CrossRef]
- Gann, E.R.; Kang, Y.; Dyhrman, S.T.; Gobler, C.J.; Wilhelm, S.W. Metatranscriptome Library Preparation Influences Analyses of Viral Community Activity During a Brown Tide Bloom. Front. Microbiol. 2021, 12, 1126. [Google Scholar] [CrossRef] [PubMed]
- Kolody, B.C.; McCrow, J.P.; Allen, L.Z.; Aylward, F.O.; Fontanez, K.M.; Moustafa, A.; Moniruzzaman, M.; Chavez, F.P.; Scholin, C.A.; Allen, E.E.; et al. Diel Transcriptional Response of a California Current Plankton Microbiome to Light, Low Iron, and Enduring Viral Infection. ISME J. 2019, 13, 2817–2833. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Reyes, A.; Semenkovich, N.P.; Whiteson, K.; Rohwer, F.; Gordon, J.I. Going Viral: Next-Generation Sequencing Applied to Phage Populations in the Human Gut. Nat. Rev. Microbiol. 2012, 10, 607–617. [Google Scholar] [CrossRef]
- Veilleux, H.D.; Misutka, M.D.; Glover, C.N. Environmental DNA and Environmental RNA: Current and Prospective Applications for Biological Monitoring. Sci. Total Environ. 2021, 782, 146891. [Google Scholar] [CrossRef]
- Legendre, M.; Bartoli, J.; Shmakova, L.; Jeudy, S.; Labadie, K.; Adrait, A.; Lescot, M.; Poirot, O.; Bertaux, L.; Bruley, C.; et al. Thirty-Thousand-Year-Old Distant Relative of Giant Icosahedral DNA Viruses with a Pandoravirus Morphology. Proc. Natl. Acad. Sci. USA 2014, 111, 4274–4279. [Google Scholar] [CrossRef] [Green Version]
- Gao, E.-B.; Gui, J.-F.; Zhang, Q.-Y. A Novel Cyanophage with a Cyanobacterial Nonbleaching Protein A Gene in the Genome. J. Virol. 2012, 86, 236–245. [Google Scholar] [CrossRef]
- Sorokovikova, E.; Belykh, O.; Krasnopeev, A.; Potapov, S.; Tikhonova, I.; Khanaev, I.; Kabilov, M.; Baturina, O.; Podlesnaya, G.; Timoshkin, O. First Data on Cyanobacterial Biodiversity in Benthic Biofilms during Mass Mortality of Endemic Sponges in Lake Baikal. J. Great Lakes Res. 2019, 46, 75–84. [Google Scholar] [CrossRef]
- Zhang, W.; Zhou, J.; Liu, T.; Yu, Y.; Pan, Y.; Yan, S.; Wang, Y. Four Novel Algal Virus Genomes Discovered from Yellowstone Lake Metagenomes. Sci. Rep. 2015, 5, 15131. [Google Scholar] [CrossRef] [PubMed]
- Mikhailov, I.S.; Zakharova, Y.R.; Bukin, Y.S.; Galachyants, Y.P.; Petrova, D.P.; Sakirko, M.V.; Likhoshway, Y.V. Co-Occurrence Networks Among Bacteria and Microbial Eukaryotes of Lake Baikal During a Spring Phytoplankton Bloom. Microb. Ecol. 2019, 77, 96–109. [Google Scholar] [CrossRef] [PubMed]
- Sorokovikova, L.M.; Popovskaya, G.I.; Belykh, O.I.; Tomberg, I.V.; Maksimenko, S.Y.; Bashenkhaeva, N.V.; Ivanov, V.G.; Zemskaya, T.I. Plankton Composition and Water Chemistry in the Mixing Zone of the Selenga River with Lake Baikal. Hydrobiologia 2012, 695, 329–341. [Google Scholar] [CrossRef]
- Belykh, O.I.; Sorokovikova, E.G. Autotrophic Picoplankton in Lake Baikal: Abundance, Dynamics, and Distribution. Aquat. Ecosyst. Health Manag. 2003, 6, 251–261. [Google Scholar] [CrossRef]
- Popovskaya, G.I. Ecological Monitoring of Phytoplankton in Lake Baikal. Aquat. Ecosyst. Health Manag. 2000, 3, 215–225. [Google Scholar] [CrossRef]
- Bondarenko, N.A.; Ozersky, T.; Obolkina, L.A.; Tikhonova, I.V.; Sorokovikova, E.G.; Sakirko, M.V.; Potapov, S.A.; Blinov, V.V.; Zhdanov, A.A.; Belykh, O.I. Recent Changes in the Spring Microplankton of Lake Baikal, Russia. Limnologica 2019, 75, 19–29. [Google Scholar] [CrossRef]
- Kolundžija, S.; Cheng, D.-Q.; Lauro, F.M. RNA Viruses in Aquatic Ecosystems through the Lens of Ecological Genomics and Transcriptomics. Viruses 2022, 14, 702. [Google Scholar] [CrossRef] [PubMed]
- Roux, S.; Solonenko, N.E.; Dang, V.T.; Poulos, B.T.; Schwenck, S.M.; Goldsmith, D.B.; Coleman, M.L.; Breitbart, M.; Sullivan, M.B.; Roux, S.; et al. Towards Quantitative Viromics for Both Double-Stranded and Single-Stranded DNA Viruses. PeerJ 2016, 4, e2777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- International Committee on Taxonomy of Viruses (ICTV). Available online: Https://Talk.Ictvonline.Org/Taxonomy/ (accessed on 5 September 2022).
- Potapov, S.A.; Tikhonova, I.; Krasnopeev, A.; Kabilov, M.R.; Tupikin, A.E.; Chebunina, N.; Zhuchenko, N.; Belykh, O.I. Characteristics of the Viromes in the Pelagic Zone of Lake Baikal. Limnol. Freshw. Biol. 2020, 4, 1013–1014. [Google Scholar] [CrossRef]
- Andreani, J.; Aherfi, S.; Khalil, J.Y.B.; di Pinto, F.; Bitam, I.; Raoult, D.; Colson, P.; la Scola, B. Cedratvirus, a Double-Cork Structured Giant Virus, Is a Distant Relative of Pithoviruses. Viruses 2016, 8, 300. [Google Scholar] [CrossRef]
- Bonza, M.C.; Martin, H.; Kang, M.; Lewis, G.; Greiner, T.; Giacometti, S.; van Etten, J.L.; de Michelis, M.I.; Thiel, G.; Moroni, A. A Functional Calcium-Transporting ATPase Encoded by Chlorella Viruses. J. Gen. Virol. 2010, 91, 2620. [Google Scholar] [CrossRef] [PubMed]
- Greiner, T.; Moroni, A.; van Etten, J.L.; Thiel, G. Genes for Membrane Transport Proteins: Not So Rare in Viruses. Viruses 2018, 10, 456. [Google Scholar] [CrossRef] [PubMed]
- Hurwitz, B.L.; U’Ren, J.M. Viral Metabolic Reprogramming in Marine Ecosystems. Curr. Opin. Microbiol. 2016, 31, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Igarashi, K.; Kashiwagi, K. Modulation of Cellular Function by Polyamines. Int. J. Biochem. Cell Biol. 2010, 42, 39–51. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, M.B.; Huang, K.H.; Ignacio-Espinoza, J.C.; Berlin, A.M.; Kelly, L.; Weigele, P.R.; DeFrancesco, A.S.; Kern, S.E.; Thompson, L.R.; Young, S.; et al. Genomic Analysis of Oceanic Cyanobacterial Myoviruses Compared with T4-like Myoviruses from Diverse Hosts and Environments. Environ. Microbiol. 2010, 12, 3035–3056. [Google Scholar] [CrossRef] [PubMed]
- Kieft, K.; Breister, A.M.; Huss, P.; Linz, A.M.; Zanetakos, E.; Zhou, Z.; Rahlff, J.; Esser, S.P.; Probst, A.J.; Raman, S.; et al. Virus-Associated Organosulfur Metabolism in Human and Environmental Systems. Cell Rep. 2021, 36, 109471. [Google Scholar] [CrossRef]
- Zhou, X.; Singh, M.; Sanz Santos, G.; Guerlavais, V.; Carvajal, L.A.; Aivado, M.; Zhan, Y.; Oliveira, M.M.S.; Westerberg, L.S.; Annis, D.A.; et al. Pharmacologic Activation of P53 Triggers Viral Mimicry Response Thereby Abolishing Tumor Immune Evasion and Promoting Antitumor Immunity. Cancer Discov. 2021, 11, 3090–3105. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.-J.; Geng, C.; Jiang, S.-Y.; Zhu, Q.; Yan, Z.-Y.; Tian, Y.-P.; Li, X.-D. A Maize Triacylglycerol Lipase Inhibits Sugarcane Mosaic Virus Infection. Plant Physiol. 2022, 189, 754–771. [Google Scholar] [CrossRef] [PubMed]
- Marine, R.L.; Nasko, D.J.; Wray, J.; Polson, S.W.; Wommack, K.E. Novel Chaperonins Are Prevalent in the Virioplankton and Demonstrate Links to Viral Biology and Ecology. ISME J. 2017, 11, 2479–2491. [Google Scholar] [CrossRef]
- Wu, R.; Davison, M.R.; Gao, Y.; Nicora, C.D.; Mcdermott, J.E.; Burnum-Johnson, K.E.; Hofmockel, K.S.; Jansson, J.K. Moisture Modulates Soil Reservoirs of Active DNA and RNA Viruses. Commun. Biol. 2021, 4, 992. [Google Scholar] [CrossRef]
- Clare, D.K.; Bakkes, P.J.; van Heerikhuizen, H.; van der Vies, S.M.; Saibil, H.R. An Expanded Protein Folding Cage in the GroEL–Gp31 Complex. J. Mol. Biol. 2006, 358, 905–911. [Google Scholar] [CrossRef]
- Campillo-Balderas, J.A.; Lazcano, A.; Becerra, A. Viral Genome Size Distribution Does Not Correlate with the Antiquity of the Host Lineages. Front. Ecol. Evol. 2015, 3, 143. [Google Scholar] [CrossRef]
- Depledge, D.P.; Mohr, I.; Wilson, A.C. Going the Distance: Optimizing RNA-Seq Strategies for Transcriptomic Analysis of Complex Viral Genomes. J. Virol. 2019, 93, e01342-18. [Google Scholar] [CrossRef]
- Urayama, S.; Takaki, Y.; Nishi, S.; Yoshida-Takashima, Y.; Deguchi, S.; Takai, K.; Nunoura, T. Unveiling the RNA Virosphere Associated with Marine Microorganisms. Mol. Ecol. Resour. 2018, 18, 1444–1455. [Google Scholar] [CrossRef]
- Bloomfield, J.A. Lakes of New York State; Elsevier: Amsterdam, The Netherlands, 1978; ISBN 9780121073015. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pithovirus sibericum | ||
|---|---|---|
| Protein | Accession | e-value |
| Glycosyltransferase | YP_009000961 | 3.1 × 10−21 |
| Adenylosuccinate synthetase | YP_009000992 | 4.2 × 10−79 |
| DNA-binding ferritin-like protein | YP_009001267 | 1.8 × 10−17 |
| ABC2 type transporter superfamily protein | YP_009001225 | 1.2 × 10−17 |
| PDZ serine protease | YP_009001035 | 2.9 × 10−9 |
| pv_324 | YP_009001226 | 3.0 × 10−12 |
| Ser/Thr protein kinase | YP_009001306 | 1.2 × 10−12 |
| Glycosyltransferase family 2 | YP_009001307 | 9.9 × 10−10 |
| Deoxycytidine triphosphate deaminase | YP_009001173 | 4.1 × 10−10 |
| Ribonucleoside-diphosphate reductase | YP_009001342 | 1.4 × 10−8 |
| Formamidopyrimidine-DNA glycosylase | YP_009001363 | 2.3 × 10−15 |
| Ran-like GTP-binding protein | YP_009001029 | 7.6 × 10−12 |
| Cedratvirus A11 | ||
| Protein | Accession | e-value |
| D-3-phosphoglycerate dehydrogenase, type2 | YP_009328950 | 1.7 × 10−99 |
| DNA-directed RNA polymerase subunit RPB2 | YP_009329295 | 2.4 × 10−14 |
| Adenylosuccinate synthetase | YP_009329210 | 7.8 × 10−14 |
| dTDPD-glucose 4,6-dehydratase | YP_009329097 | 4.2 × 10−11 |
| ABC2 type transporter superfamily protein | YP_009329403 | 3.2 × 10−26 |
| NAD dependent epimerase/dehydratase | YP_009329336 | 9.8 × 10−19 |
| Translation elongation factor EF-1 subunit alpha | YP_009329269 | 3.2 × 10−26 |
| Macrocin O-methyltransferase | YP_009329463 | 6.4 × 10−25 |
| PD-(D/E)XK nuclease | YP_009329328 | 2.5 × 10−17 |
| Hexapeptide transferase | YP_009329047 | 4.9 × 10−12 |
| 5′nucleotidase/apyrase | YP_009329013 | 8.0 × 10−18 |
| Putative serine/threonine-protein kinase/receptor | YP_009329205 | 9.7 × 10−10 |
| AMG KO Name | AMG KO | RVP1 | RVP2 | RVP3 | Enzyme | Pathway |
|---|---|---|---|---|---|---|
| GCH1 | K01495 | 1 | 0 | 0 | GTP-cyclohydrolase | Folate biosynthesis |
| gmhC | K03272 | 2 | 0 | 0 | D-beta-D-heptose 7-phosphate kinase | Lipopolysaccharide biosynthesis |
| queE | K10026 | 1 | 1 | 0 | 7-carboxy-7-deazaguanine synthase | Folate biosynthesis |
| pbsA1 | K21480 | 1 | 0 | 0 | heme oxygenase | Porphyrin metabolism |
| GAOA | K04618 | 1 | 0 | 0 | galactose oxidase | Galactose metabolism |
| glf | K01854 | 1 | 0 | 0 | UDP-galactopyranose mutase | Galactose metabolism |
| SCD | K00507 | 1 | 0 | 0 | stearoyl-CoA desaturase | Biosynthesis of unsaturated fatty acids |
| kdsD | K06041 | 1 | 0 | 0 | arabinose-5-phosphate isomerase | Lipopolysaccharide biosynthesis |
| lpxH | K03269 | 1 | 0 | 0 | UDP-2,3-diacylglucosamine hydrolase | Lipopolysaccharide biosynthesis |
| NAMPT | K03462 | 1 | 0 | 0 | nicotinamide phosphoribosyltransferase | Nicotinate and nicotinamide metabolism |
| cysC | K00860 | 1 | 0 | 0 | adenylylsulfate kinase | Purine metabolism |
| P4HA | K00472 | 1 | 0 | 0 | prolyl 4-hydroxylase | Arginine and proline metabolism |
| TGL2 | K01046 | 0 | 1 | 0 | triacylglycerol lipase | Glycerolipid metabolism |
| gpmB | K15634 | 0 | 1 | 0 | 2,3-bisphosphoglycerate-dependent phosphoglycerate mutase | Glycine, serine, and threonine metabolism |
| DNMT1 | K00558 | 0 | 1 | 0 | DNA (cytosine-5)-methyltransferase 1 | Cysteine and methionine metabolism |
| cobS | K09882 | 0 | 0 | 1 | cobaltochelatase CobS | Porphyrin metabolism |
| metK | K00789 | 0 | 0 | 1 | S-adenosylmethionine synthetase | Cysteine and methionine metabolism |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Potapov, S.; Krasnopeev, A.; Tikhonova, I.; Podlesnaya, G.; Gorshkova, A.; Belykh, O. The Viral Fraction Metatranscriptomes of Lake Baikal. Microorganisms 2022, 10, 1937. https://doi.org/10.3390/microorganisms10101937
Potapov S, Krasnopeev A, Tikhonova I, Podlesnaya G, Gorshkova A, Belykh O. The Viral Fraction Metatranscriptomes of Lake Baikal. Microorganisms. 2022; 10(10):1937. https://doi.org/10.3390/microorganisms10101937
Chicago/Turabian StylePotapov, Sergey, Andrey Krasnopeev, Irina Tikhonova, Galina Podlesnaya, Anna Gorshkova, and Olga Belykh. 2022. "The Viral Fraction Metatranscriptomes of Lake Baikal" Microorganisms 10, no. 10: 1937. https://doi.org/10.3390/microorganisms10101937
APA StylePotapov, S., Krasnopeev, A., Tikhonova, I., Podlesnaya, G., Gorshkova, A., & Belykh, O. (2022). The Viral Fraction Metatranscriptomes of Lake Baikal. Microorganisms, 10(10), 1937. https://doi.org/10.3390/microorganisms10101937