
Record-Breaking Rain Event Altered Estuarine Viral Assemblages


Abstract
:1. Introduction
2. Materials and Methods
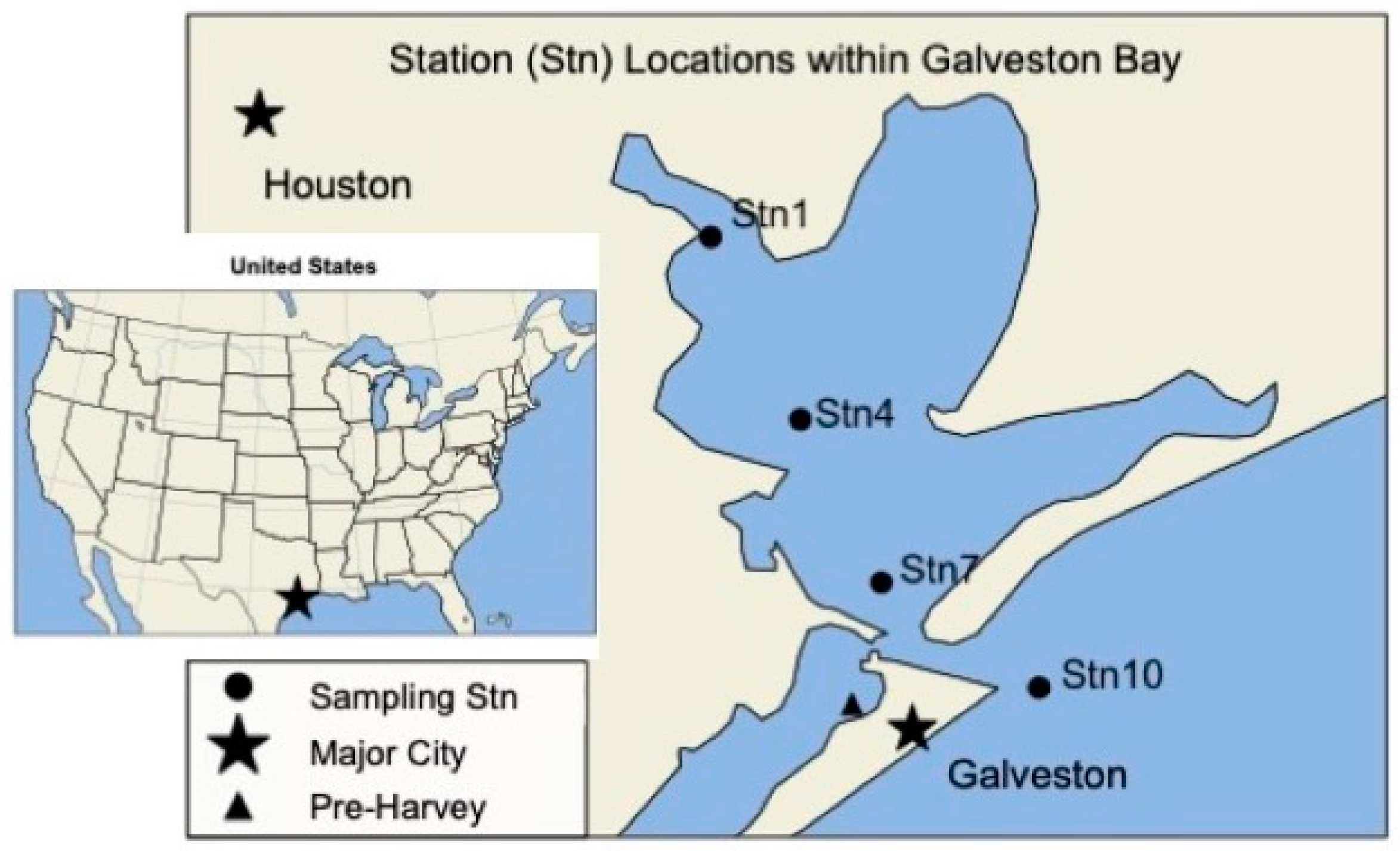
2.1. Sample Collection and Processing
2.2. DNA Extractions and Sequencing
2.3. Metagenomic Data Analysis
2.4. Identification of Putative AMGs
2.5. Viral Abundance
2.6. Phylogenetic Analysis
2.7. Data Availability
3. Results and Discussion
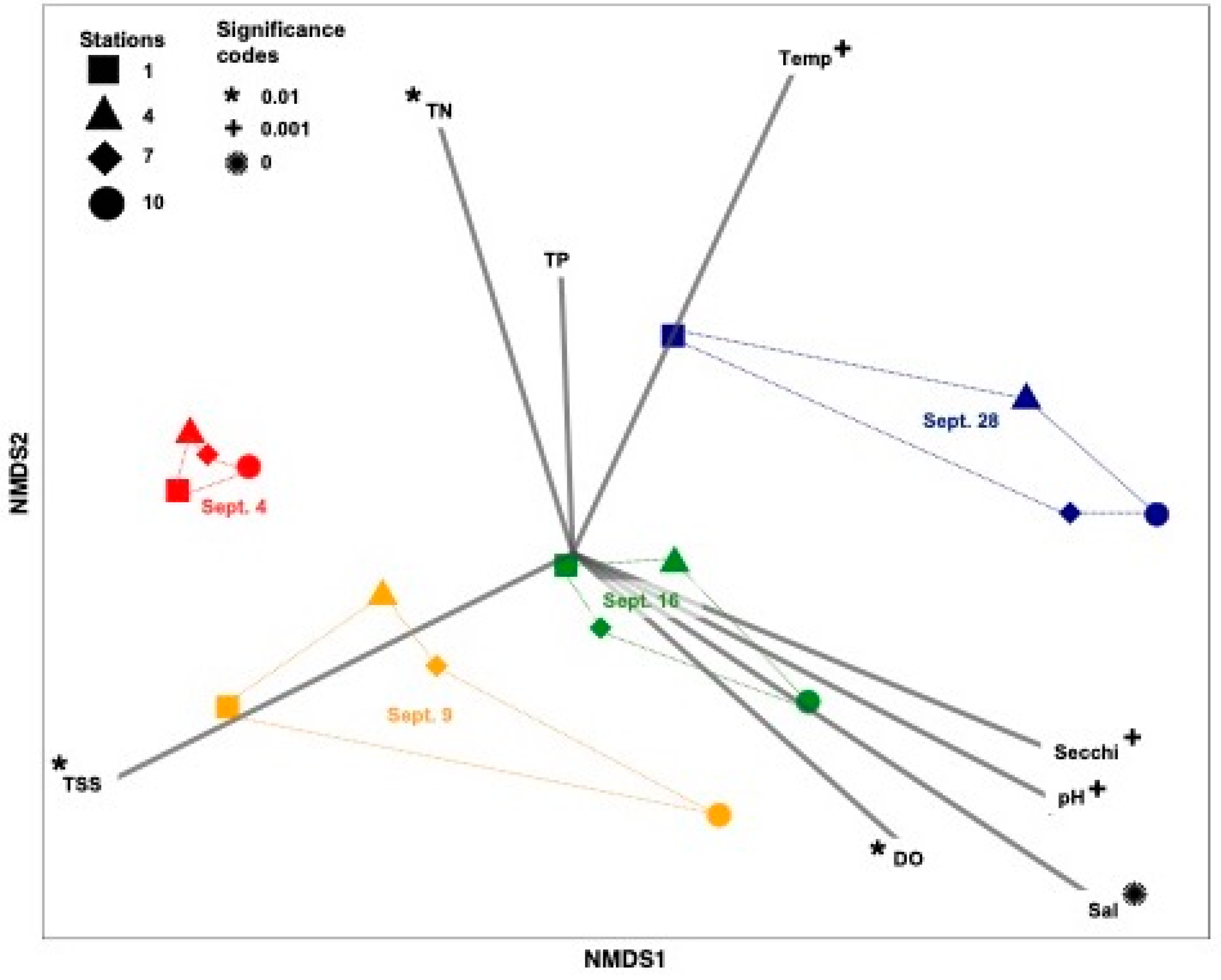
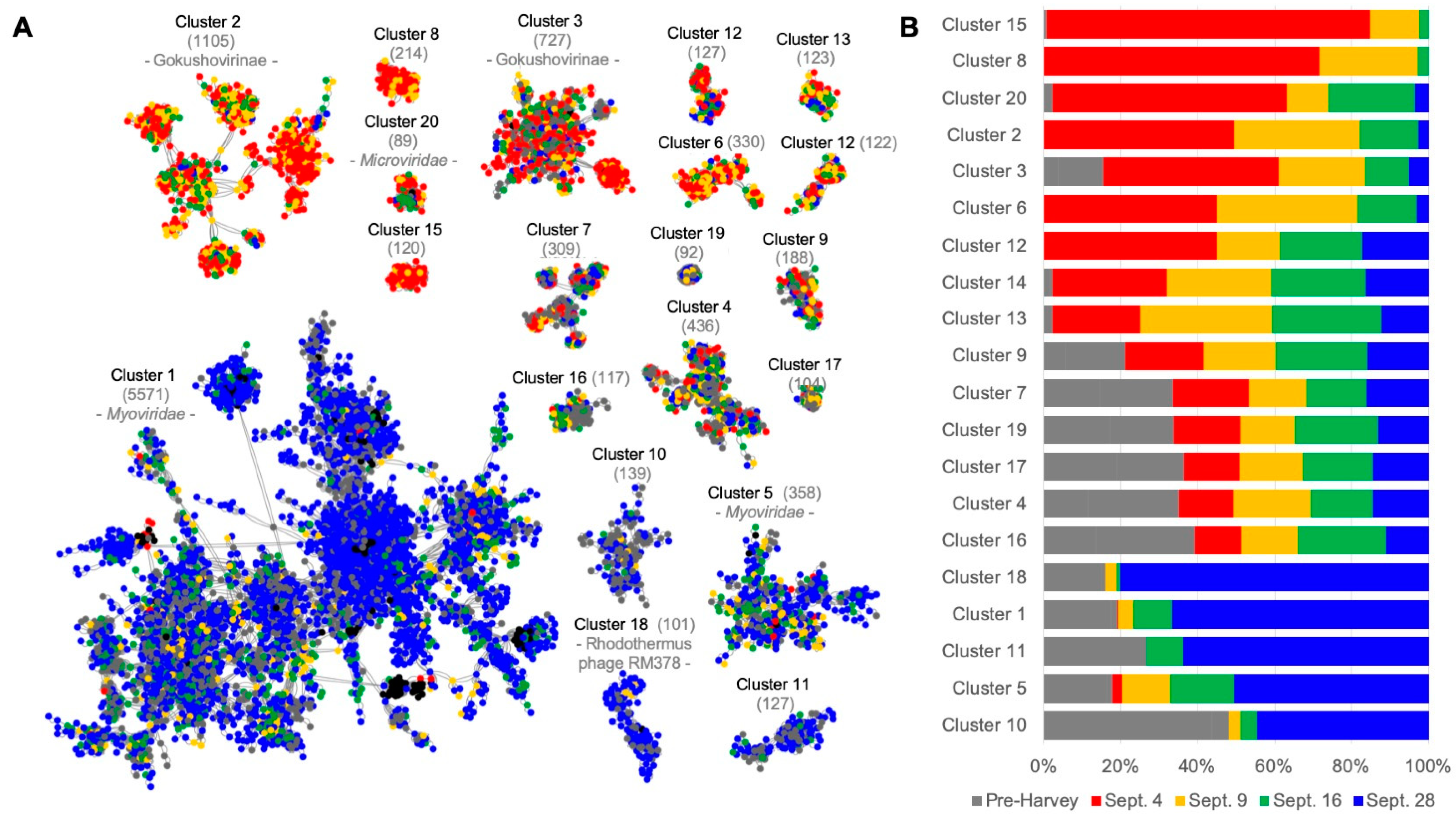
3.1. Hurricane Harvey Changed the Viral Community Diversity and Composition
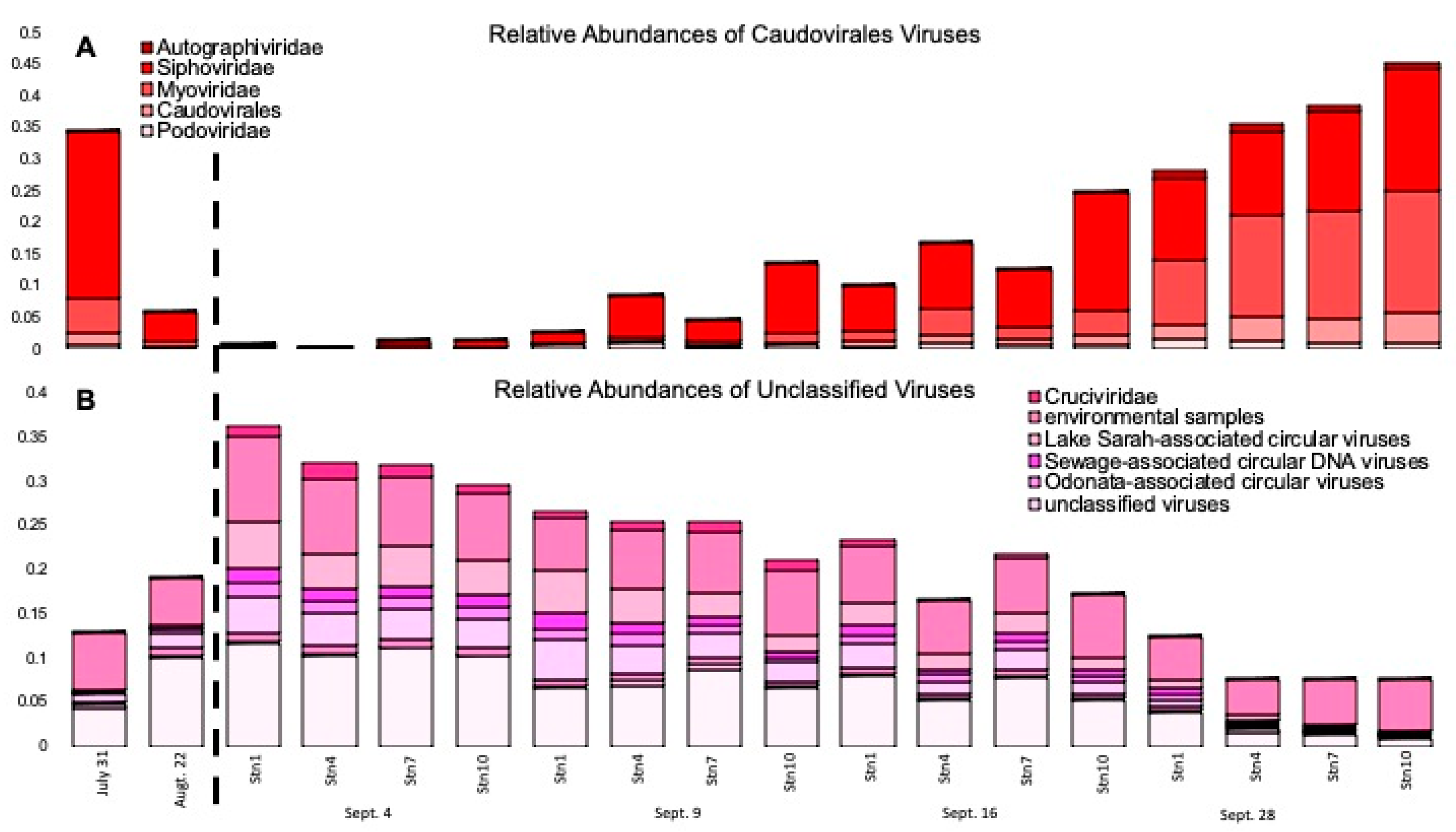
3.2. The Number of Viruses Identified in the Microbial Fraction Escalated throughout the Weeks Following Hurricane Harvey
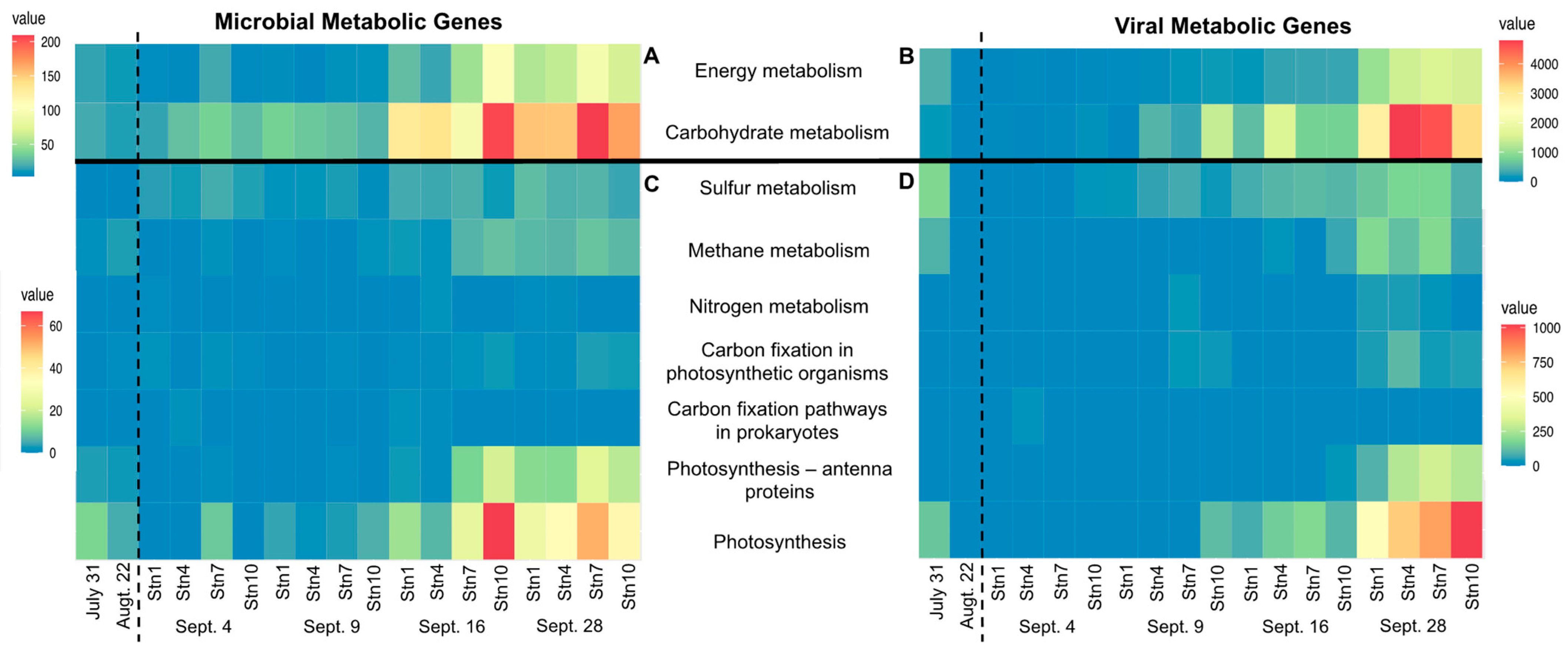
3.3. Hurricane Harvey Increased the Abundance of Virally Encoded Metabolic Genes Involved in Major Biogeochemical Cycles
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Suttle, C.A. Viruses in the sea. Nature 2005, 437, 356–361. [Google Scholar] [CrossRef] [PubMed]
- Tsiola, A.; Fodelianakis, S.; Karakassis, I.; Simboura, N.; Da, D.; Tsapakis, M. Viral Metagenomic Content Reflects Seawater Ecological Quality in the Coastal Zone. Viruses 2020, 12, 806. [Google Scholar] [CrossRef] [PubMed]
- Warwick-Dugdale, J.; Buchholz, H.H.; Allen, M.J.; Temperton, B. Host-hijacking and planktonic piracy: How phages command the microbial high seas. Virol. J. 2019, 16, 15. [Google Scholar] [CrossRef] [Green Version]
- Mojica, K.D.A.; Huisman, J.; Wilhelm, S.W.; Brussaard, C.P.D. Latitudinal variation in virus-induced mortality of phytoplankton across the North Atlantic Ocean. ISME J. 2016, 10, 500–513. [Google Scholar] [CrossRef] [Green Version]
- Suttle, C.A. The Significance of Viruses to Mortlaity in Aquatic Microbial Communities. Microbiol. Ecol. 1994, 28, 237–243. [Google Scholar] [CrossRef]
- Jover, L.F.; Effler, T.C.; Buchan, A.; Wilhelm, S.W.; Weitz, J.S. The elemental composition of virus particles: Implications for marine biogeochemical cycles. Nat. Rev. Microbiol. 2014, 12, 519–528. [Google Scholar] [CrossRef]
- Roux, S.; Brum, J.R.; Dutilh, B.E.; Sunagawa, S.; Duhaime, M.B.; Loy, A.; Poulos, B.T.; Solonenko, N.; Lara, E.; Poulain, J.; et al. Ecogenomics and biogeochemical impacts of uncultivated globally abundant ocean viruses. bioRxiv 2016, 537, 053090. [Google Scholar] [CrossRef] [Green Version]
- Suttle, C.A. Marine viruses—Major players in the global ecosystem. Nat. Rev. Microbiol. 2007, 5, 801–812. [Google Scholar] [CrossRef]
- Wilhelm, S.W.; Suttle, C.A. Viruses and Nutrient Cycles in the Sea. Bioscience 1999, 49, 781–788. [Google Scholar] [CrossRef] [Green Version]
- Weitz, J.S.; Stock, C.A.; Wilhelm, S.W.; Bourouiba, L.; Coleman, M.L.; Buchan, A.; Follows, M.J.; Fuhrman, J.A.; Jover, L.F.; Lennon, J.T.; et al. A multitrophic model to quantify the effects of marine viruses on microbial food webs and ecosystem processes. ISME J. 2015, 9, 1352–1364. [Google Scholar] [CrossRef] [Green Version]
- Fuhrman, J.A. Marine viruses and their biogeochemical and ecological effects. Nature 1999, 399, 541–548. [Google Scholar] [CrossRef] [PubMed]
- Rosenwasser, S.; Ziv, C.; van Creveld, S.G.; Vardi, A. Virocell Metabolism: Metabolic Innovations During Host???Virus Interactions in the Ocean. Trends Microbiol. 2016, 24, 821–832. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, M.B.; Lindell, D.; Lee, J.A.; Thompson, L.R.; Bielawski, J.P.; Chisholm, S.W. Prevalence and evolution of core photosystem II genes in marine cyanobacterial viruses and their hosts. PLoS Biol. 2006, 4, 1344–1357. [Google Scholar] [CrossRef] [PubMed]
- Goldsmith, D.B.; Crosti, G.; Dwivedi, B.; McDaniel, L.D.; Varsani, A.; Suttle, C.A.; Weinbauer, M.G.; Sandaa, R.A.; Breitbart, M. Development of phoH as a novel signature gene for assessing marine phage diversity. Appl. Environ. Microbiol. 2011, 77, 7730–7739. [Google Scholar] [CrossRef] [Green Version]
- Crummett, L.T.; Puxty, R.J.; Weihe, C.; Marston, M.F.; Martiny, J.B.H. The genomic content and context of auxiliary metabolic genes in marine cyanomyoviruses. Virology 2016, 499, 219–229. [Google Scholar] [CrossRef]
- Amaral-Zettler, L.A.; Rocca, J.D.; Lamontagne, M.G.; Dennett, M.R.; Gast, R.J. Changes in microbial community structure in the wake of Hurricanes Katrina and Rita. Environ. Sci. Technol. 2008, 42, 9072–9078. [Google Scholar] [CrossRef]
- Steichen, J.L.; Labonté, J.M.; Windham, R.; Hala, D.; Kaiser, K.; Setta, S.; Faulkner, P.C.; Bacosa, H.; Yan, G.; Kamalanathan, M.; et al. Microbial, Physical, and Chemical Changes in Galveston Bay Following an Extreme Flooding Event, Hurricane Harvey. Front. Mar. Sci. 2020, 7. [Google Scholar] [CrossRef]
- Mason, O.U.; Hazen, T.C.; Borglin, S.; Chain, P.S.G.; Dubinsky, E.A.; Fortney, J.L.; Han, J.; Holman, H.Y.N.; Hultman, J.; Lamendella, R.; et al. Metagenome, metatranscriptome and single-cell sequencing reveal microbial response to Deepwater Horizon oil spill. ISME J. 2012, 6, 1715–1727. [Google Scholar] [CrossRef]
- Williamson, K.E.; Harris, J.V.; Green, J.C.; Rahman, F.; Chambers, R.M. Stormwater runoff drives viral community composition changes in inland freshwaters. Front. Microbiol. 2014, 5, 105. [Google Scholar] [CrossRef] [Green Version]
- Green, J.C.; Rahman, F.; Saxton, M.A.; Williamson, K.E. Quantifying aquatic viral community change associated with stormwater runoff in a wet retention pond using metagenomic time series data. Aquat. Microb. Ecol. 2018, 81, 19–35. [Google Scholar] [CrossRef] [Green Version]
- Du, J.; Park, K.; Dellapenna, T.M.; Clay, J.M. Dramatic hydrodynamic and sedimentary responses in Galveston Bay and adjacent inner shelf to Hurricane Harvey. Sci. Total Environ. 2019, 653, 554–564. [Google Scholar] [CrossRef] [PubMed]
- Walker, L.M.; Montagna, P.A.; Hu, X.; Wetz, M.S. Timescales and Magnitude of Water Quality Change in Three Texas Estuaries Induced by Passage of Hurricane Harvey. Estuaries Coasts 2021, 44, 960–971. [Google Scholar] [CrossRef]
- Suttle, C.A.; Chan, A.M.; Cottrell, M.T. Use of ultrafiltration to isolate viruses from seawater which are pathogens of marine phytoplankton. Appl. Environ. Microbiol. 1991, 57, 721–726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Green, M.R.; Sambrook, J. Isolation of high-molecular-weight DNA using organic solvents. Cold Spring Harb. Protoc. 2017, 2017, 356–359. [Google Scholar] [CrossRef] [Green Version]
- Bushnell, B.; Rood, J.; Singer, E. BBMerge—Accurate paired shotgun read merging via overlap. PLoS ONE 2017, 12, e0185056. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Liu, C.M.; Luo, R.; Sadakane, K.; Lam, T.W. MEGAHIT: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roux, S.; Enault, F.; Hurwitz, B.L.; Sullivan, M.B. VirSorter: Mining viral signal from microbial genomic data. PeerJ 2015, 3, e985. [Google Scholar] [CrossRef]
- Guo, J.; Bolduc, B.; Zayed, A.A.; Varsani, A.; Dominguez-huerta, G.; Delmont, T.O.; Pratama, A.A.; Gazitúa, M.C.; Vik, D.; Sullivan, M.B.; et al. VirSorter2: A multi-classifier, expert-guided approach to detect diverse DNA and RNA viruses. Microbiome 2021, 9, 37. [Google Scholar] [CrossRef]
- Gruber-Vodicka, H.R.; Seah, B.K.B.; Pruesse, E. phyloFlash: Rapid Small-Subunit rRNA Profiling and Targeted Assembly from Metagenomes. mSystems 2020, 5. [Google Scholar] [CrossRef]
- Hyatt, D.; Chen, G.-L.; LoCascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 2010, 11, 119. [Google Scholar] [CrossRef] [Green Version]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2014, 12, 59–60. [Google Scholar] [CrossRef] [PubMed]
- Huson, D.H.; Beier, S.; Flade, I.; Górska, A.; El-Hadidi, M.; Mitra, S.; Ruscheweyh, H.-J.; Tappu, R. MEGAN community edition-interactive exploration and analysis of large-scale microbiome sequencing data. PLoS Comput. Biol. 2016, 12, e1004957. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oksanen, J.; Blanchet, F.G.; Friendly, M.; Kindt, R.; Legendre, P.; McGlinn, D.; Minchin, P.R.; O’Hara, R.B.; Simpson, G.L.; Solymos, P.; et al. vegan: Community Ecology Package 2020. Available online: https://cran.r-project.org/web/packages/vegan/index.html (accessed on 6 March 2022).
- Bolduc, B.; Jang, H.B.; Doulcier, G.; You, Z.Q.; Roux, S.; Sullivan, M.B. vConTACT: An iVirus tool to classify double-stranded DNA viruses that infect Archaea and Bacteria. PeerJ 2017, 2017, e3243. [Google Scholar] [CrossRef] [Green Version]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software Environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016; ISBN 978-3-319-24277-4. [Google Scholar]
- Vavrek, M.J. fossil: Palaeoecological and palaeogeographical analysis tools. Palaeontol. Electron. 2011, 14. Available online: https://www.uv.es/pe/2011_1/238/238.pdf (accessed on 6 March 2022).
- Maechler, M.; Rousseeuw, P.; Struyf, A.; Hubert, M.; Hornik, K. cluster: Cluster Analysis Basics and Extensions 2021. Available online: https://cran.r-project.org/web/packages/cluster/citation.html (accessed on 6 March 2022).
- Warnes, G.R.; Bolker, B.; Bonebakker, L.; Gentleman, R.; Liaw, W.H.A.; Lumley, T.; Maechler, M.; Magnusson, A.; Moeller, S.; Schwartz, M.; et al. Package “gplots”: Various R Programming Tools for Plotting Data, R Packag. version 2.17.0. 2016; 1–68.
- Suzuki, R.; Terada, Y.; Shimodaira, H. Package ‘pvclust’. 2019. [Google Scholar]
- Paulson, J.N.; Colin Stine, O.; Bravo, H.C.; Pop, M. Differential abundance analysis for microbial marker-gene surveys. Nat. Methods 2013, 10, 1200–1202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lang, D.T. The CRAN team RCurl: General Network (HTTP/FTP/...) Client Interface for R. R package version 1.95-4.7. 2015.
- Vázquez-Castellanos, J.F. Diversity analysis in viral metagenomes. Methods Mol. Biol. 2018, 1838, 203–230. [Google Scholar] [CrossRef] [PubMed]
- Kieft, K.; Zhou, Z.; Anantharaman, K. VIBRANT: Automated recovery, annotation and curation of microbial viruses, and evaluation of viral community function from genomic sequences. Microbiome 2020, 8, 90. [Google Scholar] [CrossRef]
- Patel, A.; Noble, R.T.; Steele, J.A.; Schwalbach, M.S.; Hewson, I.; Fuhrman, J.A. Virus and prokaryote enumeration from planktonic aquatic environments by epifluorescence microscopy with SYBR Green I. Nat. Protoc. 2007, 2, 269–276. [Google Scholar] [CrossRef] [Green Version]
- Cai, L.; Yang, Y.; Jiao, N.; Zhang, R. Evaluation of tangential flow filtration for the concentration and separation of bacteria and viruses in contrasting marine environments. PLoS ONE 2015, 10, e0136741. [Google Scholar] [CrossRef]
- John, S.G.; Mendez, C.B.; Deng, L.; Poulos, B.; Kauffman, A.K.M.; Kern, S.; Brum, J.; Polz, M.F.; Boyle, E.A.; Sullivan, M.B. A simple and efficient method for concentration of ocean viruses by chemical flocculation. Environ. Microbiol. Rep. 2011, 3, 195–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aziz, R.K.; Bartels, D.; Best, A.; DeJongh, M.; Disz, T.; Edwards, R.A.; Formsma, K.; Gerdes, S.; Glass, E.M.; Kubal, M.; et al. The RAST Server: Rapid annotations using subsystems technology. BMC Genom. 2008, 9, 75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katoh, K. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Labonté, J.M.; Hallam, S.J.; Suttle, C.A. Previously unknown evolutionary groups dominate the ssDNA gokushoviruses in oxic and anoxic waters of a coastal marine environment. Front. Microbiol. 2015, 6, 315. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Keegan, K.P.; Glass, E.M.; Meyer, F. MG-RAST, a Metagenomics Service for Analysis of Microbial Community Structure and Function BT—Microbial Environmental Genomics (MEG); Martin, F., Uroz, S., Eds.; Springer: New York, NY, USA, 2016; pp. 207–233. ISBN 978-1-4939-3369-3. [Google Scholar]
- Knutson, T.R.; Sirutis, J.J.; Zhao, M.; Tuleya, R.E.; Bender, M.; Vecchi, G.A.; Villarini, G.; Chavas, D. Global projections of intense tropical cyclone activity for the late twenty-first century from dynamical downscaling of CMIP5/RCP4.5 scenarios. J. Clim. 2015, 28, 7203–7224. [Google Scholar] [CrossRef] [Green Version]
- Weitz, J.S.; Wilhelm, S.W. Ocean viruses and their effects on microbial communities and biogeochemical cycles. F1000 Biol. Rep. 2012, 4, 17. [Google Scholar] [CrossRef] [Green Version]
- Winget, D.M.; Wommack, K.E. Diel and daily fluctuations in virioplankton production in coastal ecosystems. Environ. Microbiol. 2009, 11, 2904–2914. [Google Scholar] [CrossRef]
- Wilhelm, S.W.; Brigden, S.M.; Suttle, C.A. A dilution technique for the direct measurement of viral production: A comparison in stratified and tidally mixed coastal waters. Microb. Ecol. 2002, 43, 168–173. [Google Scholar] [CrossRef]
- Mojica, K.D.A.; Brussaard, C.P.D. Factors affecting virus dynamics and microbial host-virus interactions in marine environments. FEMS Microbiol. Ecol. 2014, 89, 495–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dayaram, A.; Galatowitsch, M.L.; Argüello-Astorga, G.R.; van Bysterveldt, K.; Kraberger, S.; Stainton, D.; Harding, J.S.; Roumagnac, P.; Martin, D.P.; Lefeuvre, P.; et al. Diverse circular replication-associated protein encoding viruses circulating in invertebrates within a lake ecosystem. Infect. Genet. Evol. 2016, 39, 304–316. [Google Scholar] [CrossRef] [PubMed]
- Ignacio-Espinoza, J.C.; Sullivan, M.B. Phylogenomics of T4 cyanophages: Lateral gene transfer in the “core” and origins of host genes. Environ. Microbiol. 2012, 14, 2113–2126. [Google Scholar] [CrossRef]
- Sullivan, M.B.; Coleman, M.L.; Weigele, P.; Rohwer, F.; Chisholm, S.W. Three Prochlorococcus cyanophage genomes: Signature features and ecological interpretations. PLoS Biol. 2005, 3, 0790–0806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Temperton, B.; Thrash, J.C.; Schwalbach, M.S.; Vergin, K.L.; Landry, Z.C.; Ellisman, M.; Deerinck, T.; Sullivan, M.B.; Giovannoni, S.J. Abundant SAR11 viruses in the ocean. Nature 2013, 494, 357–360. [Google Scholar] [CrossRef]
- Roux, S.; Enault, F.; Robin, A.; Ravet, V.; Personnic, S.; Theil, S.; Colombet, J.; Sime-Ngando, T.; Debroas, D. Assessing the diversity and specificity of two freshwater viral communities through metagenomics. PLoS ONE 2012, 7, e33641. [Google Scholar] [CrossRef] [Green Version]
- Labonté, J.M.; Suttle, C.A. Previously unknown and highly divergent ssDNA viruses populate the oceans. ISME J. 2013, 7, 2169–2177. [Google Scholar] [CrossRef]
- Angly, F.E.; Felts, B.; Breitbart, M.; Salamon, P.; Edwards, R.A.; Carlson, C.; Chan, A.M.; Haynes, M.; Kelley, S.; Liu, H.; et al. The marine viromes of four oceanic regions. PLoS Biol. 2006, 4, 2121–2131. [Google Scholar] [CrossRef]
- Marine, R.; McCarren, C.; Vorrasane, V.; Nasko, D.; Crowgey, E.; Polson, S.W.; Wommack, K.E. Caught in the middle with multiple displacement amplification: The myth of pooling for avoiding multiple displacement amplification bias in a metagenome. Microbiome 2014, 2, 3. [Google Scholar] [CrossRef] [Green Version]
- Labonté, J.M.; Suttle, C.A. Metagenomic and whole-genome analysis reveals new lineages of gokushoviruses and biogeographic separation in the sea. Front. Microbiol. 2013, 4, 404. [Google Scholar] [CrossRef]
- Tucker, K.P.; Parsons, R.; Symonds, E.M.; Breitbart, M. Diversity and distribution of single-stranded DNA phages in the North Atlantic Ocean. ISME J. 2011, 5, 822–830. [Google Scholar] [CrossRef] [Green Version]
- Williamson, S.J.; Rusch, D.B.; Yooseph, S.; Halpern, A.L.; Heidelberg, K.B.; Glass, J.I.; Pfannkoch, C.A.; Fadrosh, D.; Miller, C.S.; Sutton, G.; et al. The sorcerer II global ocean sampling expedition: Metagenomic characterization of viruses within aquatic microbial samples. PLoS ONE 2008, 3, e1456. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.M.; Labonté, J.M.; Brown, J.; Record, N.R.; Poulton, N.J.; Sieracki, M.E.; Logares, R.; Stepanauskas, R. Single Cell Genomics Reveals Viruses Consumed by Marine Protists. Front. Microbiol. 2020, 11, 524828. [Google Scholar] [CrossRef]
- Parada, V.; Herndl, G.J.; Weinbauer, M.G. Viral burst size of heterotrophic prokaryotes in aquatic systems. J. Mar. Biol. Assoc. U. K. 2006, 86, 613–621. [Google Scholar] [CrossRef]
- Thingstad, T.F. Elements of a theory for the mechanisms controlling abundance, diversity, and biogeochemical role of lytic bacterial viruses in aquatic systems. Limnol. Oceanogr. 2000, 45, 1320–1328. [Google Scholar] [CrossRef]
- Yan, G.; Labonté, J.M.; Quigg, A.; Kaiser, K. Hurricanes Accelerate Dissolved Organic Carbon Cycling in Coastal Ecosystems. Front. Mar. Sci. 2020, 7. [Google Scholar] [CrossRef] [Green Version]
- Gregory, A.C.; Zayed, A.A.; Sunagawa, S.; Wincker, P.; Sullivan, M.B.; Ferland, J.; Kandels, S.; Liu, Y.; Marec, C.; Vik, D. Marine DNA Viral Macro- and Microdiversity from Pole to Pole Article Marine DNA Viral Macro- and Microdiversity from Pole to Pole. Cell 2019, 177, 1109–1123. [Google Scholar] [CrossRef] [Green Version]
- Labonté, J.M.; Swan, B.K.; Poulos, B.; Luo, H.; Koren, S.; Hallam, S.J.; Sullivan, M.B.; Woyke, T.; Eric Wommack, K.; Stepanauskas, R. Single-cell genomics-based analysis of virus-host interactions in marine surface bacterioplankton. ISME J. 2015, 9, 2386–2399. [Google Scholar] [CrossRef]
- Hurwitz, B.L.; Brum, J.R.; Sullivan, M.B. Depth-stratified functional and taxonomic niche specialization in the “core” and “flexible” Pacific Ocean Virome. ISME J. 2015, 9, 472–484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hryniewicz, M.; Sirko, A.; Palucha, A.; Bock, A.; Hulanicka, D. Sulfate and thiosulfate transport in Escherichia coli K-12: Identificaton of a gene encoding a novel protein involved in thiosulfate binding. J. Bacteriol. 1990, 172, 3358–3366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wasmund, K.; Mußmann, M.; Loy, A. The life sulfuric: Microbial ecology of sulfur cycling in marine sediments. Environ. Microbiol. Rep. 2017, 9, 323–344. [Google Scholar] [CrossRef] [PubMed]
- Anantharaman, K.; Duhaime, M.B.; Breier, J.A.; Wendt, K.A.; Toner, B.M.; Dick, G.J. Sulfur Oxidation Genes in Diverse Deep-Sea Viruses. Science 2014, 344, 757–760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blattner, F.R.; Plunkett, G., 3rd; Bloch, C.A.; Perna, N.T.; Burland, V.; Riley, M.; Collado-Vides, J.; Glasner, J.D.; Rode, C.K.; Mayhew, G.F.; et al. The complete genome sequence of Escherichia coli K-12. Science 1997, 277, 1453–1462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, D.; Henrichs, S.M.; Guo, L. Distributions of nutrients, dissolved organic carbon and carbohydrates in the western Arctic Ocean. Cont. Shelf Res. 2006, 26, 1654–1667. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Dates in 2017 | Station | Shannon Index | Pielou Index | ACE Index |
|---|---|---|---|---|
| 31 July | TAMUG Boat Basin | 2.01 | 0.59 | 33.73 |
| 22 August | TAMUG Boat Basin | 1.57 | 0.45 | 36.91 |
| 4 September | Stn1 | 1.93 | 0.55 | 34.45 |
| Stn4 | 1.73 | 0.49 | 36.01 | |
| Stn7 | 1.72 | 0.49 | 35.13 | |
| Stn10 | 1.70 | 0.47 | 37.08 | |
| 9 September | Stn1 | 1.86 | 0.51 | 38.47 |
| Stn4 | 2.16 | 0.60 | 37.69 | |
| Stn7 | 1.92 | 0.54 | 37.44 | |
| Stn10 | 2.07 | 0.58 | 36.43 | |
| 16 September | Stn1 | 2.08 | 0.58 | 36.21 |
| Stn4 | 2.22 | 0.62 | 37.46 | |
| Stn7 | 2.09 | 0.58 | 37.42 | |
| Stn10 | 2.14 | 0.60 | 35.00 | |
| 28 September | Stn1 | 2.23 | 0.62 | 38.25 |
| Stn4 | 2.08 | 0.58 | 40.54 | |
| Stn7 | 1.96 | 0.55 | 40.75 | |
| Stn10 | 1.85 | 0.53 | 40.51 |
| Dates in 2017 | Station | Lytic Viruses | Prophages |
|---|---|---|---|
| 4 September | Stn1 | 28.7 | 0.0 |
| Stn4 | 44.6 | 0.0 | |
| Stn7 | 69.2 | 0.5 | |
| Stn10 | 47.8 | 3.0 | |
| 9 September | Stn1 | 58.6 | 0.6 |
| Stn4 | 61.3 | 1.9 | |
| Stn7 | 62.0 | 1.6 | |
| Stn10 | 37.5 | 1.6 | |
| 16 September | Stn1 | 147.1 | 1.1 |
| Stn4 | 152.7 | 5.0 | |
| Stn7 | 116.8 | 2.6 | |
| Stn10 | 199.6 | 0.6 | |
| 28 September | Stn1 | 184.8 | 2.2 |
| Stn4 | 165.3 | 4.6 | |
| Stn7 | 199.5 | 0.0 | |
| Stn10 | 144.0 | 0.2 | |
| 31 July | Pre-Harvey1 | 39.5 | 0.0 |
| 22 August | Pre-Harvey2 | 28.5 | 0.0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Woods, A.C.; Walker, J.R.; Jackson, C.D.; Labonté, J.M. Record-Breaking Rain Event Altered Estuarine Viral Assemblages. Microorganisms 2022, 10, 729. https://doi.org/10.3390/microorganisms10040729
Woods AC, Walker JR, Jackson CD, Labonté JM. Record-Breaking Rain Event Altered Estuarine Viral Assemblages. Microorganisms. 2022; 10(4):729. https://doi.org/10.3390/microorganisms10040729
Chicago/Turabian StyleWoods, Alaina C., Jordan R. Walker, Cameron D. Jackson, and Jessica M. Labonté. 2022. "Record-Breaking Rain Event Altered Estuarine Viral Assemblages" Microorganisms 10, no. 4: 729. https://doi.org/10.3390/microorganisms10040729
APA StyleWoods, A. C., Walker, J. R., Jackson, C. D., & Labonté, J. M. (2022). Record-Breaking Rain Event Altered Estuarine Viral Assemblages. Microorganisms, 10(4), 729. https://doi.org/10.3390/microorganisms10040729